Abstract
A significant portion of newly synthesized protein fails to fold properly and is quickly degraded. These defective ribosomal products (DRiPs) are substrates for the ubiquitin-proteasome system (UPS) and give rise to a large fraction of peptides presented by major histocompatibility complex class I molecules (MHCI). Here, we showed that DRiPs are also autophagy substrates, which accumulate upon autophagy inhibition in aggresome-like-induced structures (ALIS). Aggregation is critically depending on p62/SQSTM1, but occurs in the absence of activation of the NRF2 signaling axis and transcriptional regulation of p62/SQSTM1. We demonstrated that autophagy-targeted DRiPs can become UPS substrates and give rise to MHCI presented peptides upon autophagy inhibition. We further demonstrated that autophagy targeting of DRiPs is controlled by NBR1, but not p62/SQSTM1, CHIP or BAG-1. Active autophagy therefore directly modulates MHCI presentation by constantly degrading endogenous defective neosynthesized antigens, which are submitted to at least two distinct quality control mechanisms.
Keywords: :
Introduction
Cell activation is expected to increase rates of protein synthesis and the redistribution of cellular resources to new tasks. Organized protein degradation is therefore vital for maintaining cellular function and allowing rapid adaptation to new conditions. In addition, obsolete, damaged and misfolded proteins can become toxic for the cell and have to be disposed.Citation1 A large fraction of up to 30% of neosynthesized proteins is rapidly degraded (RDPs), with an average half-life of 10 min.Citation2 Since a large proportion of these neosynthetized proteins can become insoluble if their degradation is inhibited, it is assumed that they did not reach their functional conformational state and have therefore failed to pass protein quality control.Citation3 Identification of these defective proteins (DRiPs) is mostly mediated through polyubiquitination, prior degradation by the proteasome (the ubiquitin/proteasome system or UPS).Citation4 The decision between folding and degradation of nascent polypeptides requires the action of molecular chaperones such as Hsp70/HSPA8 family members.Citation5 If correct folding cannot be achieved, malconformed proteins ubiquitination can be mediated by HSPA8-interacting ubiquitin ligases such as CHIP (carboxy terminus of Hsp70-interacting protein)Citation6 and co-chaperones of the BAG (Bcl-2-associated athanogene) family.Citation7 Interestingly, UPS-mediated degradation of DRiPs/RDP generates the bulk of peptides presented by the major histocompatibility class I (MHCI) to CD8+ T cells and enables surveillance of cellular translation products by the immune system. Thus, DRiPs/RDP represent a key source of MHCI presented peptidesCitation8 and allow the rapid detection of cellular and foreign (viral) antigens, minutes after their translation and independently of the half-life of the fully functional protein.Citation9
Another endogenous protein degradation mechanism is autophagy. This term summarizes different degradation pathways, such as macroautophagy, microautophagy and chaperone-mediated autophagy, all of which result in the transfer of cytosolic material for degradation in lysosomes.Citation10 During macroautophagy (hereafter termed autophagy), cytosolic material is sequestered in double-membrane structures, forming autophagosomes. Upon fusion with lysosomes, autophagosomal content is degraded. This process can serve for bulk degradation, but also uses specific ubiquitin-dependent sorting mechanisms to target defined substrates.Citation11 Polyubiquitinylated proteins can be addressed to autophagosomes through recognition by specific adaptor proteins, which also interact with autophagosome receptors of the LC3/ATG8 family.Citation12 These adaptor proteins include NBR1 (neighbor of BRCA1 gene 1) and p62/SQSTM1 (sequestosome 1), which collaborate with autophagy-linked FYVE protein (ALFY)Citation13 to mediate the degradation of aggregated proteins. Furthermore, nuclear dot protein 52 (NDP52) has been reported to mediate autophagy of polyubiquitin-coated bacteria.Citation14
The proteasome and autophagy degradation pathways are linked with each other and share polyubiquitinylation as a common targeting signal for substrate recognition.Citation15 Autophagy inhibition has been described to reduce proteasomal degradation,Citation16 while proteasome inhibition can induce autophagy as a compensatory mechanism.Citation17 Inhibition of either system can induce accumulation and aggregation of insoluble polyubiquitinylated proteins,Citation18 while accumulation of toxic protein aggregates such as mutant huntingtin has been shown to trigger autophagy and thereby induce aggregate clearance.Citation19 Although proteasome is essential for the generation of most endogenous MHCI restricted antigenic peptides, autophagy has been shown to trigger both unconventional MHCI presentation of specific viral peptides and MHCII-restricted presentation of endogenous peptides in macrophages and dendritic cells.Citation20-Citation23
We found that a fraction of DRiPs are selected substrates for autophagy. Upon autophagy inhibition, DRiPs accumulate in p62/SQSTM1-positive aggresome-like induced structures (ALIS),Citation24 are degraded by the proteasome, and enter in the classical MHCI antigen processing and presentation pathway. As a consequence, decreased autophagic degradation of defective neosynthesized model antigens led to a specific increase in MHCI presentation. We further identified the adaptor protein NBR1 as responsible for model antigen targeting to autophagy, whereas p62/SQSTM1 was only found necessary for protein aggregation, demonstrating that the balance between these two adaptor proteins might be crucial for protein homeostasis and the control of endogenous antigen presentation. Our findings have important implications for the molecular classification of the different p62-containing aggregates, as well as understanding on how antigen presentation is impacted by the different protein quality control mechanisms and the subsequent addressing of DRiPs to different routes of degradation.
Results
Autophagy adaptors and DRiPs accumulate in ALIS
Inducible polyubiquitin aggregates have first been observed in LPS-activated dendritic cells, and were termed DALIS (for DC ALIS). Later, similar polyubiquitin-containing aggregates have been observed in HeLa cells (among others) upon exposure to different stress, such as autophagy inhibition.Citation24-Citation26 We confirmed that pharmacological inhibition of autophagy, with a 4h 3-methyladenine (3-MA) treatment, induced aggregates of polyubiquitinated proteins. These aggregates contained the autophagy adaptors p62 and NBR1 (), resisted detergent extraction before fixation (), formed independently of microtubule function (Fig. S1), and were thus considered bona fide ALIS. Interestingly, proteasome inhibition by epoxomycin caused aggregation of polyubiquitin, p62 () and NBR1 (data not shown), as well as treatment with high concentrations of puromycin. Puromycin can be mistaken by ribosomes for charged aminoacyl-tRNAs and is thus incorporated into nascent polypeptide chains, causing premature chain termination. This leads to massive induction of misfolded proteins (puro-DRiPsCitation27), which tend to aggregate. However, DALIS originally were defined as translation-dependent polyubiquitinated protein aggregates devoid of HSPA8.Citation28 Strikingly, epoxomycin- or puromycin-induced aggregates were strongly enriched for HSPA8, while 3-MA-induced ALIS were not (). These similarities suggest that high concentration of puromycin might not only induce aggregation through protein misfolding but also in combination with proteasome saturation. Our findings might indicate that autophagy and proteasome client proteins are handled by different processing machineries, and therefore HSPA8 co-aggregates with its client proteins subsequent to proteasome inhibition, but not upon autophagy inhibition. Finally, we could also observe polyubiquitin and p62 positive, HSPA8 low, ALIS induction upon autophagy inhibition by RNA silencing of the key autophagy gene atg5 (andFig. S4). These findings demonstrated that pharmacological or siRNA mediated autophagy inhibition induces ALIS, and we propose that epoxomycin- and puromycin-induced HSPA8-enriched aggregates are biochemically distinct from ALIS, but nevertheless can contain p62 and NBR1. Thus, the sole use of biochemical methods to detect insoluble ubiquitin accumulationCitation29 might not be suitable to quantify ALIS, since aggregate type and composition are not addressed.
Figure 1. ALIS form upon autophagy inhibition, and differ from proteasome inhibition-induced aggregates. (A) HeLa cells were treated for 4 h with 3-MA (5 mM) or left untreated (control) and stained for polyubiquitinated proteins (FK1 antibody, polyUbi, green), NBR1 (red) and p62 (blue) prior to analysis by confocal microscopy. (B) HeLa cells were treated for 4 h with 3-MA (5 mM), Puromycin (5 µg/ml) or Epoxomycin (5 µM). Cells were stained for ubiquitinated protein (FK2 antibody, polyUbi, blue), HSPA8 (green), and p62 (red). Arrowheads indicate ALIS in the overlay and the corresponding area in the HSPA8 staining, respectively. Pearson’s coefficient (R) is given for the colocalization of p62 and HSPA8. (C) HeLa cells were transfected with siRNA against ATG5 (siATG5) or control siRNA (siCo) and stained for ubiquitinated protein (FK2 antibody, polyUbi, blue), HSPA8 (green) and p62 (red). Bar, 10 µm.
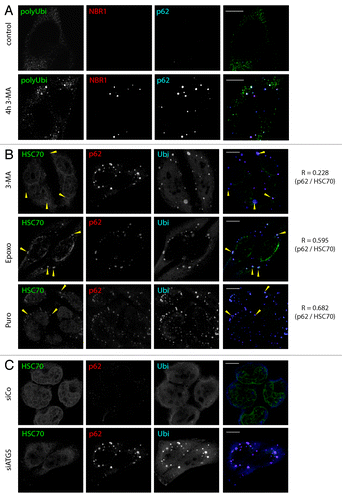
Figure 2. Puromycin DRiPs are autophagy substrates. (A) HeLa cells were treated for 4 h with 3-MA (4 h 3-MA), and puromycin (0,1 µg/µl) was added for the last 5 min (4 h 3MA + 5 min Puro). Cells were Triton X-100 permeabilized prior to fixation, and stained for ubiquitinylated protein (FK2 antibody, polyUbi, red), p62 (blue), and puromycin-containing proteins (Puro, green). Bar, 10 µm. (B) Hela cells received a short pulse of puromycin (10 min, 0.1 µg/ml), were washed and chased in the absence of puromycin in the presence or absence of proteasome inhibitor or 3-MA for the indicated times. Puromycin containing protein was resolved by SDS-PAGE and detected using anti-puromycin antibodies (top) and quantified using ImageJ (bottom), mean ± SEM from three independent experiments are shown, p values (Student’s t-test) were **p > 0.03. (C) Hela cells were stained for ubiquitinylated protein (FK1 antibody, polyUbi, green) and LC3 (red). Bar, 10 µm. (D) HeLa cells were left untreated, or treated for 30 min with puromycin (0.1 µg/µl), and stained for LC3 (red), p62 (blue) and puromycin-containing proteins (Puro, green). Bar, 10 µm.
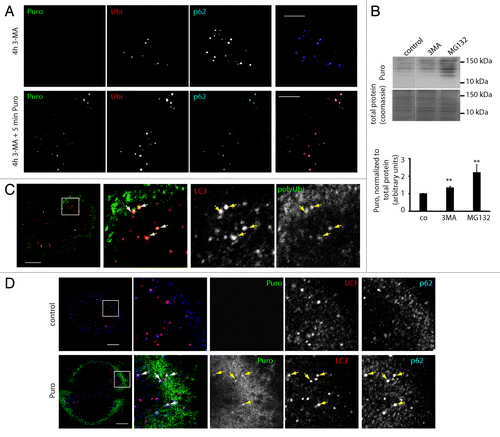
One of the key features of DALIS is their exquisite sensitivity to protein synthesis inhibition,Citation24 a consequence of the large contribution of ubiquitin-conjugated DRiPs to their formation. While the role of ubiquitin in the UPS-mediated degradation of DRiPs is evident, the role of ubiquitination in autophagy targeting, as well as the role of autophagy in the degradation of polyubiquitin conjugates is unclear. NBR1 and p62 mediated targeting of ubiquitinated proteins to autophagy has been demonstrated,Citation30 but other reports attributed polyubiquitin accumulation in autophagy deficient cells to be a secondary effect of an Nrf2-mediated stress response.Citation31 We therefore asked whether in a system of short-term autophagy inhibition, the turnover of polyubiquitinated protein, and especially DRiPs, was affected. First, we investigated whether DRiPs could be targeted to ALIS and contribute significantly to the pool of aggregated proteins formed upon autophagy inhibition. We induced DRiPs using low concentration of puromycin (puro-DRiPsCitation27) and followed their fate by microscopy in 3-MA treated cells using anti-puromycin mAb.Citation32 The puromycin concentration and treatment times in these experiments were far below the threshold of aggregate induction by puromycin itself (Fig. S2A and S2B), and Triton X-100 permeabilization before fixation was employed to remove remaining free puromycin and allow for the detection of puro-DRiPs in ALIS. We could detect neosynthesized puro-DRiPs in aggregates as early as 5 min after the pulse (). Thus, DRiPs contribute directly to the formation of ALIS and could represent a pool of previously unsuspected autophagy substrates. It should be noted that puro-DRiPs targeting to 3-MA induced aggregates did not recruit HSPA8 into those structures, in contrast again to aggregates induced by high-dose puromycin treatment (Fig. S2C). This demonstrates that low-dose puromycin treatment is suitable to induce and follow DRiPs without altering the cellular machinery handling them. To follow the biochemical fate of puro-DRiPs upon autophagy or proteasome inhibition, we exposed cells to a short (10 min) pulse of 0,5 µg/ml puromycin and chased for 2 h in the presence or absence of inhibitors. As expected, proteasome inhibition strongly reduced degradation of puromycin-containing protein, increasing the quantity of recovered protein by 120% (). Interestingly, albeit more modestly, autophagy inhibition by 3-MA also decreased puromycin-labeled protein degradation (), leading to a recovery increase of more than 30%. Autophagy targeting of polyubiquitinated protein was further corroborated by immunofluorescence analysis demonstrating localization of polyubiquitin in LC3-positive autophagosomes at steady-state (). Finally, we could demonstrate that puromycin marked protein upon low-dose treatment (0,1 µg/ml) colocalized with p62 in autophagosomes (). This suggests that while the majority of puro-DRiPs are proteasome substrates, a fraction of those can be subject to autophagy-mediated degradation shortly after synthesis and as a likely consequence, accumulate actively together with NBR1 and p62 in ALIS upon autophagy inhibition.
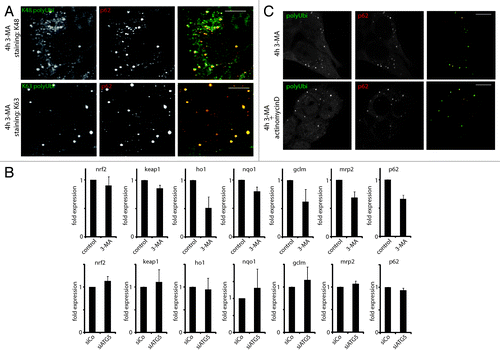
Different affinities of p62 and NBR1 have been reported for different polyubiquitin chains.Citation33 Using K48- and K63-polyubiquitin specific antibodies, we could detect both linkages abundantly in ALIS (), excluding strong specificity of either as an ALIS targeting signal. This is in line with a previous report demonstrating the cellular accumulation of all types of polyubiquitin species upon long-term genetic autophagy ablation.Citation31 In the same report, consequent to autophagy inhibition, the transcriptional activation of the NRF2-dependent antioxydative stress response pathways and of associated transcripts affecting ubiquitin metabolism was proposed to be the major cause of polyubiquitin chains aggregation. However, strongly contrasting with this hypothesis, we could not gather any evidence of NRF2 pathway activation upon short-term autophagy inhibition. Neither 3-MA treatment for 4 h nor efficient siRNA mediated silencing of ATG5 in HeLa cells, induced the transcription of nrf2 and of its well-characterized target genes ho1, nqo1, mrp2, gclm and p62 itself,Citation34 whose expressions were even moderately reduced upon 3-MA treatment (). Complementary to these observations, transcription inhibition with actinomycin did not interfere with ALIS formation in response to 3-MA (), confirming that ALIS formation is not the consequence of an indirect transcriptional response (e.g., p62 mRNA upregulation). Short-term autophagy inhibition induces therefore directly DRiPs targeting to ALIS, independently of NRF2 signaling and, probably, through the aggregative properties of nondegraded autophagy client proteins, p62 and NBR1.
Figure 3. ALIS contain different linkage type polyubiquitin, and form independently of NRF2 signaling and general transcription. (A) HeLa cells were treated for 4 h with 3-MA and stained for the indicated ubiquitin linkage type (K48 and K62 polyUbi, green) and p62 (red). (B) HeLa cells were treated or not for 4 h with 3-MA (top), or transfected with siRNA against ATG5 (siATG5) or nontargeting siRNA (siCo), and qPCR for the indicated genes was carried (mean ± SEM from three independent experiments). (C) HeLa cells were treated or not for 4 h with 3-MA in the presence or absence of Actinomycin D. Cells were stained for ubiquitinated protein (FK2 antibody, polyUbi, green) and p62 (red). Bar, 10 µm.
ALIS are dynamic structures and turn over in a UPS-dependent manner
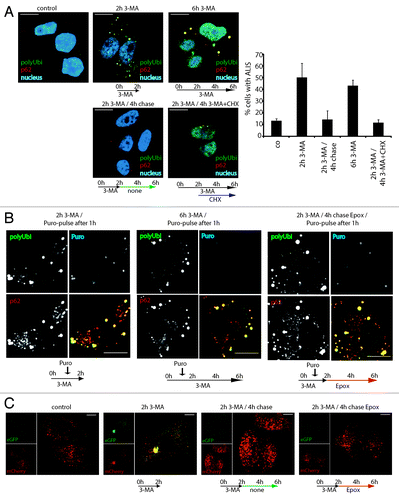
We next investigated the conditions of ALIS turnover and the molecular mechanisms which govern aggregate clearance. We followed ALIS fate in HeLa cells, following treatment with, and subsequent removal of, 3-MA. Autophagy inhibition with 3-MA (2 h) was sufficient to induce aggregates (). Upon drug removal, ALIS were lost within 4h, showing that aggregation is fully reversible. Blocking delivery of DRiPs to ALIS with the translation inhibitor cycloheximide (CHX) induced rapid aggregate clearance, even in the presence of 3-MA (), confirming that the flow of newly synthesized proteins is required to form aggregates upon autophagy inhibition, but their removal is autophagy independent. We could monitor DRiPs turnover in ALIS using puromycin-pulse labeling and chase in combination with different pharmacological inhibitors. A 15 min pulse, followed by a 1h chase in the presence of 3-MA, allowed the detection of puro-DRiPs in ALIS. As expected a 5 h-chase resulted in an almost total puro-DRiPs clearance from ALIS, but conversely to 3-MA treatment, proteasome inhibition by epoxomycin during the chase prevented efficiently this clearance (). This indicates a residency time of DRiPs in ALIS inferior to 4 h, demonstrates turnover of ALIS independently of autophagy, and suggests the involvement of the proteasome-ubiquitin system in the clearance of aggregated proteins. We confirmed that autophagy inhibition itself was indeed reversible during our experimental procedure by expressing a mCherry-eGFP-LC3 fusion protein in HeLa cells. mCherry-eGFP-LC3 allows autophagy flux quantification by monitoring red/green fluorescence intensity ratio, which is strongly influenced by eGFP quenching upon autolysosome acidification.Citation26 Whereas untreated cells contained mostly red and some double positive vesicles, 3-MA treatment reduced the overall number of autophagosomes and led to aberrant targeting of LC3 to large, nonacidic structures, possibly ALIS (). Upon removal of 3-MA, either in the absence or in the presence of epoxomycin, the number and distribution of LC3-positive vesicles appeared normal again. We therefore concluded that 3-MA treatment reversibly inhibits autophagy, allowing normalization upon drug removal and also in the presence of proteasome inhibitors. Thus, autophagy inhibition by 3-MA leads to the reversible aggregation of DRiPs in ALIS, which are turned over within few hours by the UPS independently of autophagic activity.
Figure 4. ALIS are dynamic structures forming upon autophagy inhibition and turning over by the proteasome. (A) HeLa cells were treated for 2 h or 6 h with 3-MA (2 h 3-MA / 6 h 3-MA), washed and left for further 4 h without drug (2 h 3-MA / 4 h chase), or were treated for 2 h with 3-MA and for the next 4 h, cycloheximide (CHX) was added (2 h 3-MA / 4 h 3-MA + CHX). Cells were Triton X-100 permeabilized prior to fixation, and stained for ALIS (p62 and FK2 polyubiquitin antibody) and nuclei (TOPRO-3). Cells were scored for the presence of ALIS (p62 and polyubiquitin double-positive structures outside the nucleus), mean and SEM of three independent experiments (right). (B) HeLa cells were treated for a total of 2 h or 6 h with 3-MA and received a 15 min-pulse of 0.1 µg/µl puromycin after the first hour (2 h 3MA/puro-pulse after 1 h and 6 h 3-MA / puro-pulse after 1 h, respectively), or were treated for 2 h with 3-MA (receiving a 15 min-puro-pulse after 1 h), washed and then treated for the remaining 4 h with Epoxomycin (2 h 3-MA / 4 h chase Epox / puro-pulse after 1 h). Cells were Triton X-100 permeabilized, fixed and stained for polyubiquitinylated protein (FK2 antibody, polyUbi, green), p62 (red) and puromycin-containing proteins (Puro, blue). Treatment schemes are represented at the bottom of the images. (C) Hela cells expressing mCherry-GFP-LC3 were left untreated (control), or treated with 3-MA for 2 h (2 h 3-MA), or treated with 3-MA for 2 h followed by a 4 h chase in the presence (2 h 3-MA + chase Epox) or absence (2 h 3-MA + chase) of epoxomycin. Autophagosome formation and acidification was monitored by confocal microscopy of eGFP and mCherry fluorescence. Treatment schemes are represented at the bottom of the images. All images are representative for at least three independent experiments. Bar, 10 µm.
DRiPs retargeting to proteasomal degradation upon autophagy inhibition
DRiPs are key substrates for MHC I-restricted presentation, thus offering a quantitative assessment of the impact of autophagy on DRiPs degradation. We generated a model cell line (iHeLa) expressing the mouse H-2Kb heavy chain, the reverse tetracycline-controlled transactivator (rtTA), and the cytosolic eGFP-SL8 (SIINFEKL) model antigen under the control of a doxycycline/rtTA-inducible promoter. Induction of eGFP-SL8 expression led to H-2Kb restricted SL8 peptide presentation, which could be quantified using direct eGFP fluorescence measurement and the 25D1.16 antibody specific for H-2Kb/SL8, respectively (). As expected, SL8 presentation in iHeLa was strictly dependent on proteasomal degradation and neosynthesis. Protein synthesis inhibition by CHX blocked any further increase in antigen expression as well as 25D1.16 staining, and proteasome inhibition with MG-132 abolished antigen presentation completely (). Therefore, we considered antigen presentation in this system to be dependent on DRiPs production and subsequent proteasomal degradation. Interestingly, proteasome inhibition increased the accumulation of fluorescent, properly conformed GFP-SL8 by nearly 40%, suggesting that a subset of newly synthesized proteasome-degraded GFP might not be irreversibly misfolded and can become fluorescent if provided with enough time to fold.Citation35 Conversely, autophagy inhibition only weakly increased fluorescent GFP-SL8 levels, while it enhanced antigen presentation by almost 40% (). The effect of autophagy inhibition on antigen presentation was completely blocked by simultaneous protein synthesis inhibition (), as well as upon simultaneous proteasome inhibition (data not shown). The effect of autophagy induction on antigen presentation could not reliably be assessed using this system. Even though rapamycin treatment moderately decreased antigen presentation levels, antigen expression also was affected, and translation was severely reduced (Fig. S3). These findings suggest again a direct processing of DRiPs by autophagy, which are redirected to the UPS pathway upon 3-MA treatment. Autophagy inhibition did not influence overall surface levels of loaded H-2Kb, nor did it increase general protein translation (data not shown). Similar effects on antigen expression and presentation were obtained using siRNA specific for ATG5 and Beclin 1 to inhibit autophagy (). Lysosome inhibition by bafilomycin, however, decreased H-2Kb and endogenous MHCI, indicating detrimental effects on protein trafficking under these conditions (data not shown). ATG5 and Beclin 1 silencing increased SL8 presentation by more than 40% and 20%, respectively, while GFP levels were only modestly increased (up to 15%) (). This demonstrates that regardless of the mode of autophagy inhibition, antigen can be diverted from autophagic to proteasomal processing, promoting their subsequent MHCI presentation.
Figure 5. Autophagy targeted DRiPs can be re-targeted to the proteasome upon autophagy inhibition, increasing MHCI presentation. HSPA8, BAG-3 and BAG-6 are important for GFP DRiPs processing. (A) iHela model cell line: schematic representation of the constitutive expression cassettes for H-2Kb and rtTA, and the inducible model antigen expression cassette, consisting of the TRE promoter and eGFP-SL8 (left). FACS analysis of GFP-SL8 expression and SL8 presentation on H-2Kb (25D1.16) in iHeLa cells upon doxycycline treatment for 16 h (right). (B) iHeLa cells were induced with doxycycline for 6 h and treated (arrow) with MG132 or cycloheximide (CHX), and analyzed for GFP-SL8 expression (left) and Kb/SL8 complexes appearance with the 25-D1.16 antibody staining (right) at the indicated time. (C) iHeLa cells were induced for 6 h, treated with 3-MA alone or in combination with CHX (arrow), and analyzed for GFP-SL8 expression (left) and Kb/SL8 complexes appearance with the 25-D1.16 antibody staining (right) at the indicated time. In (B and C), representative result from at least four independent experiments is shown. (D) iHeLa cells were transfected either with control siRNA (si co), or siRNAs directed against Atg5 (siATG5) or Beclin 1 (siBeclin 1). Forty-eight hours after transfection of indicated siRNA, cells were induced for 16 h and analyzed Kb/SL8 surface appearance (left) and for GFP-SL8 expression (right). Mean ± SEM from at least four independent experiments are shown, p values (Student’s t-test) were **p < 0.001, *p < 0.03. (E) iHeLa cells were transfected either with control siRNA (si co) or siRNAs directed against HSPA8 (HSPA8). Forty-eight hours after transfection, cells were induced for 16 h and analyzed Kb/SL8 surface appearance (left) and for GFP-SL8 expression (right). Mean ± SEM from at least four independent experiments are shown, p values (Student’s t-test) were *p < 0.03. (F) iHeLa cells were transfected either with control siRNA (si co), or siRNAs directed against CHIP (siCHIP), BAG-1 (siBAG-1), BAG-3 (siBAG-3) or BAG-6 (siBAG-6). Forty-eight hours after transfection of indicated siRNA, cells were induced for 16 h and analyzed Kb/SL8 surface appearance (left) and for GFP-SL8 expression (right). Mean ± SEM from at least four independent experiments are shown, p values (Student’s t-test) were *p < 0.03.
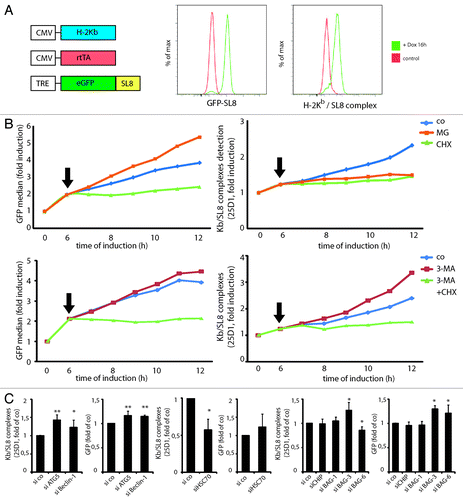
HSPA8, CHIP and BAG co-chaperones modulate differently DRiPs presentation
We then wanted to further validate the iHeLa model system and evaluate the potential of RNAi approaches to unravel the function of specific proteins in MHC I-restricted presentation, and to precisely define the degradation pathways of GFP-SL8 in iHeLa cells. Ubiquitination of misfolded HSPA8 clients involves the E3 ligase CHIP,Citation6 and is modulated by co-chaperones of the Bcl-2-associated athanogene (BAG) family, although the precise role of HSPA8 and these associated molecules in DRiPs processing and subsequent MHCI-restricted presentation has not been extensively addressed.Citation36,Citation37 BAG proteins have been reported to facilitate the degradation of Hsp70/HSPA8 substrates by the proteasome or through autophagy,Citation7,Citation29,Citation38-Citation41 and are therefore likely to also participate in the quality control mechanisms regulating GFP-SL8 DRiPs folding and degradation. We silenced different mRNAs coding for HSPA8Citation42 and associated BAG co-chaperones.Citation29,Citation43 Upon efficient HSPA8 depletion (Fig. S4), GFP-SL8 induction was slightly increased (), while H-2Kb/SL8 levels were strongly reduced. H-2Kb expression and cell survival were not affected (data not shown) during the course of the experiments, excluding general detrimental effects of HSPA8 loss and confirming the major role played by HSPA8 for proteasome targeted DRiPs processing and subsequent antigen presentation. Similar experiments were performed to address the role of CHIP, BAG-1, BAG-3 and BAG-6 in DRiPs processing and antigen presentation. Surprisingly, depletion of CHIP or BAG-1 had no effect on the level of SL8 presentation, suggesting that these two previously characterized enhancers of proteasome degradationCitation29,Citation40 interact with a different set of misfolded HSPA8 clients than GFP-SL8 DRiPs (). BAG-3 depletion however resulted in increased GFP-SL8 antigen expression and H-2Kb/SL8 antigen presentation, in agreement with a putative role of BAG-3 in triggering degradation of DRiPs and other insoluble substrates through autophagy.Citation39 Conversely BAG-6 silencing resulted in weakly, but significantly decreased antigen presentation (), a phenotype compatible with a role in addressing DRiPs to proteasomal degradation as recently suggested by Minami et al.Citation41
Misfolded GFP-SL8 represents both an autophagy and proteasome substrate
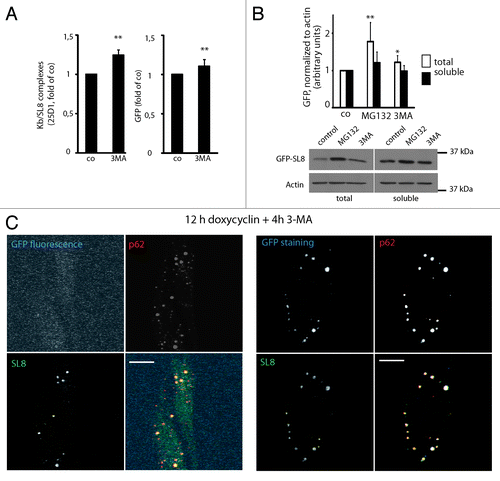
Autophagy inhibition by 3-MA treatment for 4 h in iHeLa cells augmented SL8 antigen presentation by almost 25% and GFP-SL8 fluorescence levels by 10% (). To confirm that GFP-SL8 DRiPs can be bona fide autophagy substrates, besides being subject to proteasome degradation, we quantified GFP-SL8 levels upon autophagy and proteasome inhibition in both total and Triton X-100 soluble fractions. Conversely to proteasome inhibition, which strongly induced both insoluble (78% increase) and soluble (22% increase) GFP accumulation, autophagy inhibition only impacted significantly on total (22% increase) GFP levels, but not on detergent-soluble fractions (). Thus, a fraction GFP-SL8 DRiPs are autophagy substrates and their degradation is likely influenced by their folding state (aggregation prone), which in GFP’s case is reflected by a decrease in its fluorescence potential and accumulation in detergent insoluble fractions. Supporting this conclusion, GFP-SL8 could be abundantly detected by confocal microscopy in Triton X-100 resistant ALIS, using both an anti-GFP antibody, and an antibody raised against the C-terminal SL8 tag, thus confirming the primary sequence integrity of the antigenic construct (). However, GFP fluorescence remained undetectable in the ubiquitinylated aggregates suggesting that the antigenic protein was intact, but inadequately folded. Thus, autophagy degrades a considerable fraction of severely misfolded newly synthesized proteins, which accumulate in ALIS upon autophagy inhibition and can subsequently be degraded by the proteasome to generate MHCI-presented peptides. Furthermore, since autophagy inhibition increased proteasome dependent antigen presentation, we could rule out negative effects on the proteasome by autophagy inhibition in this experimental system.Citation16
Figure 6. GFP-SL8 DRiPs are autophagy substrates, and full-length but misfolded GFP-SL8 accumulates in ALIS. (A) iHela cells were induced with doxycyclin (1 µg/ml) for 12 h and treated for further 4 h with 3-MA. Cells were analyzed Kb/SL8 surface appearance (left) and for GFP-SL8 expression (right). Mean ± SEM from seven independent experiments are shown, p values (Student’s t-test) were **p < 0.01. (B) iHela cells were induced with doxycyclin (1 µg/ml) for 12 h and treated for further 4 h with MG132 or 3-MA. GFP levels were assessed by western blot in both the Triton X-100 soluble extracts (25 µg protein) and in total protein (5 × 104 cells) (bottom). GFP levels were quantified using ImageJ (top, expressed as relative units, normalized to the respective actin control), p values (Student’s t-test) were **p < 0.03. (C) iHeLa cells induced for 16 h prior to treatment with 3-MA (4 h) were permeabilized to remove soluble GFP-SL8, and then fixed. Cells were subsequently analyzed for GFP direct fluorescence, and stained for GFP-SL8 (SL8) and p62 (left), or additionally for total GFP protein localization by anti-GFP antibody staining (right). Scale bar, 10 µm.
NBR1 but not p62 participates in GFP DRiPs addressing to autophagy
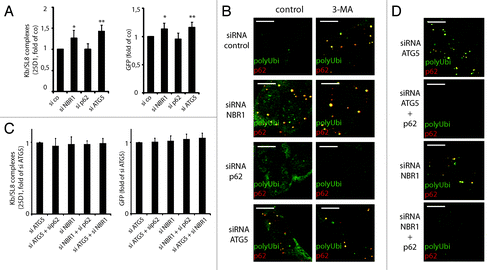
Using siRNA, we then examined, which of the LC3/ATG8 interacting proteins were participating to DRiPs recruitment to autophagy. NBR1 and p62/SQSTM1, which serve as a bridge between ubiquitin and LC3/ATG8 family members, have been reported to shuttle ubiquitinylated proteins to autophagosomes.Citation12 We therefore silenced p62 and NBR1 in iHeLa cells (Fig. S4) and followed Kb/SL8 appearance. Knockdown of NBR1 enhanced Kb/SL8 surface appearance and GFP-SL8 fluorescence almost to the same level that did ATG5 silencing (). In agreement with this observation, loss of p62 did not cause aggregation, and even prevented it upon 3-MA treatment (). NBR1 silencing, on the contrary, induced polyubiquitinated protein accumulation in ALIS, similarly to pharmacological- or ATG5 silencing-mediated inhibition. Thus, although p62 is the driving force leading to aggregation,Citation31 NBR1 is likely to be the adaptor primarily responsible for targeting GFP DRiPs to autophagy.
Figure 7. NBR1 is required for DRiPs targeting to autophagy, while p62 is required for aggregation. (A) iHeLa cells were transfected either with control siRNA (si co), or siRNAs directed against NBR1 (siNBR1), p62 (sip62) or ATG5 (siATG5). Forty-eight hours after transfection of indicated siRNA, cells were induced for 16 h and analyzed Kb/SL8 surface appearance (left) and for GFP-SL8 expression (right). Mean ± SEM from at least four independent experiments are shown, p values (Student’s t-test) were *p < 0.03, **p < 0.001. (B) Cells were transfected with indicated siRNA as above (left), and treated or not with 3-MA for 4 h. Aggregates were visualized by polyubiquitin (FK2 antibody, polyUbi, green) and p62 (red) staining. (C) iHeLa cells were transfected either with control siRNA (si co), or siRNAs directed against ATG5 (siATG5) or NBR1 (siNBR1), alone or in combination with siRNA against p62. Forty-eight hours after transfection of indicated siRNA, cells were induced for 16 h and analyzed Kb/SL8 surface appearance (left) and for GFP-SL8 expression (right). Mean ± SEM from at least four independent experiments are shown. (D) Cells were transfected with indicated siRNA (left), and treated or not with 3-MA for 4 h. Aggregates were visualized by polyubiquitin staining and confocal microscopy.
Proteasome re-targeting of DRiPs does not depend on aggregation
Finally, we wanted to determine if p62 knockdown was dominantly repressing ALIS formation upon ATG5 depletion, and if the macro-aggregation of autophagic DRiPs in ALIS was absolutely required for proteasome addressing. Therefore, we performed double knockdown for ATG5 and p62, and for NBR1 and p62. Silencing of p62 did not decrease the effect of either ATG5 or NBR1 knockdown (). However, it efficiently prevented the large ALIS formation induced by ATG5 or NBR1 siRNA (). This demonstrated that even though p62 drives polyubiquitinated protein in visually detectable aggregates, this extreme phenomenon is not an absolute prerequisite for re-targeting autophagic DRiPs to the proteasome and their subsequent MHC-I restricted presentation. This confirms that accumulation of DRiPs in insoluble fractions is not necessarily followed by macro-aggregate formation, which is clearly indicative of active protein synthesis and of autophagy inhibition, but is likely to also reflect abundant expression of p62.Citation31
Discussion
Aggregation of polyubiquitinated proteins has been observed in different cell types upon exposure to various stressing agents.Citation24-Citation26,Citation44 All aggregates of polyubiquitinated proteins are not equivalent biochemically, and clear definition of the folding state of the aggregated proteins as well as their formation mechanisms is required to understand their cell protective or detrimental functions.Citation45 A subset of cytosolic aggregates, which have been termed ALIS,Citation24 containing autophagy adaptors and polyubiquitinated proteins have been observed following autophagy inhibition.Citation26,Citation33,Citation46-Citation48 ALIS are particularly sensitive to protein synthesis inhibition and are clearly different from the recently characterized insoluble protein deposit (IPOD) and ‘juxta-nuclear quality control’ compartment (JUNQ), notably in their insensitivity to microtubule depolymerizing drugsCitation24 and the nature of aggregated proteins found in the deposits. We also found that proteasome inhibition induced aggregates are strongly enriched for HSPA8, in contrast to ALIS, demonstrating that purely biochemical methods quantitating ubiquitin and p62 solubility are not sufficient to differentiate substrate origin and mechanisms of aggregation.Citation29 In this report, we demonstrated that newly synthesized defective proteins are not only substrates for proteasomal degradation, but are also autophagy substrates. Using conditional expression of a model antigen, we showed that autophagy inhibition re-routes autophagic DRiPs toward ALIS and the proteasome. We identified NBR1 as a key adaptor protein responsible for targeting GFP DRiPs to autophagy. ALIS are thus not a dead-end for DRiPs and represent an intermediate addressing step prior delayed proteasome degradation and subsequent MHCI-restricted presentation. Although ALIS formation is p62/SQSTM1 dependent, p62 does not interfere with the addressing of DRiPs toward autophagosome and is not required for their re-routing toward the UPS pathway upon autophagy inhibition. This set of data also suggests that p62 does not primarily deal with neosynthetized defective proteins or does not function as an autophagy adaptor as recently proposed.Citation31,Citation49
We have extended our current understanding of ALIS formation by demonstrating that a fraction of puro-DRiPs are normally degraded in autophagosomes and are rapidly aggregated with NBR1 and p62 upon autophagy inhibition. Ubiquitination has been shown to target cargo to autophagy,Citation11,Citation12,Citation33 but the precise ubiquitin linkage type, which serves as targeting signal remains unknown. UPS degradation of pre-existing intact proteins, such as p53 or Iκb, and directly ubiquitin-fused protein might differ from bulk degradation of neosynthesized misfolded proteins. Interestingly, we have shown that CHIP or BAG-1 depletion does not impact GFP-DRiPs fate, although these two molecules have been shown to play a major role in quality control of multiple severely misfolded client proteins, such as CFTRΔF508, polyglutamine-expanded huntingtin, ataxin-1 and ataxin-3Citation50 and control antigen presentation in cells infected with adenoviruses.Citation29 Accumulation of prototypical proteasome substrates has also been reported under condition of autophagy inhibition.Citation16 This has been interpreted as a block in UPS flux, mostly due to p62 accumulation and active retention of ubiquitin-linked proteins. Thus different quality control pathways specialized in different type of substrates are at work in parallel and could be influenced differently by p62 accumulation and protein neosynthesis (Fig. S5).
Using MHCI restricted antigen presentation as a quantitative readout, we did not detect any impact of autophagy inhibition on DRiPs proteasomal degradation. MHCI presentation was on the contrary increased in these conditions, likely due to a general raise in proteasome substrate availability. This observation also implies that DRiPs are degraded by both the UPS and autophagy at the same time and that a substrate conversion upon inhibition of one of the two degradative pathways can be implemented (Fig. S5). The impact on MHCI presentation of inducing autophagy by rapamycin treatment or starvation could not be assessed, since both decreased translation, making it impossible to reliably determine an effect on antigen presentation. In the case of the puro-DRiPs used in this study, we could demonstrate rapid accumulation in ALIS upon autophagy inhibition, followed by relatively rapid but not immediate proteasome degradation. K63 polyubiquitin linkage was shown to have the highest affinity for p62 and suggested to serve as an autophagic degradation signal for misfolded proteins.Citation33 However, multiple polyubiquitin chain topologies have been shown to accumulate upon genetic ablation of autophagy,Citation31 thus leading to the conclusion that there is no prevalence of K63 polyubiquitin linkage as an autophagic degradation signal. We confirmed accumulation of both K48- and K63- linked polyubiquitin chains in ALIS, although we could rule out a contribution of de novo transcription of p62 and other nrf2 target genes in this process.Citation31 This is most likely due to key differences between the experimental system used in this report and ours, namely the kinetics of autophagy inhibition and protein aggregation. While we investigated the effects of autophagy inhibition over a few hours up to two days, Nrf2 mediated stress responses were observed after several days to weeks of autophagy ablation.Citation31 Our report therefore demonstrates that short-term autophagy inhibition alone is sufficient to induce ALIS and affect DRiPs turnover. Given the change of DRiPs fate from autophagic to proteasome degradation, a massive reorganization of ubiquitin linkages is also likely to be required to achieve this conversion and could occur in ALIS. This hypothesis is supported by the high turnover of ubiquitin observed in dendritic cells-specific ALIS (DALIS) by FRAP.Citation27 Thus the accumulation of different polyubiquitin chains upon autophagy inhibition could be the result of the cell attempts to dispatch the excess of autophagic substrates in the different degradation pathways still available.
The precise role of the adaptors p62 and NBR1, are currently under intense scrutiny.Citation12 We could demonstrate that p62 is dispensable for autophagic DRiPs degradation, but not for accumulation in ALIS. Furthermore, we could identify NBR1 as the adaptor responsible for addressing DRiPs to autophagy, which is supported by the much faster lysosomal turnover of NBR1 in comparison to p62.Citation33 These data also indicate that polyubiquitinated protein accumulation is a direct consequence of autophagy shutdown, and not of proteasome inhibition.Citation16 The fact that NBR1 has been shown to heterodimerize with p62 and that its depletion induces ALIS formation suggests that the function of p62 cannot be considered independently of NBR1 fate and the stoichiometric ratio of the two molecules might be of great importance to control polyubiquitinated protein aggregation.Citation26 Our previous and current observations also support that DRiPs aggregation after autophagy inhibition occurs in different steps (insolubility, micro-aggregation),Citation27 the latest being the microscopy visible macro-aggregation (ALIS), which is strictly p62-dependent, but is not absolutely required for redirecting autophagic DRiPs toward UPS mediated degradation and MHC I presentation.
A still missing biochemical step in allowing DRiPs recognition by NBR1 is the identification of the ubiquitin-ligases and regulatory proteins responsible for the ubiquitination tagging of these defective proteins allowing their autophagic degradation. The Bcl-2 associated athanogene (BAG) proteins modulate the chaperone activities of HSPA8 and provide a physical link between the chaperone system and the different degradation pathways. Thus BAG proteins are likely candidates to control DRiPs sorting to the different degradation pathways. We confirmed that BAG-6 control DRiPs addressing to the UPS,Citation41 while demonstrating that BAG-3 is probably involved in their autophagy targeting.Citation39 Surprisingly, BAG-1 and CHIP silencing did not impact DRiPs processing nor aggregation, suggesting that, at least three different folding quality control pathways are at work to promote the degradation of defective proteins (Fig. S5).
Interestingly polyubiquitinated oxidized protein aggregates have been observed upon interferon-γ stimulation of cells.Citation44 We are unsure of the nature of these aggregates, since they form with much slower kinetics than ALIS, and require immunoproteasomes for clearance. Interferon-γ has been shown to increase autophagic activity,Citation51 thus ALIS formation is unlikely to depend on the production of reactive oxygen species. Similarly, subsequent autophagic DRiPs processing is unlikely to be immunoproteasome dependent since HeLa cells in steady-state express the immunoproteasome catalytic subunits only at very low levels.Citation52 Our observations clearly imply that autophagy modulates the MHCI presentation pathway by controlling the flow of antigens available for proteasome degradation. Thus the variations in autophagy activity observed in cells exposed to different immunologically relevant stimuli are likely to impact directly on the pool of cytosolic antigens available for MHCI-restricted presentation and change the outcome of the immune response.
Materials and Methods
Cell culture
HeLa cells were maintained in DMEM (Gibco Invitrogen, 41965) supplemented with 10% fetal calf serum (Hyclone, SV30160.03), at 37°C and 5% CO2. iHeLa cells were generated by transfection with pcDNA3.1-H-2Kb (G418 selection), pcDNA-Zeo-rtTA (Zeocin selection) and co-transfection of pTRE-eGFP-SL8 and pTK-Hyg (Clontech, Hygromycin selection). Chemicals used were: 3-methyladenine (Sigma, M9281), puromycin (Sigma, P8833), cycloheximide (Sigma, C7698), Epoxomycin (Tebu, EW8700), MG132 (Tebu, ZW8440), bafilomycin A1 (Applichem, 7823). SiRNA against Beclin 1 (SignalSilence Beclin 1 siRNA I, Cell Signaling), ATG5 (Hs_APG5L_6), p62 (Hs_SQSTM1_1_HP), NBR1 (Hs_M17S2_2), HSC 70 (Hs_HSPA8_11), CHIP (Hs_STUB1_1), BAG-1 (Hs_BAG1_10), BAG-3 (Hs_BAG3_2), BAG-6 (Hs_BAT3_3) or unspecific control (all from Qiagen) were transfected using Lipofectamine2000 (Invitrogen, 11668).
Immunodetection
Twenty-five to 50 µg of TX-100 soluble material, or total protein from 5x104 cells (resuspended directly in 2x reducing sample buffer, boiled and sonicated) was separated by 3% to 15% gradient or 12% nongradient SDS-PAGE prior immunoblotting and chemiluminescence detection. For immunofluorescence, cells on coverslips were fixed with 3.5% PFA and permeabilized with 0,1% Triton X-100. Where indicated, cells were permeabilized (0.2% Triton X-100, 5 min, on ice) prior to fixation (3.5% PFA), in order to remove soluble protein and decrease background signal. Antibodies used were p62 rabbit polyclonal (Santa Cruz Biotechnology, sc-25575), anti-actin (Sigma AC-15, A1978), rabbit anti-ATG5 (n-term, Sigma, A0731), anti-LC-3 (2G6 and 5F10, nanotools), anti-NBR1 (6B11, AbD Serotec), anti-conjugated ubiquitin (clones FK1 and FK2, Biomol), anti-HSPA8 (B6, Santa Cruz), anti-CHIP (rabbit polyclonal, kind gifts from R. TakahashiCitation53 and C. PattersonCitation54), anti-BAG1 (rabbit polyclonal, Santa Cruz), anti-SIINFEKL (IF10.2.2,Citation55), anti-GFP (JL-8, Clontech) and anti-Puromycin (12D10,Citation32). Secondary antibodies were from Jackson Immunoresearch and from Molecular Probes.
Imaging
Immunofluorescence and confocal microscopy was performed with a Zeiss LSM 510 using a 40× and 63× objective and accompanying imaging software. For DALIS scoring, total cell number was determined by TOPRO-3 staining of nuclei, and cells with DALIS (p62 and FK2 positive, extranuclear structures) were counted. Pearson’s coefficient was calculated using ImageJ and the JACoP plugin (using Costes’ automatic threshold and randomization, excluding the nucleus).
Plasmids
All plamids were generated using standard cloning, PCR and fusion PCR techniques. The H-2Kb cDNA (kind gift from F. Momburg, Heidelberg) was inserted into pcDNA3.1. mCherry-eGFP-LC3 cDNA (kind gift from T. JohansenCitation46) was subcloned into pCDNA3.1. The rtTA (Clontech) was subcloned into pcDNA/Zeo. GFP-SL8 was subcloned into pTRE (Clontech).
qPCR
RNA was isolated from iHela cells (RNeasy, Qiagen 74104) and cDNA produced according to standard protocols (Invitrogen). qPCR was performed (Power SYBR green, Applied Biosystems 4367659) using primers hBag3.fwd AACCGA TGTGTGCTTTAGGG, hBag3.rev TTTGCCTCCACCCAA GTTAC, hBag6.fwd ATCTTTGAGCCTGGAGCTGA, hBag6.rev CCGTTGTAGTGGCTGGAAAT. Primers for NRF2 signaling targets were fromCitation34: hNrf2.for ACACGG TCCACAGCTCATC, hNrf2.rev TGTCAATCAAATCCA TGTCCTG, hKeap1.for ATTGGCTGTGTGGAGTTGC, hKeap1.rev CAGGTTGAAGAACTCCTCTTGC, hHO-1.for AACTTTCAGAAGGGCCAGGT, hHO-1.rev CTGGGCTCT CCTTGTTGC, hNQO1.for ATGTATGACAAAGGACCC TTCC, hNQO1.rev TCCCTTGCAGAGAGTACATGG hGCLM.for GACAAAACACAGTTGGAACAGC, hGCLM.rev CAGTCAAATCTGGTGGCATC, hMRP2.for TGAGCATGC TTCCCATGAT, hMRP2.rev CTTCTCTAGCCGCTCTGTGG.
Flow cytometry
Anti-KB/SL8 25D1.16Citation56 and anti H-2Kb (HB-176) were purified and fluorochrome-coupled using the AlexaFluor647 protein labeling kit (Invitrogen, A20173). Data was acquired on a FACS LSRII or CantoII (BD Biosciences) and analyzed using FlowJo (Treestar).
Additional material
Download Zip (553.3 KB)Acknowledgments
The laboratory is supported by la Ligue Nationale Contre le Cancer (‘‘Equipe Labelisée La Ligue’’), l’Agence Nationale de la Recherche ANR 07-MIME-005 “DC-TRANS,” Institut National du Cancer (INCA). T.W. is a Feodor-Lynen-Fellow of the Alexander von Humboldt-foundation and a INCA postdoctoral fellow. S.T. is a Uehara Memorial Foundation fellow and Fondation de la Recherche Médicale fellow. N.Ca. is supported by Association Pour la Recherche Contre le Cancer. We also thank the PICsL Imaging and the CIML Flow Cytometry core facilities for expert technical assistance.
Declaration of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008; 454:1088 - 95; http://dx.doi.org/10.1038/nature07195; PMID: 18756251
- Yewdell JW, Nicchitta CV. The DRiP hypothesis decennial: support, controversy, refinement and extension. Trends Immunol 2006; 27:368 - 73; http://dx.doi.org/10.1016/j.it.2006.06.008; PMID: 16815756
- Princiotta MF, Finzi D, Qian SB, Gibbs J, Schuchmann S, Buttgereit F, et al. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 2003; 18:343 - 54; http://dx.doi.org/10.1016/S1074-7613(03)00051-7; PMID: 12648452
- Navon A, Ciechanover A. The 26 S proteasome: from basic mechanisms to drug targeting. J Biol Chem 2009; 284:33713 - 8; http://dx.doi.org/10.1074/jbc.R109.018481; PMID: 19812037
- Young JC. Mechanisms of the Hsp70 chaperone system. Biochem Cell Biol 2010; 88:291 - 300; http://dx.doi.org/10.1139/O09-175; PMID: 20453930
- McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones 2003; 8:303 - 8; http://dx.doi.org/10.1379/1466-1268(2003)008<0303:CALBTC>2.0.CO;2; PMID: 15115282
- Kabbage M, Dickman MB. The BAG proteins: a ubiquitous family of chaperone regulators. Cell Mol Life Sci 2008; 65:1390 - 402; http://dx.doi.org/10.1007/s00018-008-7535-2; PMID: 18264803
- Yewdell JW. Plumbing the sources of endogenous MHC class I peptide ligands. Curr Opin Immunol 2007; 19:79 - 86; http://dx.doi.org/10.1016/j.coi.2006.11.010; PMID: 17140786
- Khan S, de Giuli R, Schmidtke G, Bruns M, Buchmeier M, van den Broek M, et al. Cutting edge: neosynthesis is required for the presentation of a T cell epitope from a long-lived viral protein. J Immunol 2001; 167:4801 - 4; PMID: 11673482
- Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res 2010; 20:748 - 62; http://dx.doi.org/10.1038/cr.2010.82; PMID: 20548331
- Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA 2008; 105:20567 - 74; http://dx.doi.org/10.1073/pnas.0810611105; PMID: 19074260
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259 - 69; http://dx.doi.org/10.1016/j.molcel.2009.04.026; PMID: 19450525
- Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010; 6:330 - 44; http://dx.doi.org/10.4161/auto.6.3.11226; PMID: 20168092
- Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 2009; 10:1215 - 21; http://dx.doi.org/10.1038/ni.1800; PMID: 19820708
- Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol 2009; 10:659 - 71; http://dx.doi.org/10.1038/nrm2767; PMID: 19773779
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517 - 27; http://dx.doi.org/10.1016/j.molcel.2009.01.021; PMID: 19250912
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007; 447:859 - 63; http://dx.doi.org/10.1038/nature05853; PMID: 17568747
- Cuervo AM, Wong ES, Martinez-Vicente M. Protein degradation, aggregation, and misfolding. Mov Disord 2010; 25:Suppl 1 S49 - 54; http://dx.doi.org/10.1002/mds.22718; PMID: 20187257
- Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol 2006; 172:719 - 31; http://dx.doi.org/10.1083/jcb.200510065; PMID: 16505167
- English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol 2009; 10:480 - 7; http://dx.doi.org/10.1038/ni.1720; PMID: 19305394
- Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007; 26:79 - 92; http://dx.doi.org/10.1016/j.immuni.2006.10.018; PMID: 17182262
- Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci USA 2005; 102:7922 - 7; http://dx.doi.org/10.1073/pnas.0501190102; PMID: 15894616
- Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010; 32:227 - 39; http://dx.doi.org/10.1016/j.immuni.2009.12.006; PMID: 20171125
- Pierre P. Dendritic cells, DRiPs, and DALIS in the control of antigen processing. Immunol Rev 2005; 207:184 - 90; http://dx.doi.org/10.1111/j.0105-2896.2005.00300.x; PMID: 16181336
- Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, et al. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2006; 2:189 - 99; PMID: 16874109
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Lelouard H, Ferrand V, Marguet D, Bania J, Camosseto V, David A, et al. Dendritic cell aggresome-like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J Cell Biol 2004; 164:667 - 75; http://dx.doi.org/10.1083/jcb.200312073; PMID: 14981091
- Lelouard H, Gatti E, Cappello F, Gresser O, Camosseto V, Pierre P. Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature 2002; 417:177 - 82; http://dx.doi.org/10.1038/417177a; PMID: 12000969
- Kettern N, Rogon C, Limmer A, Schild H, Hohfeld J. The Hsc/Hsp70 co-chaperone network controls antigen aggregation and presentation during maturation of professional antigen presenting cells. PLoS ONE 2011; 6:e16398; http://dx.doi.org/10.1371/journal.pone.0016398; PMID: 21283720
- Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009; 8:1986 - 90; http://dx.doi.org/10.4161/cc.8.13.8892; PMID: 19502794
- Riley BE, Kaiser SE, Shaler TA, Ng AC, Hara T, Hipp MS, et al. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol 2010; 191:537 - 52; http://dx.doi.org/10.1083/jcb.201005012; PMID: 21041446
- Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 2009; 6:275 - 7; http://dx.doi.org/10.1038/nmeth.1314; PMID: 19305406
- Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505 - 16; http://dx.doi.org/10.1016/j.molcel.2009.01.020; PMID: 19250911
- Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 2010; 30:3275 - 85; http://dx.doi.org/10.1128/MCB.00248-10; PMID: 20421418
- Qian SB, Reits E, Neefjes J, Deslich JM, Bennink JR, Yewdell JW. Tight linkage between translation and MHC class I peptide ligand generation implies specialized antigen processing for defective ribosomal products. J Immunol 2006; 177:227 - 33; PMID: 16785518
- Yewdell JW. Not such a dismal science: the economics of protein synthesis, folding, degradation and antigen processing. Trends Cell Biol 2001; 11:294 - 7; http://dx.doi.org/10.1016/S0962-8924(01)02030-X; PMID: 11413040
- Kunisawa J, Shastri N. The group II chaperonin TRiC protects proteolytic intermediates from degradation in the MHC class I antigen processing pathway. Mol Cell 2003; 12:565 - 76; http://dx.doi.org/10.1016/j.molcel.2003.08.009; PMID: 14527404
- Alberti S, Esser C, Hohfeld J. BAG-1–a nucleotide exchange factor of Hsc70 with multiple cellular functions. Cell Stress Chaperones 2003; 8:225 - 31; http://dx.doi.org/10.1379/1466-1268(2003)008<0225:BNEFOH>2.0.CO;2; PMID: 14984055
- Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J 2009; 28:889 - 901; http://dx.doi.org/10.1038/emboj.2009.29; PMID: 19229298
- Gehring U. Biological activities of HAP46/BAG-1. The HAP46/BAG-1 protein: regulator of HSP70 chaperones, DNA-binding protein and stimulator of transcription. EMBO Rep 2004; 5:148 - 53; http://dx.doi.org/10.1038/sj.embor.7400083; PMID: 14755308
- Minami R, Hayakawa A, Kagawa H, Yanagi Y, Yokosawa H, Kawahara H. BAG-6 is essential for selective elimination of defective proteasomal substrates. J Cell Biol 2010; 190:637 - 50; http://dx.doi.org/10.1083/jcb.200908092; PMID: 20713601
- Rohde M, Daugaard M, Jensen MH, Helin K, Nylandsted J, Jaattela M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev 2005; 19:570 - 82; http://dx.doi.org/10.1101/gad.305405; PMID: 15741319
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 2005; 62:670 - 84; http://dx.doi.org/10.1007/s00018-004-4464-6; PMID: 15770419
- Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schroter F, et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010; 142:613 - 24; http://dx.doi.org/10.1016/j.cell.2010.07.036; PMID: 20723761
- Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 2010; 11:777 - 88; http://dx.doi.org/10.1038/nrm2993; PMID: 20944667
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149 - 63; http://dx.doi.org/10.1016/j.cell.2007.10.035; PMID: 18083104
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204
- Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol 2011; 192:17 - 27; http://dx.doi.org/10.1083/jcb.201009067; PMID: 21220506
- Sha Y, Pandit L, Zeng S, Eissa NT. A critical role for CHIP in the aggresome pathway. Mol Cell Biol 2009; 29:116 - 28; http://dx.doi.org/10.1128/MCB.00829-08; PMID: 18955503
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119:753 - 66; http://dx.doi.org/10.1016/j.cell.2004.11.038; PMID: 15607973
- Heink S, Ludwig D, Kloetzel PM, Kruger E. IFN-gamma-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc Natl Acad Sci USA 2005; 102:9241 - 6; http://dx.doi.org/10.1073/pnas.0501711102; PMID: 15944226
- Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI, et al. CHIP is associated with Parkin, a gene responsible for familial Parkinson's disease, and enhances its ubiquitin ligase activity. Mol Cell 2002; 10:55 - 67; http://dx.doi.org/10.1016/S1097-2765(02)00583-X; PMID: 12150907
- Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, et al. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol 1999; 19:4535 - 45; PMID: 10330192
- Hilton CJ, Dahl AM, Rock KL. Anti-peptide antibody blocks peptide binding to MHC class I molecules in the endoplasmic reticulum. J Immunol 2001; 166:3952 - 6; PMID: 11238640
- Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity 1997; 6:715 - 26; http://dx.doi.org/10.1016/S1074-7613(00)80447-1; PMID: 9208844