Abstract
Recent studies have suggested that autophagy plays a prosurvival role in ischemic preconditioning (IPC). This study was taken to assess the linkage between autophagy and endoplasmic reticulum (ER) stress during the process of IPC. The effects of IPC on ER stress and neuronal injury were determined by exposure of primary cultured murine cortical neurons to 30 min of OGD 24 h prior to a subsequent lethal OGD. The effects of IPC on ER stress and ischemic brain damage were evaluated in rats by a brief ischemic insult followed by permanent focal ischemia (PFI) 24 h later using the suture occlusion technique. The results showed that both IPC and lethal OGD increased the LC3-II expression and decreased p62 protein levels, but the extent of autophagy activation was varied. IPC treatment ameliorated OGD-induced cell damage in cultured cortical neurons, whereas 3-MA (5–20 mM) and bafilomycin A1 (75–150 nM) suppressed the neuroprotection induced by IPC. 3-MA, at the dose blocking autophagy, significantly inhibited IPC-induced HSP70, HSP60 and GRP78 upregulation; meanwhile, it also aggregated the ER stress and increased activated caspase-12, caspase-3 and CHOP protein levels both in vitro and in vivo models. The ER stress inhibitor Sal (75 pmol) recovered IPC-induced neuroprotection in the presence of 3-MA. Rapamycin 50–200 nM in vitro and 35 pmol in vivo 24 h before the onset of lethal ischemia reduced ER stress and ischemia-induced neuronal damage. These results demonstrated that pre-activation of autophagy by ischemic preconditioning can boost endogenous defense mechanisms to upregulate molecular chaperones, and hence reduce excessive ER stress during fatal ischemia.
Introduction
In recent years, the mechanisms of ischemic preconditioning (IPC) have been systematically studied and several molecular regulatory pathways participating in preconditioning have been discovered, but definitive mechanisms underlying IPC remain incompletely understood to date.Citation1-Citation4 Autophagy is a regulated process for degrading and recycling of cellular constituents, participating in organelle turnover and bioenergetic management during starvation.Citation5-Citation8 Autophagy and proteasomes may work together to degrade misfolded proteins that have been retained in the cytoplasm or endoplasmic reticulum (ER).Citation9,Citation10 However, autophagy sometimes concurs with apoptosis and plays important roles in both normal tissue development and a variety of diseases.Citation11 Recent studies have reported the activation of autophagy following ischemia insult and preconditioning, but the contribution of autophagy to neuronal death/survival is still being debated.Citation12-Citation14 In earlier studies, we found that both IPC and fatal cerebral ischemia induced autophagy activation, but the extent and persistence of autophagy activation were varied. Treatment with autophagy inhibitors before IPC significantly suppressed the neuroprotection induced by IPC, while rapamycin, an autophagy activator, could mimic the IPC’s neuroprotection. These results demonstrate that autophagy activation during IPC offers remarkable tolerance to subsequent fatal ischemic insult.Citation15,Citation16 However, to date, no evidence has shown if autophagy takes part in the oxygen glucose deprivation (OGD) preconditioning in primary cultured cortical neurons and, if so, whether autophagy regulates ER stress during IPC.
Cellular stress conditions, such as glucose deprivation, depletion of ER Ca2+ stores, exposure to free radicals, and accumulation of unfolded/misfolded proteins, disrupt the proper function of the ER and initiates cell stress response, the unfolded protein response (UPR).Citation17-Citation19 UPR is characterized by the activation of three ER transmembrane effector proteins: PKR-like ER kinase (PERK), inositol requiring enzyme 1 (IRE1), and the activating transcription factor-6 (ATF-6). One major pathway of UPR is the suppression of most protein translations through phosphorylation of eukaryotic translation initiation factor 2 subunit a (eIf2a) by PERK. Another pathway is the upregulated expression of ER-localized molecular chaperones, such as glucose-regulated protein 78 (GRP78/Bip), GRP94 and other molecular chaperones like heat shock proteins (HSPs). However, prolonged ER stress may activate an ER stress-dependent apoptotic pathway by induction of caspases-12 and CHOP (C/EBP homologous protein, growth arrest and DNA damage inducible gene 153, GADD153).
Mounting evidence shows that ER stress plays an important role in the pathological process of fatal cerebral ischemia and preconditioning. Fatal cerebral ischemia induces ER stress characterized by caspase-12 activation, and many caspase-12-positive cells exhibited DNA fragmentation.Citation20,Citation21 On the other hand, IPC might regulate ER molecular chaperones or HSPs to reduce severe ER stress occurring in PFI.Citation22-Citation25 All these reports support the hypothesis that severe ER stress is involved in the injury of neurons during cerebral ischemia by inducing ER-stress dependent apoptosis, while IPC may upregulate molecular chaperones to lessen severe ER stress in fatal ischemia.
Several studies suggested a crosstalk between autophagy and ER function in different pathological processes.Citation26-Citation28 However, no relationship has been assessed between autophagy and ER stress during the process of IPC. This study sought to evaluate if activation of autophagy during IPC reduced fatal ischemia-induced excessive ER stress.
Results
Preconditioning-induced autophagy activation contributes to neuroprotection
To examine autophagic activity after OGD preconditioning, we examined the protein levels of LC3 and p62 in the cortical neurons. Western blot analysis revealed that LC3-II (16 kD) was progressively upregulated after IPC 1, 3 and 6 h. OGD also elicited a minor elevation of LC3-II and reached statistical significance 6 h after OGD (; p < 0.05 or p < 0.01 vs. the control group). In contrast, IPC+OGD induced more robust increases in LC3-II levels at 1 and 3 h but not at 6 h (p < 0.05 or p < 0.01 vs. the OGD alone group).
Figure 1. Preconditioning-induced autophagy activation contributes to neuroprotection in primary cultured cortical neurons. In IPC groups, cortical neurons were subjected to OGD for 30 min and then reperfused for 1–6 h. In OGD groups, neurons were deprived of oxygen and glucose for 1–6 h. In IPC+OGD groups, neurons were preconditioned for 30 min and exposed to OGD 24 h later for 1?6 h. The protein levels of LC3 and p62 were detected with immunoblotting. Levels of β-actin protein were used as the loading control. Quantitative analysis was performed with Sigma Scan Pro 5. (A) Protein expression of LC3. (B) Protein expression of p62. (C and D) 3-MA abolished IPC mediated neuroprotection in primary cultured cortical neurons. Cortical neurons were pre-incubated with 3-MA (5–20 mM) or BAF (37.5–150 nM) 30 min before the onset of IPC. After 24 h of reperfusion, a lethal OGD was induced by deprivation of oxygen and glucose for 6 h. The cell injury was evaluated by morphological changes and LDH leakage and MTT assay. (C) Cell morphological changes were observed with phase contrast microscopy. Bar = 50 μm. (D) 3-MA increased the LDH leakage of the cortical neurons. 3-MA (S) = 3-MA 5 mM; 3-MA (M) = 3-MA 10 mM; 3-MA (L) = 3-MA 20 mM. (E) Bafilomycin A1 (BAF) abolished the IPC elicited neuroprotection in cortical neurons. BAF (S) = BAF 37.5 nM; BAF (M) = BAF 75 nM; BAF (L) = BAF 150 nM. Bar represents mean ± SD, n = 6. *p < 0.05, **p < 0.01 vs. control group; #p < 0.05, ##p < 0.01 compared with the OGD group at each time point; $$p < 0.01 vs. IPC+OGD group.
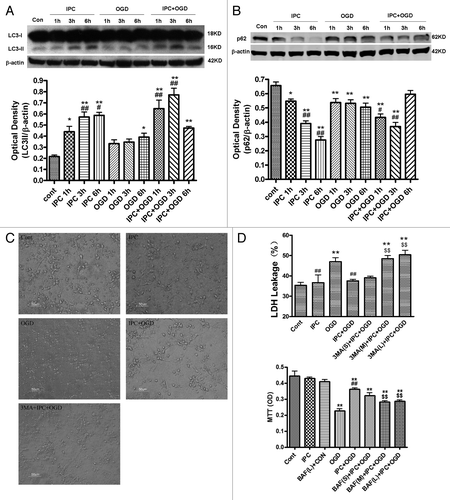
p62 is associated with mature autophagic vesicles and is degraded within autophagosomes. Western blot analysis revealed that the p62 protein levels were progressively reduced after IPC 1–6 h. OGD also slightly decreased p62 levels (; p < 0.05 or p < 0.01 vs. the control group). In contrast, IPC+OGD induced a more dramatic reduction in p62 levels at 1 and 3 h but not at 6 h (; p < 0.05 or p < 0.01 vs. the OGD alone group). These results suggest that autophagy was activated after both preconditioning and lethal OGD condition, but the extent of autophagy activation in preconditioning is stronger than that induced by lethal OGD.
To determine the contribution of autophagic mechanisms to the neuroprotection elicited by ischemic preconditioning, we examined the effects of 3-MA (5–20 mM) and BAF (37.5–150 nM) on the OGD preconditioning model in primary cultured cortical neurons. Lethal OGD for 6 h induced significant cell injury and marked LDH leakage in the culture medium (p < 0.01 vs. the control group; ). IPC greatly attenuated lethal OGD induced cell injury and LDH release (p < 0.01 vs. the OGD group), whereas 3-MA, 10 mM and 20 mM, or BAF, 75 and 150 nM pretreatment recovered the OGD-induced LDH leakage and aggravated cell damage (p < 0.01 vs. the IPC+OGD group). We then determined if 3-MA has a toxic effect on cortical neurons under normal conditions, the results showed that exposure of neurons to 3-MA 10 mM for 48 h has no effect on cell viability (data not shown).
To examine if 3-MA blocks the autophagy activation after IPC treatment, we examined the protein levels of LC3 and p62 in cortical neurons after 3-MA treatment. In and, the amount of LC3-I (18 kD) appears to be saturated, so we normalized the LC3-II (16 kD) levels to β-actin and compared LC3 levels in different groups rather than use the ratio of LC3-II to LC3-I. Western blot analysis revealed that the LC3-II was significantly upregulated after IPC and IPC+OGD 3 h, but not in OGD alone group (; p < 0.01 vs. the control group and OGD group). The induction of LC3-II by preconditioning in cortical neurons was significantly blunted by 3-MA (p < 0.01 vs. IPC+OGD group). Similarly, p62 was downregulated only in IPC and IPC+OGD groups, but not in OGD group (; p < 0.01 vs. the control group and OGD group), while 3-MA attenuated IPC and elicited p62 downregulation (p < 0.01 vs. IPC+OGD group).
Figure 2. 3-MA inhibited IPC-induced autophagy activation in cortical neurons. Cortical neurons were pre-incubated with 3-MA (10 mM) 30 min before the onset of IPC (OGD for 30 min). After 24 h of reperfusion, a lethal OGD was induced by deprivation of oxygen and glucose for 3 h. The protein levels of LC3 and p62 were detected with immunoblotting. Levels of β-actin protein were used as the loading control. Quantitative analysis was performed with Sigma Scan Pro 5. (A) Protein expression of LC3. (B) Protein expression of p62. Bar represents mean ± SD, n = 6. *p < 0.05, **p < 0.01 compared with the control group; #p < 0.05, ##p < 0.01 compared with the OGD group; $$p < 0.05, $$p < 0.01 compared with IPC+OGD group. (C) Cortical neurons were fixed with 4% paraformaldehyde and processed for immunofluorescence. Representative images of cortical neurons were stained with DAPI (blue), and antibodies against LC3 (red) and MAP-2 (green). Note, LC3 immunostaining was upregulated in IPC and IPC+OGD groups. Bar = 10 μm. (D): The formation of autophagic vacuoles is suggested by punctate MDC staining. Cells were labeled with MDC and observed with a fluorescent microscope. Bar = 10 μm. Microphotographs were shown as representative results from three independent experiments.
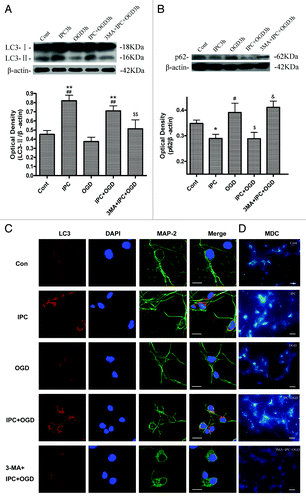
We further evaluated autophagy activation by analysis of the LC3 immunofluorescence and MDC-labeled vacuoles in the cortical neurons. In the control group, LC3 immunoreactivity in cortical neurons was very low. There was a marked induction of punctate LC3 immunoreactivity in some cortical neurons in IPC 3 h and IPC+OGD 3 h groups, suggesting induction of autophagy in these groups, but in the 3-MA+IPC+OGD 3 h group, the LC3 immunoreactivity declined toward basal levels (). The autofluorescent substance MDC has been shown to be a specific marker for autophagic vacuoles (AVs). When cells are viewed with a fluorescence microscope, AVs stained by MDC appear as distinct dot-like structures distributed within the cytoplasm or localizing in the perinuclear regions.Citation29,Citation30 In the IPC and IPC+OGD groups, an increase in the MDC labeled vesicles was observed as indicated by punctate MDC fluorescence (), suggesting an induction of AV formation after preconditioning. But in the 3-MA+IPC+OGD 3 h group, the MDC-labeled vesicles declined toward basal levels.
All these results suggest that autophagy was activated after preconditioning. Inhibiting autophagy activation during IPC by pretreatment of 3-MA abolishes IPC induced neuroprotection.
Preconditioning reduces ER stress-dependent apoptosis through autophagy
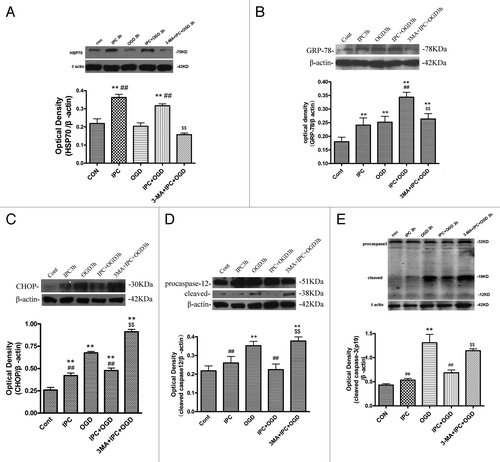
We then evaluated the effects of autophagy on IPC-elicited induction of ER molecular chaperones and HSPs both in vitro and in vivo. In IPC 3 h and IPC+OGD 3 h groups, the protein levels of HSP70 and GRP78 in cortical neurons were higher than that in the control groups (, p < 0.01). The induction of HSP70 and GRP78 in IPC+OGD groups was also stronger than that in the OGD alone group (p < 0.01). Inhibition of autophagy with 3-MA significantly prevented IPC-induced increases in HSP70 and GRP78 in vitro (p < 0.01 vs. the IPC+OGD group). EIF2α, CHOP, caspase-12 and caspase-3 have been proposed as important markers of ER stress-dependent apoptosis, so the present study evaluated the effects of autophagy on eIF2α, CHOP, caspase-12 and caspase-3 after IPC. In OGD 3 h groups, the protein levels of CHOP, cleaved caspase-12 and caspase-3 in cortical neurons were significantly upregulated compared with that in the control groups (, p < 0.01), but in IPC+OGD group, these protein levels significantly decreased compared with OGD alone group (p < 0.01). However, 3-MA treatment significantly resumed CHOP, cleaved caspase-12 and caspase-3 levels in cortical neurons (p < 0.01 vs. the IPC+OGD group). To determine the incidence of apoptosis, we stained the nuclei of treated cortical neurons with Hoechst 33258. As shown in Figure S1, OGD induced condensation of chromatin and fragmentation of the nuclei of cortical neurons (p < 0.01 vs. control group). IPC significantly improved the morphological changes and reduced the apoptotic rate in cortical neurons (p < 0.01 vs. OGD group), while 3-MA aggravated cell apoptosis in cortical neurons (p < 0.01 vs. IPC+OGD group).
Figure 3. 3-MA decreased HSP70 and GRP78 expression, while increasing CHOP, cleaved caspase-12 and caspase-3 expression in cortical neurons. Cortical neurons were treated as described in the legend to . The protein levels of HSP70, GRP78, CHOP, caspase-12 and caspase-3 were detected with immunoblotting. Levels of β-actin protein were used as the loading control. Quantitative analysis was performed with Sigma Scan Pro 5. (A): Protein expression of HSP70. (B) Protein expression of GRP78. (C) Protein expression of CHOP. (D) Protein expression of caspase-12. (E) Protein expression of caspase-3. Bar represents mean ± SD, n = 6. *p < 0.05, **p < 0.01 compared with the control group; #p < 0.05, ##p < 0.01 compared with the OGD group; $p < 0.05, $$p < 0.01 compared with IPC+OGD group.
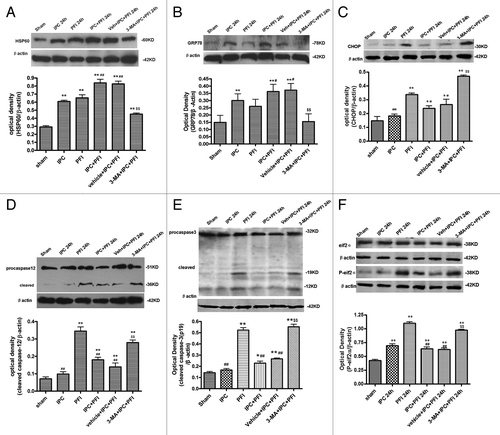
Similar to in vitro results, 24 h after ischemia, the protein levels of HSP60 and GRP78 in ischemic cortex were higher than that in the sham-operated rats (p < 0.01, ). The expression of HSP60 and GRP78 in IPC+PFI and vehicle+IPC+PFI groups further increased compared with PFI alone group (p < 0.01). However, 3-MA significantly prevented IPC-induced increases in HSP60 and GRP78 (p < 0.01 vs. the vehicle+IPC+PFI group). Twenty-four hours after ischemia, the protein levels of p-eIF2α, CHOP, cleaved caspase-12 and caspase-3 in PFI rats were higher than that in the sham-operated rats (, p < 0.05 or 0.01), but these proteins in IPC+PFI and vehicle+IPC+PFI groups significantly decreased compared with that in PFI alone group (p < 0.01). Moreover, inhibiting autophagy with 3-MA significantly prevented IPC-induced downregulation of p-eIF2α, CHOP, cleaved caspase-12 and caspase-3 (p < 0.01 vs. the vehicle+IPC+PFI group). The results from both in vivo and in vitro studies suggested that an autophagic mechanism is involved in ER stress response in IPC and PFI.
Figure 4. 3-MA decreased HSP60 and GRP78 expression, while increasing CHOP, caspase-12 and caspase-3 expression in a rat model of ischemic preconditioning. Rats were treated with a single i.c.v. of 3-MA 200 nmol 5 min before the onset of IPC (10 min MCA occlusion). A lethal ischemia was induced by PFI. Rats were killed 24 h after ischemia. Extracts from the ischemic and sham-operated cortex were separated on SDS-PAGEL and protein levels were detected with immunoblotting. Levels of β-actin protein were used as the loading control. Quantitative analysis was performed with Sigma Scan Pro 5. (A) Protein expression of HSP60. (B) Protein expression of GRP78. (C) Protein expression of CHOP. (D) Protein expression of caspase-12. (E) Protein expression of caspase-3. (F) Protein expression of P-eIF2α and eIF2α. Bar represents mean ± SD, n = 6. *p < 0.05, **p < 0.01 compared with the sham-operated group; #p < 0.05, ##p < 0.01 compared with the PFI group; $p < 0.05, $$p < 0.01 compared with the vehicle+IPC+PFI group.
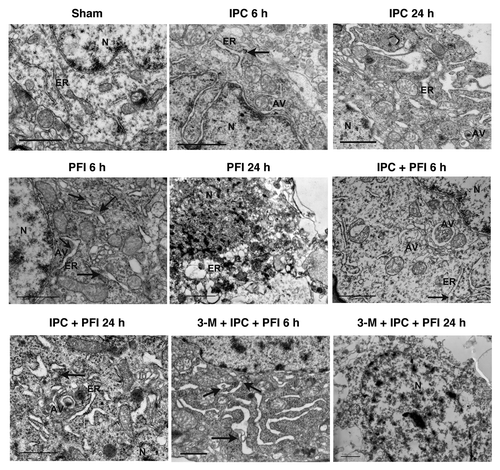
Electron microscopy was then used to evaluate the ER ultrastructural changes in the neurons 6 h and 24 h after IPC, PFI and IPC+PFI treatment. Neurons in the control cortex appeared to be normal with relatively healthy-looking ER, mitochondria and nuclei (). Six and 24 h after IPC, the mitochondria and the nuclei in neurons were also normal without appreciable pathological change, but some swollen ER with medium densities in the ER lumen were consistent with mild protein aggregates (arrows). Some autophagosomes, C-shaped double-membrane structures, and engulfment of cytoplasmic materials by autophagosomes were found, suggesting concurrence of mild ER stress and autophagy activation in IPC groups. In contrast, the neurons in the ischemic cortex of PFI groups displayed apparent injured morphology. Six hours after PFI, the neurons showed dilated and distorted ER, swollen mitochondria, formation of autophagosomes and condensation of chromatin; more protein aggregates were seen within the ER lumen than that observed in IPC groups, indicating severe ER stress in this group. Twenty-four hours after PFI, however, the neurons showed shrunken nuclei, disrupted cell membranes, fragmented ER and formation of numerous vacuoles in the cytoplasm, a mixed feature of apoptosis and necrosis. In the IPC+PFI 6 h and 24 h groups, the neurons displayed dilated ER and swollen mitochondria, yet the cell damage seems much less severe compared with the PFI group. Some protein aggregates within the ER lumen and increased autophagosomes were also found. In addition, we examined the EM morphological changes after drug treatment. In 3-MA+IPC+PFI 6 h groups (3-MA 200 nmol), the neurons showed swollen ER with apparently increased protein aggregates in the ER lumen. Moreover, as ischemia prolonged, in 3-MA+IPC+PFI 24 h groups, the neurons showed disrupted cell membranes, damaged organelles and swollen nuclei. The injurious morphology was similar to that of PFI group. These results suggest that although both preconditioning and fatal ischemia induce ER stress accompanied by autophagy activation at the early phase of ischemia, fatal ischemia induces prolonged and more severe ER stress than preconditioning.
Figure 5. ER ultrastructural changes in the neurons of ischemic cortex after ischemic preconditioning. Rats were treated with a single i.c.v. injection of 3-MA 200 nmol 5 min before the onset of IPC. Lethal ischemia was induced by PFI. Rats were killed 6 h or 24 h after ischemia. Rats were perfused with 4% paraformaldehyde and 0.25% glutaraldehyde and were processed for electron microscopic examination. Photomicrographs represent samples taken from 3 rats in each group. Scale bars: 1 μm. N: nucleus; ER: rough endoplasmic reticulum; AV: autophagosomes; arrows indicate protein aggregates within ER lumen or associated with ER.
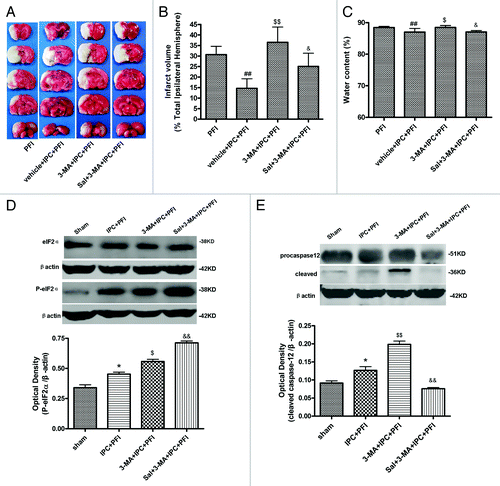
Modulation of ER stress via autophagy produces preconditioning-like effects
The previous and present studies indicated that inhibition of autophagy with 3-MA prevented IPC-induced neuroprotection both in vivoCitation16 and in vitro. To test if this is caused by resumed ER stress after 3-MA treatment, we examined the effects of an ER stress inhibitor Sal (75 pmol) on 3-MA-induced enhancement in neuronal damage in the IPPC+PFI model. The results showed that the IPC treatment significantly reduced the infarct volume and water content following subsequent PFI, as shown in vehicle+IPC+PFI group (, p < 0.01 vs. the PFI group); 3-MA 200 nmol pretreatment completely abolished the protective effects of IPC on infarct volume and brain edema (p < 0.05 or p < 0.01 vs. the vehicle+IPC+PFI group). Sal alone has no significant effect on infarct volume compared with IPC+OGD group (p > 0.05, Fig. S2). In contrast, Sal pretreatment significantly reduced the infarct volume and brain edema (p < 0.05 or p < 0.01 vs. the 3-MA+IPC+PFI group) in the presence of 3-MA. Sal significantly inhibited the dephosphorylation of eIF2α, and abolished the upregulation of cleaved caspase-12 elicited by 3-MA (, p < 0.01 vs. the 3-MA+IPC+PFI group). These results suggest that blocking autophagy increases ischemia-induced ER stress, and thus increases ischemic damage, whereas inhibition of ER stress might lessen 3-MA-induced injury in the IPC model.
Figure 6. Salubrinal (Sal) inhibited 3-MA-induced neuronal damage. Rats were treated with a single i.c.v injection of Sal (75 pmol); 10 min later, 3-MA (200 nmol) was administered 5 min before the onset of IPC (10 min MCA occlusion). Lethal ischemia was induced by PFI. Rats were killed 24 h after ischemia. (A) TTC staining of brain sections. Infarct brain regions display white after TTC staining. (B) Quantitative analysis of brain infarct volume. (C) Sal pretreatment decreased brain edema. (D and E) Extracts from the ischemic and sham-operated cortex were separated on SDS-PAGEL and protein levels were detected with immunoblotting. Levels of β-actin protein were used as the loading control. Quantitative analysis was performed with Sigma Scan Pro 5. (D) Protein expression of P-eIF2α and eIF2α. (E) Protein expression of caspase-12. Bar represents mean ± SD, n = 6. *p < 0.05, **p < 0.01 compared with the sham-operated group; #p < 0.05, ##p < 0.01 compared with PFI group. $p < 0.05, $$p < 0.01 compared with the IPC+PFI group; &p < 0.05, &&p < 0.01 compared with the 3-MA+IPC+PFI group.
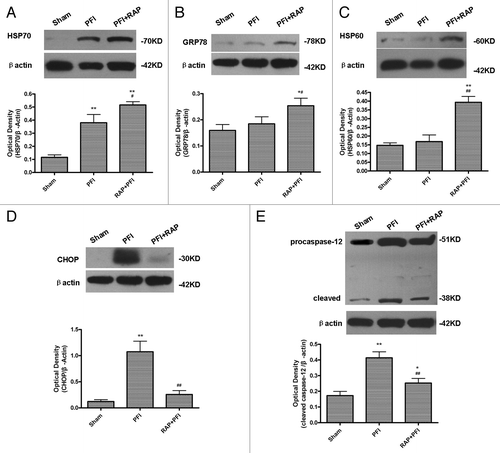
We have previously showed that the autophagy inducer rapamycin could produce pharmacological preconditioning. To explore if rapamycin could mimic the effects of IPC on ER stress, we determined the effects of an autophagy inducer rapamycin (35 pmol in vivo, 50–200 nM in vitro) administered 24 h before the onset of lethal cerebral ischemia or OGD. In cortical neurons, lethal OGD for 6 h induced marked LDH leakage in the culture medium (p < 0.01 vs. the control group, Fig. S3). Rapamycin 50–200 nM dose-dependently attenuated the OGD induced LDH release in the culture medium (p < 0.01 vs. the OGD group). We then examined rapamycin’s effects on ER stress induced by lethal ischemia. The results from in vivo experiments showed that rapamycin alone had no significant effects on protein expression of HSP70, HSP60, GRP78, CHOP and caspase-12 in sham-operated rats (Fig. S4). In the PFI group, HSP70, CHOP and caspase-12 were greatly increased compared with the sham-operated group (p < 0.01 vs. the sham-operated group, ). A single i.c.v. injection of rapamycin 35 pmol before the onset of PFI also increased the expression of HSP70, HSP60, GRP78 (p < 0.01 vs. the PFI group), while it significantly reduced the expression of cleaved caspase-12 and CHOP (p < 0.01 vs. the PFI group). These results imply that autophagy inducers might upregulate molecular chaperones and lessen excessive ER stress to elicit neuroprotection on lethal cerebral ischemia.
Figure 7. Rapamycin increased HSP70, HSP60, GRP78 expression, while decreasing CHOP and cleaved caspase-12 expression in ischemic cortex. Rats were treated with a single i.c.v. injection of rapamycin 35 pmol 24 h before the onset of PFI. PFI animals received an i.c.v. injection of same volume of vehicle. Rats were killed 24 h after ischemia. The extracts from the ischemic and sham-operated cortex were prepared and separated on SDS-PAGEL and protein levels were detected with immunoblotting. (A) Protein expression of HSP70. (B) Protein expression of GRP78. (C) Protein expression of HSP60. (D) Protein expression of CHOP. (E) Protein expression of caspase-12. Levels of β-actin protein were used as the loading control. Quantitative analysis was performed with Sigma Scan Pro 5. Bar represents mean ± SD, n = 6. *p < 0.05, **p < 0.01 compared with the sham–operated group; #p < 0.05, ##p < 0.01 compared with PFI group.
Discussion
Autophagy is an evolutionarily conserved pathway that involves the sequestration and delivery of cytoplasmic materials to the lysosomes, where proteins are degraded and recycled. Autophagy is a double-edged sword in cell survival.Citation9,Citation31 Autophagy activation could promote cell death through apoptotic-dependent and independent cascades,Citation32 but in some other conditions, autophagy activation may be associated with neuroprotection by degrading misfolded proteins.Citation9,Citation33,Citation34 Some recent reports suggest an association between preconditioning and autophagy both in the heart and brain.Citation16,Citation35-Citation40 In the earlier study, we demonstrated that the neuroprotection induced by IPC is, at least partly, mediated by activation of autophagy in a rat model of ischemic preconditioning, and pharmacological induction of autophagy can mimic IPC’s neuroprotection.Citation16 In the present study, we verify a similar role of autophagy in an in vitro cortical neuron OGD preconditioning model. Furthermore, we found that preconditioning induced autophagy plays a role in modulating ER stress response during IPC and PFI. Inhibition of autophagy results in excessive ER stress to aggravate ischemic neuronal damage, while autophagy activation elicited by ischemic preconditioning or pharmacological preconditioning may lessen brain damage by inhibiting excessive ER stress.
Activation of autophagy was first determined in the cortical neurons with immunoblotting of LC3 and p62. LC3, the microtubule-associated protein 1A light chain 3, consists of cytosolic type I (LC3-I) and membrane-bound type II (LC3-II). During maturation of autophagy, LC-I converts to LC3-II after cleavage of a few amino acid at the C-terminus, so autophagic activity, defined in terms of autophagosome amounts, is directly correlated with LC3-II amount or the ratio of LC3-II(16 kD)/LC3-I(18 kD).Citation41 Another method for monitoring autophagy flux is to determine the levels of p62/SQSTM1, as p62 is associated with mature autophagic vesicles and is degraded within autophagosomes.Citation42 The results showed that both preconditioning and OGD could upregulate LC3-II and downregulate p62, suggesting that autophagy was activated after both preconditioning and OGD conditions, but the extent of autophagy activation in IPC is stronger than that induced by lethal OGD alone.
To determine the involvement of an autophagic mechanism in the neuroprotection offered by IPC, we investigated the effects of the autophagy inhibitors 3-MA and BAF on the OGD preconditioning model in cultured cortical neurons. 3-MA blocks autophagy through inhibiting class III phosphatidylinositol 3-kinases (PtdIns3K), whose activity is required for the membrane dynamics involved in autophagic vesicle trafficking, and thus, it can inhibit the formation of autophagosomes.Citation43,Citation44 BAF, a vacuolar H+-ATPase inhibitor, prevents lysosomal function, thus blocking maturation of autophagy.Citation45 In previous studies, intracerebral ventricle injection of 3-MA 100–400 nmol in vivo and 3-MA 5–20 mM or BAF 40–200 nM in vitro have been shown to effectively block autophagic activity.Citation16,Citation43,Citation45 The results showed that IPC treatment significantly attenuated OGD induced cell injury and LDH release in the medium, whereas 3-MA (5–20 mM) and BAF (75, 150 nM) pretreatment suppressed the protective effects of IPC. 3-MA pretreatment also canceled preconditioning-mediated reduction in OGD-induced LDH release and caspase-3 activation in cortical neurons, suggesting that 3-MA-mediated increases in OGD-induced cell death in preconditioned cortical neurons is a mixture of apoptosis and necrosis.Citation13 This can also be supported from morphological observations ( and Fig. S1).
We then evaluated if 3-MA blocked the IPC-induced autophagy activation. Western blot analysis revealed that 3-MA pretreatment significantly attenuated LC3-II upregulation and p62 downregulation induced by IPC. The punctate immunofluorescence of LC3 and MDC-labeled vacuoles in the cortical neurons were also inhibited by 3-MA. 3-MA is a PtdIns3K inhibitor and may have other effects besides autophagy blockage. However, in our previously published studies,Citation16 we have verified that bafilomycin produced the same effects as 3-MA in rat preconditioning model. Some recent studies also reported that inhibition of autophagy with Tat-ATG5 (K130R, knockdown of Atg5)Citation35 or wortmanninCitation36 could reduce the cardioprotection by IPC in rat hearts. In light of these studies, the present results suggest that autophagic mechanism is involved in the IPC elicited neuroprotection in primary cultured cortical neurons.
During cerebral ischemia, hypoxia, glucose deprivation, formation of free radicals and calcium overload disturb the proper function of the ER and initiate ER stress. The signal pathway in ER stress is complex. Cells first activate the unfolded protein response (UPR), characterized by the activation of three ER transmembrane effector proteins: PERK, IRE1 and ATF-6. One major pathway of UPR is the suppression of most protein translations through phosphorylation of eIf2α by PERK. Another pathway is the upregulated expression of ER-localized molecular chaperones, GRP78/Bip, GRP94 and HSPs. However, prolonged ER stress may activate an ER stress-dependent apoptotic pathway by induction of caspases-12 and CHOP. In this study, we explored the role of autophagy and ER stress on cellular fate during preconditioning and lethal ischemia, the ER stress responsive proteins GRP78, HSPs, CHOP and caspase-12 were the focus of our study.
ER, mitochondria and cytoplasm each respond to the accumulation of unfolded proteins by compartment-specific signaling pathways.Citation46-Citation48 Accumulation of unfolded proteins in the ER lumen will induce main ER molecular chaperone GRP78/Bip, which regulates protein folding and facilitates protein translocation in the ER and protein secretion. Previous reports show that induction of GRP78 prevents the neuronal death induced by severe ER stress both in vitro and in vivo.Citation23,Citation24,Citation49,Citation50 Accumulating unfolded proteins in the cytoplasm and mitochondria also induce expression of molecular chaperones such as HSP70 and HSP60. HSP70 is the major stress-induced cytoplasmic chaperonee and has been intensively studied in brain ischemia and ischemic tolerance.Citation51-Citation54 Recent research also indicates that HSP70 might interrelate with autophagy to exert beneficial effects on neurodegenerative diseases.Citation55,Citation56 HSP60 is a major molecular chaperonee predominantly residing in the mitochondrial matrix. It has been demonstrated that the HSP60 gene is selectively induced by mitochondrial accumulation of unfolded proteins,Citation47,Citation57 and induction of HSP60 could protect the neurons from ischemic damage.Citation57 In agreement with previous findings,Citation23,Citation58 our results showed that the HSP70, HSP60 and GRP78 expression were significantly upregulated after IPC both in vivo and in vitro. However, 3-MA significantly prevented IPC-induced increases in HSP60, HSP70 and GRP78. These results suggest that an autophagic mechanism is involved in IPC–induced HSP70, HSP60 and GRP78 upregulation. However, signaling mechanisms by which autophagy promote chaperonee upregulation remain to be determined.
Opposite to the effects on these beneficial molecular chaperones, CHOP, caspase-12 and caspase-3 are involved in ER-stress dependent apoptosis. The proapoptotic transcription factor CHOP plays a key role in ER stress-induced apoptosis.Citation59,Citation60 Downregulation of CHOP activity compromises cell viability, and cells lacking CHOP are significantly protected from the lethal ER stress.Citation61 Caspase-12, a murine protein associated with the ER membrane, normally exists in an inactive pro-caspase form.Citation62 During ER stress, caspase-12 dissociates from the ER membrane and is cleaved to a fragment, and then it is activated and initiates downstream apoptotic pathways. Caspase-12-deficient mice are resistant to ER stress-induced apoptosis. Our results showed that protein levels of p-eIF2α, CHOP, cleaved caspase-12 and caspase-3 were greatly upregulated after fatal ischemia, whereas IPC reduced the expression of p-eIF2α, CHOP, cleaved caspase-12 and caspase-3. 3-MA treatment canceled the inhibitory effects of IPC on p-eIF2α, CHOP, cleaved caspase-12 and increased OGD-induced apoptosis in cortical neurons. Ultrastructural studies showed mild ER stress and autophagy induction were induced in both IPC and IPC+PFI groups. However, the neurons in the fatal ischemic cortex displayed much more severe ER stress and signs of cell injury. Inhibition of autophagy with 3-MA exacerbated ER stress. All these results from both in vivo and in vitro studies strongly suggest that fatal ischemia seems to induce prolonged and severe ER stress leading to cell death, while IPC induces minor ER stress to elicit beneficial ER response and to limit excessive subsequent ER stress.
Although GRP78, CHOP and caspase-12 are all biomarkers of ER stress, they respond to various degrees of ER stress and have different durations of expression and functions during ER stress. At the early phase of ER stress, cells exhibit a self-protective signal-transcription pathway by upregulated expression of ER-localized molecular chaperones or other chaperones, such as Bip/GRP78, GRP94, HSP60 and HSP70, which contribute to repairing of unfolded proteins. However, at the late phase of ER stress, excessive ER stress may activate an apoptotic pathway by activating caspases-12 and CHOP that induce cell apoptosis. Some studies show that increased expression of GRP78 attenuated the induction of CHOP, caspase-12 and subsequential neuronal death during excessive ER stress.Citation23,Citation24,Citation49,Citation50 Lithium can induce autophagy, and thereby, enhances the clearance of mutant huntingtin and α-synucleins by inhibiting inositol monophosphatase.Citation63 Many reports including our own show that lithium could exert neuroprotection through upregulation of cytoprotective HSP70, GRP78, HSP60 protein levels in fatal cerebral ischemia.Citation64-Citation66 We thus assume autophagy might have an effect on expression of GRP78, HSP 60 and 70 during ischemia. In addition, 3-MA is demonstrated to not only suppress ER stress-induced autophagy, but also suppress UPR activation.Citation67 Thus blockage of autophagy with 3-MA might reduce the expression of GRP78 and HSPs by suppression of UPR process. Another reason is that GRP78, HSP70 and HSP60 may also exert neuroprotective effects by antagonizing other apoptotic and necrotic cell death mechanisms during cerebral ischemia, such as maintaining proper protein folding, degradation of unfolded protein, modulation of inflammation, preservation of mitochondrial function, regulating signal-transduction proteins, and so on.Citation68-Citation70 Thus 3-MA might aggravate neuronal injury and reduce the expression of GRP78 and HSPs through affecting cell survival. It is expected that during IPC, autophagy reduces ER stress, thus preventing activation of ER stress-dependent apoptosis. In this study, inhibition of autophagy with 3-MA resulted in excessive ER stress, leading to increases in CHOP and caspase-12. However, why autophagy activation has differential effects on ER chaperones and ER stress-related cell death signaling remains to be studied.
To further evaluate if modulating ER stress can produce neuroprotection in PFI, the present study examined the effects of an ER stress inhibitor Salubrinal (Sal) on 3-MA-induced antagonism against IPC-induced neuroprotection. Sal can counteract ER stress-induced cell degeneration by selectively inhibiting eIF2α dephosphorylation both in vitro and in vivo.Citation71,Citation72 Our results showed that 3-MA completely abolished the protective effects of IPC on infarct volume and brain edema. By contrast, Sal pretreatment significantly resumed IPC-elicited neuroprotective effects in the presence of 3-MA. Although Sal has some other effects, such as inhibition of NFκB activation, transglutaminase 2 expression and modulation of JNK activation, it is the most commonly used ER stress inhibitor. The protective effect of Sal in this study was apparently mediated via the inhibition of ER stress as shown by its effect on caspase-12, and dephosphorylation of eIF2α. Our results were also supported by the previous study showing that Sal is able to afford protection against cerebral ischemia/reperfusion injury and kainic acid-induced excitotoxicity (the same dose used in the present study) via inhibition of ER stress.Citation72,Citation73 Taken together, the results from 3-MA and Sal suggest that preconditioning reduces ER stress-dependent apoptosis via autophagic activation.
Recently, a correlation has been observed between induction of ER stress and autophagic activation in neurodegeneration.Citation27,Citation28 Several lines of evidence demonstrate that ER stress contributes to autophagy activation.Citation26-Citation28 In yeast, ER stress was found to stimulate the assembly of the phagophore assembly site, formation of autophagosomes and transport them to the vacuole.Citation26 A study performed in the myocardial ischemic/reperfusion (I/R) model showed that induction of autophagy by therapeutic levels of ER stress inducers would ameliorate subsequent lethal injury. Importantly, only the autophagy produced with lower doses of ER stress inducer resulted in reduction of the I/R injury and cardiomyocyte apoptosis, which is accompanied by induction of GRP78 and Bcl-2. The higher doses of ER stress inducer, however, were detrimental for the heart and were associated with induction of CHOP.Citation74 Recent results indicate that the accumulation of protein aggregate-associated organelles seen following ischemia is likely to be due to failure of the autophagy pathway. The resulting protein aggregation on subcellular organelle membranes could lead to multiple organelle damage and to delayed neuronal death after cerebral ischemia.Citation75 Combining these reports and our present results, we predict that mild ER stress might be potent stimuli for autophagy during preconditioning. ERAD II (ER associated degradation II; autophagy /lysosome pathway) induced by ER stress then upregulates molecular chaperones for degradation of misfolded/unfolded protein within ER lumen. By this mechanism, autophagy during preconditioning prevents severe ER stress and protein aggregation during subsequent fatal ischemia.
In an earlier study, we found that rapamycin (8.8–35 pmol) significantly reduced infarct volume, brain edema and neurological deficits in PFI rats.Citation16 In the present study, we found that rapamycin (35 pmol) significantly increased the expression of HSP70, HSP60 and GRP78, while reducing the expression of cleaved caspase-12 and CHOP in PFI rats. Thus, rapamycin seems to be able to boost expression of protective molecular chaperones and reduce the expression of ER associated pro-apoptotic proteins; the effects elicited by mild ER stress seen in IPC. In addition, rapamycin showed protection on fatal OGD injury in cortical neurons. We believe these results may reveal a link between autophagy activation and reduction of ER stress-dependent apoptosis during preconditioning. These results strongly support the hypothesis that drugs activating autophagy may be used for pharmacological preconditioning to reduce damage of fatal cerebral ischemia and provide novel strategies for clinical prevention or treatment of ischemic stroke.
In conclusion, our present results support the role that autophagy plays in the neuroprotection induced by ischemic preconditioning in primary cultured cortical neurons. Pre-activation of autophagy by ischemic preconditioning or pharmacological preconditioning can boost endogenous defense mechanisms to upregulate some molecular chaperones, and hence reduce excessive ER stress during fatal ischemia. These findings would suggest a novel strategy for development of potential novel therapies for ischemic cerebrovascular diseases.
Materials and Methods
Oxygen glucose deprivation (OGD) preconditioning model in primary cultured cortical neurons
Primary neuronal culture was prepared from the cortex of E18 embryonic mice as described previously.Citation76 First, the cortices were dissected out into pre-cooled PBS. After removal of meninges and blood vessels, the tissue was minced and digested with 2.5% trypsin solution at 37°C for 15 min. The sediments were washed three times with PBS, and then incubated with DMEM/F12 containing 10% fetal bovine serum and 0.5 mg/ml DNase (for 3 min. The cell suspensions were disassociated by pipetting up and down and centrifuged at 500 × g for 5 min. The pellets were resuspended in Neurobasal medium (NBM, Invitrogen, 21103) containing 10% B27 (Gibco, 17504-044), 25 μM glutamate, 1% penicillin G and streptomycin. Then the cells were plated onto poly (d-lysine)-coated plates at the density of 1.5 × 106 cells/ml. Twenty-four hours later, the whole medium was replaced by NBM containing 10% B27, 0.5 mM glutamine, 1% penicillin G and streptomycin. Thereafter, half of the medium was replaced every 2 d. The cultures were treated with 10 µM arabinosylcytosin (Sigma, C6645) to remove glial cells at day 3 or 4 after plating. The cortical neurons mature at day 8~10 d and then were used for further experiments.
To induce OGD, the cultures were washed three times with Hepes balanced salt solution (HBSS: 140 mM NaCl, 3.5 mM KCl, 12 mM MgSO4, 5 mM NaHCO3, 1.7 mM CaCl2, 0.4 mM KH2PO4, 10 mM Hepes), and then placed in a modular incubator chamber (Billups-Rothenberg, MC-101) aerated with an anaerobic gas mixture (95% N2/5%CO2).Citation77 To terminate OGD, cells were removed from the anaerobic chamber, and then changed with normal medium. For preconditioning, the cortical neurons were deprived of oxygen and glucose for 30 min, an insult that did not induce neuronal death, as measured by lactate dehydrogenase (LDH) release in preliminary experiments. Twenty-four hours after the preconditioning stimulus, the cultures were again deprived of oxygen and glucose for 3 or 6 h to cause lethal OGD. The controls included IPC group (subjected to OGD for 30 min without subsequent lethal OGD) and OGD group (underwent lethal OGD treatment without prior IPC). The morphological changes of cells were observed using a phase contrast microscopy.
Lactate dehydrogenase (LDH) assay
Following treatment, the culture medium was harvested from the plates. The attached cells and culture medium were lysed in PBS containing 1% Triton X-100 (Fluka, 93443) at 37°C for 30 min, respectively. The LDH activities in both the cell lysates and the culture mediums were assayed with an assay kit (Nanjing Jiancheng Bioengineering Institute, A020) following the manufacturer’s instructions. Absorbance value at 440 nm was determined with an automatic multiwell spectrophotometer (Bio-Rad). LDH leakage was calculated as follows: LDH leakage (%) = LDH culture medium/(LDH culture medium + LDH cell lysates) × 100%.Citation29
MTT assay
Cell death was quantified with MTT at 24 h later. Neurons were incubated in 80 μg/ml Thiazolyl Blue Tetrazolium Bromide (MTT, Sigma, M2128) at 37°C for 2 h. Culture medium was aspirated and cells were lysed in 250 μl DMSO. Color intensity was measured at 570 nm using a Victor3V plate reader.
Rat focal ischemic preconditioning model
Male Sprague-Dawley rats weighing 280–300 g were purchased from the Center for Experimental Animals of Soochow University (certificate No 20020008, Grade II). The NIH Guidelines for Care and Use of the Laboratory Animals were followed in all animal procedures. Focal ischemic preconditioning (IPC) and/or permanent focal cerebral ischemia (PFI) models were performed using the suture occlusion technique.Citation78,Citation79 Rats were anesthetized with intraperitoneal injection of 4% chloral hydrate (Sinopharm Chemical Reagent Co. Ltd., 30037517) 350 mg/kg. Through a ventral midline incision, the right common carotid artery (CCA), external carotid artery (ECA) and internal carotid artery (ICA) were isolated. A 30-mm length of monofilament nylon suture (Φ0.22–0.24 mm), with its tip rounded by heating near a flame, was inserted from the right CCA to the internal carotid artery (ICA) through the stump of the ECA and then advanced to the Circle of Willis to occlude the origin of the right middle cerebral artery. Transient focal ischemia of 10 min duration was used to induce IPC. After IPC induction, the suture was gently withdrawn to permit reperfusion. Then the ECA was ligated and the wound was closed. Twenty-four hours later, the suture was re-introduced into the ICA to induce permanent focal cerebral ischemia (PFI, permanent middle cerebral artery occlusion, pMCAO). The IPC group underwent transient focal ischemia of 10 min without subsequent pMCAO, whereas the PFI group underwent pMCAO surgery without prior IPC. Sham-operated rats underwent identical surgery but the suture was not inserted. Laser-Doppler flowmetry (LDF, ML191 Laser Doppler Blood FlowMeter) was used to monitor blockage of cerebral blood flow. Body temperature was closely monitored with a rectal probe and maintained in the range of 37.0 ± 0.5°C with a heating pad (CAMS, BME-412A ANIMAL REGULATOR, Institute of Biomedical Engineering) during and after surgery until recovery from anesthesia. Rats showing tremor and seizure after surgery were excluded from further experiments.
Transmission electron microscopic examination
Six or 24 h after IPC and/or PFI, rats were perfused with pre-cooled phosphate-buffered saline solution (PBS, pH 7.4) followed by PBS containing 4% paraformaldehyde (Sinopharm Chemical Reagent Co. Ltd., 80096628) and 0.25% glutaraldehyde (Sinopharm Chemical Reagent Co. Ltd., 30092436) after anesthetization. The brains were removed and kept in 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M PBS (pH 7.4). Then the brains were cut with a vibratome into 50-mm thick slices. The parietal lobe cortex in the ischemic core area was selected for analysis, and selected areas were processed by postfixation in 1% osmium tetroxide for 1 h, dehydrated in graded ethanol, and embedded in epoxy resin. Polymerization was performed at 80°C for 24 h. Blocks were cut on a Reichert ultramicrotome into ultrathin sections (60–70 nm), which were post-stained with uranyl acetate and lead citrate, and viewed under a Hitachi 7100 electron microscopy (Nikon).
Evaluation of infarct volume and brain water content
Twenty-four hours after ischemia, the rats were killed and the brains were dissected and sliced in a plastic module (Harvard Apparatus, 3-mm thickness) and stained with 4% 2, 3, 5-triphenyltetrazolium chloride (TTC, Sinopharm Chemical Reagent Co. Ltd., 30187713) for 30 min and then fixed with 4% paraformaldehyde. The total wet weight of the TTC stained brains was quantified with an electronic scale (Mettler-Toledo Group). The wet red and white brain regions of the TTC-stained brains were collected separately. Infarct volume was analyzed using 5 slices of 3 mm coronal sections from each brain and calculated with the following formula: infarct volume = (total wet weight–red weight)/ total wet weight × 100%.
After the wet weight of the brains was quantified, the red and white parts of these brains were desiccated at 105°C for 48 h until the weight was constant. The total weight of the dried TTC-stained brains was obtained, and the water content of each brain was measured as follows: water content = (wet weight–dried weight)/wet weight × 100%.Citation80
Immunoblotting
The cortical neurons were washed twice with ice-cooled PBS and lysed in a buffer containing TRIS-HCl (pH 7.4) 10 mM, NaCl 150 mM, 1% Triton X-100, 1% sodium deoxycholate (Sinopharm Chemical Reagent Co. Ltd., 69022780), 0.1% SDS, edetic acid (Sinopharm Chemical Reagent Co. Ltd., 10009617) 5 mM, phenylmethylsulfonyl fluoride (PMSF, Sigma, P7626), 1 protease inhibitor cocktail tablet (Roche, 05892791001)/10ml. The ischemic cortex of the right middle cerebral artery territory and the corresponding area of sham-operated rats were homogenized and proteins were also extracted with this lysis buffer.Citation81 Protein concentrations were determined (SmartSpec3000 Spectrophotometer, Bio-Rad) using a BCA kit (Pierce, 23227). A 50 µg (in vivo) or 30 µg (in vitro) aliquot of proteins from each sample was separated by SDS-PAGE and subsequently transferred to a nitrocellulose membrane. Afterwards, the membranes were incubated with the specific antibodies against LC3 (1:1000; Abcam, ab62721), p62 (1: 1000; American Research Product, 12-1107), HSP70 (1:200; Santa Cruz Biotechnology, sc-24), GRP78 (1:1000; Stressgen Bioreagents, SPA-826), HSP60 (1:100; Sigma, H 4149), CHOP (1:100; Santa Cruz Biotechnology, sc-7351), caspase-12 (1:1000; Chemicon, AB3613), caspase-3 (1:1000; Sigma, C8487), eIF2α (1:1000; Cell Signaling, 9722) or Phospho-eIF2a (Ser51, 1:1000; Cell Signaling, 9721) at 4°C for overnight, then incubated with a horseradish peroxidase-conjugated secondary antibody (1:5,000; Jackson Immuno Research, anti-rabbit 715-035-150; anti-mouse 711-035-152) at room temperature for 1 h. Immunoreactivity was detected by enhanced chemiluminescent autoradiography (Pierce, 34080) in accordance with the manufacturer’s instructions. The membranes were reprobed with β-actin (1:5,000; Sigma, A5441) after striping (TBST with 2% β-mecaptoethanol, 65°C, 1 h). The films were used for final determination of protein expression with Sigma Scan Pro 5.0 and normalized to the loading control.
Immunofluorescence
Cortical neurons were grown onto cover glasses in a 24-well plate. After treatment, the cells were fixed for 15 min using 4% paraformaldehyde, and then incubated with PBS containing 0.1% Triton X-100 for 30 min. After blocking with 2% BSA, the cells were then incubated with antibodies against LC3 (1:500; Abcam, ab62721), MAP-2 antibody (1:1000, Sigma, M 9942) at 4°C for 24 h. The cells were sequentially incubated with FITC-conjugated anti-mouse IgG (1:400; Jackson ImmunoResearch, 715-091-150) and Cy3-conjugated anti-rabbit IgG (1:400; Jackson ImmunoResearch, 711-165-152) for 2 h. Afterwards, the cells were incubated with 0.5 μg/ml 4,6-diamidino-2- phenylindole (DAPI, Sigma, D9564) for 10 min. Then the cells were mounted on slides and images of fluorescence were acquired using a laser confocal microscope (Nikon D-Eclipse C1).
Visualization of MDC-labeled vacuole
Autophagic vacuoles were labeled with MDC,Citation29,Citation30 by incubating cells with 20 µM MDC (Sigma, D4008) at 37°C for 30 min. After incubation, cells were washed three times with PBS and immediately analyzed with a fluorescence microscope (Nikon Eclipse TE 300) equipped with a filter system (V-2A excitation filter: 380/420 nm, barrier filter: 450 nm).
Nuclear staining with Hoechst 33258
Cortical neurons were fixed in 4% paraformaldehyde, rinsed with PBS, and then incubated for 20 min at room temperature with Hoechst 33258 (0.5 μg/ml, Sigma, 94403). After rinsing with PBS, the cells were mounted on slides and images of fluorescence were acquired using a fluorescence microscope (Nikon Eclipse TE 300). Apoptotic cells were recognized by the condensed, fragmented and degraded nuclei. At least 200 cells were counted in each group of three independent experiments and all the counting was performed in a blinded fashion.
Drug treatment
To determine the effects of autophagy inhibitor 3-methyladenine (3-MA) on autophagy activation and ER stress in vitro and in vivo, the cortical neurons were pretreated with 3-MA (Sigma, M9281) 5–20 mM or bafilomycin A1 (BAF, LC lab, B-1080) 37.5–150 nM 30 min before the onset of IPC. Neurons continued to be incubated with 3-MA or BAF during the preconditioning episode. To determine if 3-MA has toxic effects on cortical neurons, the neurons were incubated with 3-MA 10 mM for 48 h. Rats were treated with intracerebral ventricle (i.c.v.) injection of 3-MA 200 nmol (1 µl) 5 min before the onset of IPC. 3-MA was dissolved in normal saline by heating solution to 60–70°C immediately before treatment. BAF was first dissolved in DMSO and then diluted to the final concentration with normal saline and culture medium (the final DMSO concentration < 0.1%). To explore if rapamycin, an autophagy inducer, has some effects on cell survival and ER stress, the cortical neurons were pretreated with rapamycin (Sigma, R0395) for 24 h before the onset of a lethal OGD for 6 h and rapamycin presence was continued during OGD episode. Rats were treated with i.c.v. injection of rapamycin 35 pmol (4 µl) 24 h before the onset of PFI. Rapamycin was first dissolved in ethanol and then diluted to the final concentration with normal saline (the final ethanol concentration < 2%). To explore the linkage between autophagy and ER stress during the IPC process, rats were given i.c.v. injections of salubrinal (Sal; Calbiochem, 324895) 75 pmol (1 µl). Ten minutes later, rats were treated with an additional i.c.v. injection of 3-MA 200 nmol 5 min before the onset of IPC. Sal was dissolved in dimethylsulfoxide (DMSO) and further diluted with saline (the final DMSO concentration < 0.2%). The optimal effective concentration and time of administration of 3-MA, rapamycin and Sal were chosen from published reports and pilot experiments.Citation13,Citation16,Citation72,Citation82 Control animals received i.c.v. injections of same volume of vehicle.
Statistics analysis
Statistical analysis was performed by one-way anova. The intergroup comparisons (post-hoc analysis) among the data with equal variances were performed with the least significant difference (LSD) method, while Tamhane’s T2 method was used for the data with unequal variances. p < 0.05 was considered to be significant.
Additional material
Download Zip (1.2 MB)Acknowledgments
This work was supported by grants from the Priority Academic Program of Jiangsu Higher Education Institutions (PAPD), the Natural Science Foundation of China (30801391, 30930035, 81173057), the Natural Science Foundation of Jiangsu Province (BK 2007548), Suzhou social progress foundation (SS0729), and Jiangsu Government Scholarship for Overseas Studies.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Dirnagl U, Meisel A. Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning?. Neuropharmacology 2008; 55:334 - 44; http://dx.doi.org/10.1016/j.neuropharm.2008.02.017; PMID: 18402985
- Obrenovitch TP. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev 2008; 88:211 - 47; http://dx.doi.org/10.1152/physrev.00039.2006; PMID: 18195087
- Perez-Pinzon MA. Mechanisms of neuroprotection during ischemic preconditioning: Lessons from anoxic tolerance. Comp Biochem Physiol A Mol Integr Physiol 2007; 147:291 - 9; http://dx.doi.org/10.1016/j.cbpa.2006.08.032; PMID: 17045830
- Liu XQ, Sheng R, Qin ZH. The neuroprotective mechanism of brain ischemic preconditioning. Acta Pharmacol Sin 2009; 30:1071 - 80; http://dx.doi.org/10.1038/aps.2009.105; PMID: 19617892
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717 - 21; http://dx.doi.org/10.1126/science.290.5497.1717; PMID: 11099404
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/10.1038/nature04723; PMID: 16625205
- McCray BA, Taylor JP. The role of autophagy in age-related neurodegeneration. Neurosignals 2008; 16:75 - 84; http://dx.doi.org/10.1159/000109761; PMID: 18097162
- Yen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 2008; 23:248 - 62; http://dx.doi.org/10.1152/physiol.00013.2008; PMID: 18927201
- Qin ZH, Wang Y, Kegel KB, Kazantsev A. Autophagy regulates processing of amino terminal huntingtin fragments. Hum Mol Genet 2003; 12:3231 - 44; http://dx.doi.org/10.1093/hmg/ddg346; PMID: 14570716
- Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, et al. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/ proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum Mol Genet 2007; 16:618 - 29; http://dx.doi.org/10.1093/hmg/ddm002; PMID: 17331981
- Lockshin RA, Zakeri Z. Apoptosis, autophagy and more. Int J Biochem Cell Biol 2004; 36:2405 - 19; http://dx.doi.org/10.1016/j.biocel.2004.04.011; PMID: 15325581
- Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol 2006; 169:566 - 83; http://dx.doi.org/10.2353/ajpath.2006.051066; PMID: 16877357
- Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis 2008; 32:329 - 39; http://dx.doi.org/10.1016/j.nbd.2008.07.022; PMID: 18760364
- Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol 2008; 172:454 - 69; http://dx.doi.org/10.2353/ajpath.2008.070876; PMID: 18187572
- Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, et al. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 2008; 4:762 - 9; PMID: 18567942
- Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 2010; 6:482 - 94; http://dx.doi.org/10.4161/auto.6.4.11737; PMID: 20400854
- Ma Y, Hendershot LM. The unfolding tale of the unfolded protein response. Cell 2001; 107:827 - 30; http://dx.doi.org/10.1016/S0092-8674(01)00623-7; PMID: 11779459
- DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem 2004; 91:1 - 8; http://dx.doi.org/10.1111/j.1471-4159.2004.02703.x; PMID: 15379881
- Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ 2006; 13:363 - 73; http://dx.doi.org/10.1038/sj.cdd.4401817; PMID: 16397583
- Shibata M, Hattori H, Sasaki T, Gotoh J, Hamada J, Fukuuchi Y. Activation of caspase-12 by endoplasmic reticulum stress induced by transient middle cerebral artery occlusion in mice. Neuroscience 2003; 118:491 - 9; http://dx.doi.org/10.1016/S0306-4522(02)00910-7; PMID: 12699784
- Morimoto N, Oida Y, Shimazawa M, Miura M, Kudo T, Imaizumi K, et al. Involvement of endoplasmic reticulum stress after middle cerebral artery occlusion in mice. Neuroscience 2007; 147:957 - 67; http://dx.doi.org/10.1016/j.neuroscience.2007.04.017; PMID: 17590517
- Lehotský L, Urban P, Pavlikova M, Tatarkova Z, Kaminska B, Kaplan P. Molecular Mechanisms Leading to Neuroprotection/Ischemic Tolerance: Effect of Preconditioning on the Stress Reaction of Endoplasmic Reticulum. Cell Mol Neurobiol 2009; 29:917 - 25; http://dx.doi.org/10.1007/s10571-009-9376-4; PMID: 19283468
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Chan PH. Induction of GRP78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronal cell death. J Cereb Blood Flow Metab 2003; 23:949 - 61; http://dx.doi.org/10.1097/01.WCB.0000077641.41248.EA; PMID: 12902839
- Yuan Y, Guo Q, Ye Z, Pingping X, Wang N, Song Z. Ischemic postconditioning protects brain from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis through PI3K-Akt pathway. Brain Res 2011; 1367:85 - 93; http://dx.doi.org/10.1016/j.brainres.2010.10.017; PMID: 20940001
- Kishi S, Shimoke K, Nakatani Y, Shimada T, Okumura N, Nagai K, et al. Nerve growth factor attenuates 2-deoxy-D-glucose-triggered endoplasmic reticulum stress-mediated apoptosis via enhanced expression of GRP78. Neurosci Res 2010; 66:14 - 21; http://dx.doi.org/10.1016/j.neures.2009.09.003; PMID: 19766678
- Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem 2006; 281:30299 - 304; http://dx.doi.org/10.1074/jbc.M607007200; PMID: 16901900
- Matus S, Lisbona F, Torres M, León C, Thielen P, Hetz C. The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration. Curr Mol Med 2008; 8:157 - 72; http://dx.doi.org/10.2174/156652408784221324; PMID: 18473817
- Sakaki K, Kaufman RJ. Regulation of ER stress-induced macroautophagy by protein kinase C. Autophagy 2008; 4:841 - 3; PMID: 18670192
- Yan CH, Liang ZQ, Gu ZL, Yang YP, Reid P, Qin ZH. Contributions of autophagic and apoptotic mechanisms to CrTX-induced death of K562 cells. Toxicon 2006; 47:521 - 30; http://dx.doi.org/10.1016/j.toxicon.2006.01.010; PMID: 16542694
- Munafó DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci 2001; 114:3619 - 29; PMID: 11707514
- Shintani T, Klionsky DJ. Autophagy in health and disease: A double-edged sword. Science 2004; 306:990 - 5; http://dx.doi.org/10.1126/science.1099993; PMID: 15528435
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004; 304:1500 - 2; http://dx.doi.org/10.1126/science.1096645; PMID: 15131264
- Cuervo AM, Stefanis L, Fredenburg R. Impaired degradation of mutant alpha-synuclein by chaperonee-mediated autophagy. Science 2004; 305:1292 - 5; http://dx.doi.org/10.1126/science.1101738; PMID: 15333840
- Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, et al. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 2005; 1:11 - 22; http://dx.doi.org/10.4161/auto.1.1.1513; PMID: 16874045
- Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM Jr., et al. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res 2010; 3:365 - 73; http://dx.doi.org/10.1007/s12265-010-9189-3; PMID: 20559777
- Gurusamy N, Lekli I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med 2009; 13:373 - 87; http://dx.doi.org/10.1111/j.1582-4934.2008.00495.x; PMID: 18793351
- Yan L, Sadoshima J, Vatner DE, Vatner SF. Autophagy in ischemic preconditioning and hibernating myocardium. Autophagy 2009; 5:709 - 12; http://dx.doi.org/10.4161/auto.5.5.8510; PMID: 19398895
- Yitzhaki S, Huang C, Liu W, Lee Y, Gustafsson AB, Mentzer RM Jr., et al. Autophagy is required for preconditioning by the adenosine A1 receptor selective agonist CCPA. Basic Res Cardiol 2009; 104:157 - 67; http://dx.doi.org/10.1007/s00395-009-0006-6; PMID: 19242639
- Park HK, Chu K, Jung KH, Lee ST, Bahn JJ, Kim M, et al. Autophagy is involved in the ischemic preconditioning. Neurosci Lett 2009; 451:16 - 9; http://dx.doi.org/10.1016/j.neulet.2008.12.019; PMID: 19103253
- Wu BX, Darden AG, Laser M, Li Y, Crosson CE, Hazard ES 3rd, et al. The rat Apg3p/Aut1p homolog is upregulated by ischemic preconditioning in the retina. Mol Vis 2006; 12:1292 - 302; PMID: 17110912
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Chakrabarti L, Eng J, Ivanov N, Garden GA, La Spada AR. Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol Brain 2009; 2:24; http://dx.doi.org/10.1186/1756-6606-2-24; PMID: 19640278
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/ lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 1982; 79:1889 - 92; http://dx.doi.org/10.1073/pnas.79.6.1889; PMID: 6952238
- Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25:1025 - 40; http://dx.doi.org/10.1128/MCB.25.3.1025-1040.2005; PMID: 15657430
- Hung SY, Huang WP, Liou HC, Fu WM. Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy 2009; 5:502 - 10; http://dx.doi.org/10.4161/auto.5.4.8096; PMID: 19270530
- Paschen W. Shutdown of translation: lethal or protective? Unfolded protein response versus apoptosis. J Cereb Blood Flow Metab 2003; 23:773 - 9; http://dx.doi.org/10.1097/01.WCB.0000075009.47474.F9; PMID: 12843781
- Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 2004; 117:4055 - 66; http://dx.doi.org/10.1242/jcs.01275; PMID: 15280428
- DeGracia DJ, Hu BR. Irreversible translation arrest in the reperfused brain. J Cereb Blood Flow Metab 2007; 27:875 - 93; PMID: 16926841
- Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperonee protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem 2003; 278:20915 - 24; http://dx.doi.org/10.1074/jbc.M212328200; PMID: 12665508
- Oida Y, Izutaa H, Oyagia A, Shimazawaa M, Kudob T, Imaizumic K, et al. Induction of BiP, an ER-resident protein, prevents the neuronal death induced by transient forebrain ischemia in gerbil. Brain Res 2008; 1208:217 - 24; http://dx.doi.org/10.1016/j.brainres.2008.02.068; PMID: 18395193
- Chen J, Graham SH, Zhu RL, Simon RP. Stress proteins and tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab 1996; 16:566 - 77; http://dx.doi.org/10.1097/00004647-199607000-00006; PMID: 8964795
- Currie RW, Ellison JA, White RF, Feuerstein GZ, Wang X, Barone FC. Benign focal ischemic preconditioning induces neuronal Hsp70 and prolonged astrogliosis with expression of Hsp27. Brain Res 2000; 863:169 - 81; http://dx.doi.org/10.1016/S0006-8993(00)02133-8; PMID: 10773205
- Dhodda VK, Sailor KA, Bowen KK, Vemuganti R. Putative endogenous mediators of preconditioning-induced ischemic tolerance in rat brain identified by genomic and proteomic analysis. J Neurochem 2004; 89:73 - 89; http://dx.doi.org/10.1111/j.1471-4159.2004.02316.x; PMID: 15030391
- Nishino K, Nowak TS Jr. Time course and cellular distribution of hsp27 and hsp72 stress protein expression in a quantitative gerbil model of ischemic injury and tolerance: thresholds for hsp72 induction and hilar lesioning in the context of ischemic preconditioning. J Cereb Blood Flow Metab 2004; 24:167 - 78; http://dx.doi.org/10.1097/01.WCB.0000100853.67976.8B; PMID: 14747743
- Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperonee-mediated autophagy. Proc Natl Acad Sci USA 2006; 103:5805 - 10; http://dx.doi.org/10.1073/pnas.0507436103; PMID: 16585521
- Meriin AB, Sherman MY. Role of molecular chaperones in neurodegenerative disorders. Int J Hyperthermia 2005; 21:403 - 19; http://dx.doi.org/10.1080/02656730500041871; PMID: 16048838
- Truettner JS, Hu K, Liu CL, Dietrich WD, Hu B. Subcellular stress response and induction of molecular chaperones and folding proteins after transient global ischemia in rats. Brain Res 2009; 1249:9 - 18; http://dx.doi.org/10.1016/j.brainres.2008.10.032; PMID: 18996359
- Hwang IK, Ahn HC, Yoo KY, Lee JY, Suh HW, Kwon YG, et al. Changes in immunoreactivity of HSP60 and its neuroprotective effects in the gerbil hippocampal CA1 region induced by transient ischemia. Exp Neurol 2007; 208:247 - 56; http://dx.doi.org/10.1016/j.expneurol.2007.08.017; PMID: 17931626
- Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol 2002; 318:1351 - 65; http://dx.doi.org/10.1016/S0022-2836(02)00234-6; PMID: 12083523
- Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, et al. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol Cell Biol 1996; 16:4273 - 80; PMID: 8754828
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 1998; 12:982 - 95; http://dx.doi.org/10.1101/gad.12.7.982; PMID: 9531536
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000; 403:98 - 103; http://dx.doi.org/10.1038/47513; PMID: 10638761
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170:1101 - 11; http://dx.doi.org/10.1083/jcb.200504035; PMID: 16186256
- Shao L, Sun X, Xu L, Young LT, Wang JF. Mood stabilizing drug lithium increases expression of endoplasmic reticulum stress proteins in primary cultured rat cerebral cortical cells. Life Sci 2006; 78:1317 - 23; http://dx.doi.org/10.1016/j.lfs.2005.07.007; PMID: 16236328
- Sheng R, Zhang LS, Han R, Gao B, Liu XQ, Qin ZH. Combined prostaglandin E1 and lithium exert potent neuroprotection in a rat model of cerebral ischemia. Acta Pharmacol Sin 2011; 32:303 - 10; http://dx.doi.org/10.1038/aps.2010.211; PMID: 21258357
- Xu XH, Zhang HL, Han R, Gu ZL, Qin ZH. Enhancement of neuroprotection and heat shock protein induction by combined prostaglandin A1 and lithium in rodent models of focal ischemia. Brain Res 2006; 1102:154 - 62; http://dx.doi.org/10.1016/j.brainres.2006.04.111; PMID: 16797496
- Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ 2008; 15:1460 - 71; http://dx.doi.org/10.1038/cdd.2008.81; PMID: 18551133
- Arrigo AP. Small stress proteins: chaperones that act as regulators of intracellular redox state and programmed cell death. Biol Chem 1998; 379:19 - 26; PMID: 9504712
- Benn SC, Woolf CJ. Adult neuron survival strategies–slamming on the brakes. Nat Rev Neurosci 2004; 5:686 - 700; http://dx.doi.org/10.1038/nrn1477; PMID: 15322527
- Hishiya A, Takayama S. Molecular chaperones as regulators of cell death. Oncogene 2008; 27:6489 - 506; http://dx.doi.org/10.1038/onc.2008.314; PMID: 18955975
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2a dephosphorylation protects cells from ER stress. Science 2005; 307:935 - 9; http://dx.doi.org/10.1126/science.1101902; PMID: 15705855
- Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, et al. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci 2007; 27:901 - 8; http://dx.doi.org/10.1523/JNEUROSCI.4289-06.2007; PMID: 17251432
- Nakka VP, Gusain A, Raghubir R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox Res 2010; 17:189 - 202; http://dx.doi.org/10.1007/s12640-009-9110-5; PMID: 19763736
- Petrovski G, Das S, Juhasz B, Kertesz A, Tosaki A, Das DK. Cardioprotection by endoplasmic reticulum stress-induced autophagy. Antioxid Redox Signal 2011; 14:2191 - 200; http://dx.doi.org/10.1089/ars.2010.3486; PMID: 20726815
- Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem 2010; 115:68 - 78; http://dx.doi.org/10.1111/j.1471-4159.2010.06905.x; PMID: 20633207
- Zhang J, Qian H, Zhao P, Hong SS, Xia Y. Rapid hypoxia preconditioning protects cortical neurons from glutamate toxicity through δ-opioid receptor. Stroke 2006; 37:1094 - 9; http://dx.doi.org/10.1161/01.STR.0000206444.29930.18; PMID: 16514101
- Meloni BP, Majda BT, Knuckey NW. Evaluation of preconditioning treatments to protect near-pure cortical neuronal cultures from in vitro ischemia induced acute and delayed neuronal death. Brain Res 2002; 928:69 - 75; http://dx.doi.org/10.1016/S0006-8993(01)03361-3; PMID: 11844473
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989; 20:84 - 91; http://dx.doi.org/10.1161/01.STR.20.1.84; PMID: 2643202
- Lee SH, Kim YJ, Lee KM, Ryu S, Yoon BW. Ischemic preconditioning enhances neurogenesis in the subventricular zone. Neuroscience 2007; 146:1020 - 31; http://dx.doi.org/10.1016/j.neuroscience.2007.02.058; PMID: 17434685
- Rosen GD, Harry JD. Brain volume estimation from serial section measurements: a comparison of methodologies. J Neurosci Methods 1990; 35:115 - 24; http://dx.doi.org/10.1016/0165-0270(90)90101-K; PMID: 2283883
- Qin ZH, Chen RW, Wang Y, Nakai M, Chuang DM, Chase TN. Nuclear factor kappar B nuclear translocation upregulates c-Myc and p53 expression during NMDA receptor- mediated apoptosis in rat stratium. J Neurosci 1999; 19:4023 - 33; PMID: 10234031
- Qin AP, Liu CF, Qin YY, Hong LZ, Xu M, Yang L, et al. Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy 2010; 6:738 - 53; http://dx.doi.org/10.4161/auto.6.6.12573; PMID: 20574158