Abstract
Initially described as a nonspecific degradation process induced upon starvation, autophagy is now known also to be involved in the degradation of specific ubiquitinated substrates such as mitochondria, bacteria and aggregated proteins, ensuring crucial functions in cell physiology and immunity. We report here that the deubiquitinating enzyme USP36 controls selective autophagy activation in Drosophila and in human cells. We show that dUsp36 loss of function autonomously inhibits cell growth while activating autophagy. Despite the phenotypic similarity, dUSP36 is not part of the TOR signaling pathway. Autophagy induced by dUsp36 loss of function depends on p62/SQSTM1, an adaptor for delivering cargo marked by polyubiquitin to autophagosomes. Consistent with p62 requirement, dUsp36 mutant cells display nuclear aggregates of ubiquitinated proteins, including Histone H2B, and cytoplasmic ubiquitinated proteins; the latter are eliminated by autophagy. Importantly, USP36 function in p62-dependent selective autophagy is conserved in human cells. Our work identifies a novel, crucial role for a deubiquitinating enzyme in selective autophagy.
Introduction
Autophagy is a generic term, which describes the lysosomal degradation of cytoplasmic material. Three different types of autophagy occur: (1) macroautophagy, in which autophagosomes engulf portions of cytoplasm and eventually fuse with lysosomes; (2) microautophagy, in which lysosomes directly sequester cytosolic components; and (3) chaperone-mediated autophagy, in which unfolded proteins are translocated to lysosomes. Macroautophagy, hereafter referred to as autophagy, is the major lysosomal degradation pathway in cells. It is involved in several physiological and pathological processes such as cellular homeostasis, cell survival, development, neurodegeneration, cancer and defense against infections.Citation1,Citation2 Originally described as a nonspecific degradation process limited to bulk cytosol in response to starvation, recent research has identified selective autophagy pathways for the targeting of specific substrates to autophagy, including senescent organelles, bacteria, viral proteins and aggregated proteins.Citation3-Citation7
The overall molecular mechanisms of autophagy are relatively well understood, due to the discovery of yeast autophagy-related genes (atg) and the core autophagy machinery in all eukaryotic cells from yeast to humans.Citation8,Citation9 During autophagy, a phagophore is formed under the control of the autophagosome formation complex which includes the class III P(I)3-kinase (VPS34) and BECLIN 1 (the ortholog of yeast ATG6).Citation10,Citation11 Elongation of this membrane involves two conjugation systems. The first system results in the association of the cytosolic microtubule-associated protein 1 light-chain 3-I (LC3-I) with phosphatidylethanolamine to generate a lipidated LC3-II form. The second system forms the ATG12–ATG5-ATG16 macromolecular complexes. Both conjugation systems contribute to the completion of the double-membrane-containing autophagosomes which then fuse with lysosomes to generate the degradative single-membraned autolysosomes.
Autophagy is induced upon starvation through a pathway dependent on the TOR (target of rapamycin) kinase.Citation12,Citation13 Under nutrient-rich conditions, the TOR pathway is activated, which prevents activation of the Ser/Thr kinase ATG1 in yeast or ULK1/2 (the orthologs of ATG1) in mammalian cells. Nutrient privation and rapamycin treatment downregulate TOR kinase activity, which results in activation of the ATG1 kinase to induce autophagy.
Recent studies identified ubiquitination as a key signal for selective autophagy. Ubiquitin functions as a tag targeting protein aggregates, bacteria and mitochondria to autophagic degradation.Citation14 The first evidence for the role of ubiquitination in autophagy came from the p62/SQSTM1 protein identified as an adaptor for delivering cargo marked by polyubiquitin to autophagosomes.Citation15,Citation16 p62/SQSTM1 binds to polyubiquitinated protein cargo via a ubiquitin-associated (UBA) domain and to the autophagy machinery via a LC3-interacting region (LIR). It thus functions as a bridge between polyubiquitinated cargo and autophagosomes. Two other proteins, NDP52 and NBR1, were described later, and found to have similar functions to p62/SQSTM1.Citation17,Citation18 Recently ALFY (autophagy-linked FYVE domain) was shown to orchestrate the assembly of autophagosomes to p62/SQSTM1-captured cargo by binding to phosphatidylinositol 3-phosphate (PtdIns(3)P), ATG12-ATG5-ATG16 complex, LC3 and p62.Citation19,Citation20
The mechanisms responsible for the ubiquitination of specific autophagy substrates remain largely unknown except for mitochondria, which have been shown to be polyubiquitinated on VDAC1 by the PARKIN E3 ubiquitin ligase.Citation21 It is likely that ubiquitination of other substrates for selective autophagy is achieved by specific, unknown E3 ligases and highly regulated by deubiquitinating enzymes. These enzymes remove ubiquitin residues from ubiquitinated substrates and function as antagonists of E3 ligases.Citation22 Some of them may regulate the ubiquitination status of selective autophagy substrates. We previously demonstrated that in Drosophila the deubiquitinating enzyme dUSP36 acts as a negative regulator of the NFκB dependent immune deficiency (imd) pathway and deubiquitinates the IMD protein.Citation23 dUsp36 (also known as emperor’s thumb and scrawny) was independently shown to be required for apoptosis regulationCitation24 and for stem cells maintenance through deubiquitination of the Histone H2B protein.Citation25 We report here that dUsp36 loss of function inhibits cell growth and activates autophagy. Although these phenotypes are the hallmark of TOR pathway mutants, we show that dUsp36 is not part of this signaling pathway. Autophagy induced by dUsp36 loss of function is not the cause of the observed growth defects and strictly depends on the p62 adaptor protein. This suggests that dUSP36 prevents the induction of selective ubiquitin-mediated autophagy. Accordingly, dUsp36 inactivation results in nuclear aggregates of ubiquitinated proteins, including ubiquitinated Histone H2B. Cytoplasmic ubiquitinated aggregates are also formed during dUsp36 inactivation, and eliminated by autophagy. This USP36 requirement to prevent selective autophagy is conserved in human cells: silencing of hUSP36 triggers autophagy via a p62-dependent pathway. Our work identifies for the first time a crucial role for a deubiquitinating enzyme in selective autophagy inhibition.
Results
Usp36 loss of function inhibits larval growth and activates autophagy
We previously reported that dUsp36 null mutants are lethal at larval stages.Citation23 To examine the cause of this lethality, larvae homozygous for a null dUsp36 mutationCitation23 were collected. Compared with wild-type larvae (, left), mutant larvae displayed a significantly reduced size without gross morphological defects (, middle), and eventually died, 5 d after egg laying (AEL). Larval length measurements at different time points after hatching showed that dUsp36 mutant larvae grew more slowly than wild-type larvae (). This growth defect is specific to dUsp36 loss of function mutants since it can be completely rescued by ubiquitous expression of the dUSP36 protein (, right).
Figure 1.dUsp36 null mutation impairs organismal growth and activates autophagy. (A) Wild-type (left), dUsp36 null mutant (middle, dUsp36Δ43/dUsp36Δ43) and rescued (right, UAS-dUsp36/+; daGal, dUsp36Δ43/dUsp36Δ43) larvae. (B) Larval length measurements at different time points. Continuous line: wild-type larvae, dotted line: dUsp36 null mutant larvae. Fluorescent microscope imaging of unfixed fat bodies from wild-type (C) or dUsp36 null mutant larvae stained with Lysotracker Red (red) and Hoechst 33342 (blue). Scale bar: 10 µm.
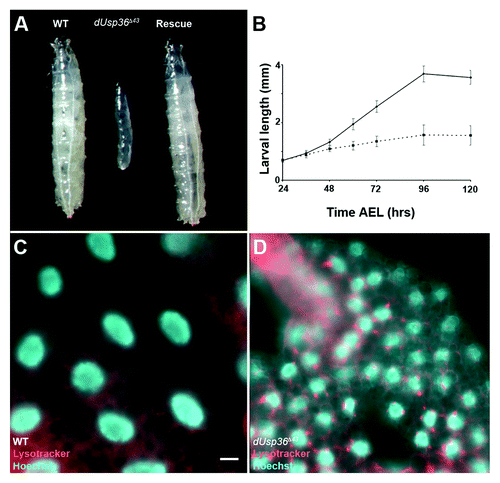
Constitutive activation of autophagy has been reported to cause growth defects in larvae.Citation26 Thus, we monitored autophagy by staining the larval fat body with the Lysotracker Red dye that identifies acidified lysosomes and autolysosomes.Citation13 Due to the low baseline level of lysosomal activity in well-fed early third instar larvae (4 d AEL), this dye did not accumulate in fat body cells of wild-type larvae (). However, in age-matched dUsp36 mutant larvae, numerous acidified lysosomes were observed in the fat body cells suggesting robust autophagy activity ().
dUsp36 function is cell autonomous
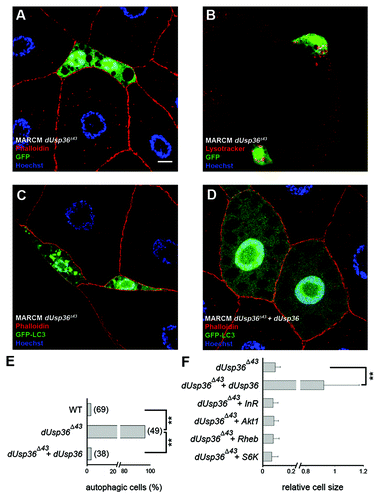
We next asked whether the role dUSP36 plays in cell growth and autophagy is cell autonomous. We induced clones of dUsp36 null mutant fat body cells using the MARCM system,Citation27 which enables the targeted expression of transgenes in cells that have undergone mitotic recombination. In a first set of experiments, we generated homozygous dUsp36 null mutant cells that expressed nuclear GFP. These cells were much smaller than their heterozygous neighbors (relative cell size = 0.09, ) and were positive for LysoTracker staining (). In a second set of experiments, we modified MARCM to allow for the expression of the GFP-LC3 reporter gene following mitotic recombination.Citation28 This reporter encodes a fusion protein between green fluorescent protein and human LC3.Citation29 The expression of GFP-LC3 is diffused in the cytoplasm when autophagy is not induced. However GFP-LC3 is recruited to autophagosomes upon autophagy induction by starvation or rapamycin treatment.Citation13 This experiment revealed that, in mid third instar larvae, 96% of dUsp36 null mutant cells contained autophagosomes compared with 3% of wild-type cells (n = 49 and 69 cells, respectively; p < 0.0001, ). Finally, MARCM induced expression of the dUSP36 protein in dUsp36 mutant cells restored cell size and autophagy to wild-type levels (). We have also performed clonal analysis in the wing imaginal disc and observed that dUsp36 mutant clones are smaller than wild-type clones (Fig. S1), indicating that dUsp36 function is also required in mitotic tissues. Taken together, these results clearly demonstrate that the deubiquitinating enzyme dUSP36 is required cell autonomously to promote cell growth and/or proliferation and to prevent autophagy activation.
Figure 2. Clonal analysis of dUsp36 function in the fat body. dUsp36 null mutant cells expressing nuclear GFP were generated by MARCM and stained with Alexa 546-phalloidin (red) and DAPI (blue) (A) or Lysotracker Red (red) (B). MARCM was also used to express the GFP-LC3 autophagy reporter in dUsp36 null mutant (C) and rescued cells (D). (E) Quantification of autophagy in wild-type, dUsp36 mutant or rescued cells. Autophagy was considered to be activated in a particular cell if at least one GFP-LC3 vesicle was observed. Bracketed numbers represent the number of individual cells examined. (F) Quantification of the relative cell size of dUsp36 mutant, rescued cells, and of dUsp36 mutant cells expressing various members of the TOR pathway. Area measurements of mutant cells relative to surrounding wild-type cells are shown. (**p < 0.001, independence chi-square test). Genotypes: (A and B) y,w,hsFLP/+; Cg-Gal4, UAS-GFPnls/+; FRT80 dUsp36Δ43/FRT80 TubGal80 (C) y,w,hsFLP/+; Cg-Gal4, UAS-GFP-LC3/+; FRT80 dUsp36Δ43/FRT80 TubGal80 (D) y,w,hsFLP/UAS-dUSP36; Cg-Gal4, UAS-GFP-LC3/+; FRT80 dUsp36Δ43/FRT80 TubGal80. Scale bar: 10 µm.
dUSP36 does not function in the TOR pathway
In Drosophila, inhibition of the TOR pathway, either by rapamycin treatment or genetic mutations prevents cell growth and activates autophagy.Citation13,Citation30 The TOR kinase independently controls cell growth partly through the S6 Kinase (S6K) and autophagy through the ATG1 kinase. The phenotypic similarities between TOR and dUsp36 loss of functions suggested that both proteins may be involved in the same signaling pathway, and that dUSP36 may act upstream or at the level of TOR.
As a readout of TOR pathway activity we first monitored the levels of active phosphorylated S6K (P-S6K) in age-matched larvae (Fig. S2A). In young second instar (48 h AEL) larvae we observe no difference between control and dUsp36 silenced larvae whereas a decrease in P-S6K is observed in mid-L2 (60 h AEL) dUsp36 silenced larvae. We think that this late decrease is a consequence of the growth inhibition; indeed, a decrease in P-S6K is also observed by inhibiting the dATAD3A mitochondrial protein,Citation31 which is required for cell growth independently of the TOR pathway. Furthermore comparison of P-S6K levels between 48 h AEL control larvae and 60 h AEL dUsp36 silenced larvae (which have approximately the same size) does not display significant differences. These results thus suggest that dUsp36 loss of function does not directly inhibit the TOR pathway
In order to get functional data we expressed various activators of the TOR pathway in dUsp36 mutant cells by MARCM and examined their ability to rescue dUsp36 phenotypes. We expressed insulin receptor (InR), AKT1 or RHEB proteins, each of which is sufficient to activate the TOR pathway and to increase cell size.Citation13,Citation32 Strikingly, none of these proteins rescued the size defects of dUsp36 mutant cells indicating that dUSP36 does not act upstream of RHEB, the most downstream component of the TOR pathway tested ( and Fig. S2C–E). Since RHEB is a direct activator of the TOR complex, these results suggest that dUSP36 may act either at the level of TOR activation or independently of the TOR pathway. Overexpression of the TOR kinase has detrimental effectsCitation33 and thus could not been assayed directly, so we attempted to rescue the dUsp36 mutant phenotype by expressing the TOR substrate S6 Kinase (S6K). Overexpression of this kinase has been shown to partially rescue the cell growth defect of dTOR mutant cells.Citation13 In contrast, we observed that S6K overexpression did not rescue the cell growth defect of dUsp36 mutant cells indicating that dUSP36 does not function upstream of S6K ( and Fig. S2F). Together, these results indicate that dUSP36 is not part of the TOR pathway.
Cell growth defects and autophagy activation are not mediated by the IMD pathway
Our previous work identified dUSP36 as a negative regulator of the IMD pathway.Citation23 We showed that dUSP36 interacts with and deubiquitinates the IMD protein. dUsp36 silencing results in the constitutive activation of this NFκB dependent signaling pathway and consequently of the connected JNK pathway. Therefore, we asked whether the cell growth inhibition and autophagy activation that we observed upon dUsp36 silencing would depend on inappropriate activation of the IMD pathway. We used the FLPout GAL4/UAS methodCitation34 to clonally activate expression of the GFP-LC3 reporter and transgenes containing specific inverted repeats (IR). This induced RNAi-dependent gene silencing in GFP-LC3 expressing clones. When dUsp36-IR transgene was used in mid third instar larvae to silence dUsp36, we observed localization of GFP-LC3 to autophagosomes in more than 30% of the knocked-down cells (), compared with 96% of the null mutant cells (). This result indicates that RNAi-mediated dUsp36 silencing results in a hypomorphic phenotype. As RNAi-induced phenotypes are known to require more time to develop, autophagy activation was also monitored in late third instar larvae. At this developmental stage autophagy is activated in wild-type cellsCitation28 and we detected autophagosomes in 20% of them (). In contrast, we observed that more than 80% of the knocked-down cells contained autophagosomes (). We also observed that dUsp36-IR expressing cells displayed a strong size reduction (). These results show that silencing dUsp36 by RNAi recapitulates the null mutant phenotype.
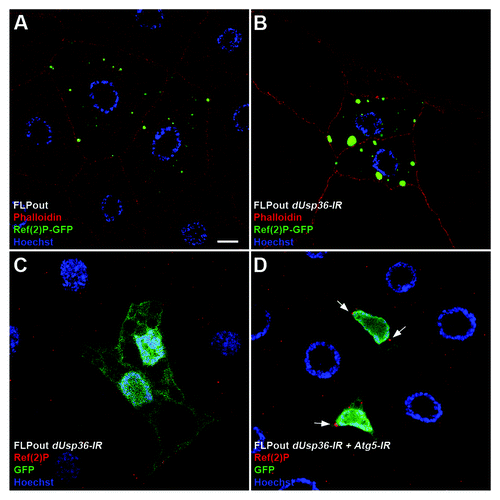
Figure 3. Autophagy activation in dUsp36 silenced cells depends on Atg5 and ref(2)P. (A–C) Mid third instar larvae. Expression of the GFP-LC3 autophagy reporter was clonally activated in fat body cells alone (A) or in combination with a dsRNA trangene targeting dUsp36 (B) using the FLPout method. (C) Quantification of autophagy in cells expressing GFP-LC3 alone (WT) or in combination with the dsRNA trangene targeting dUsp36 (dUsp36-IR). (D–H) Late third instar larvae. The FLPout method was used to express the GFP-LC3 reporter and the dUsp36-IR transgene alone (D) or in combination with the Atg5-IR (E) or ref(2)P-IR (F) transgenes. (G) Quantification of autophagy in wild-type cells, in cells expressing the dUsp36-IR transgene alone, or in combination with the imd-IR, Atg5-IR or ref(2)P-IR transgenes. (H) Quantification of the relative cell size of cells expressing the dATA3A-IR transgene or the dUsp36-IR transgene alone or in combination with the imd-IR, Atg5-IR or ref(2)P-IR transgenes. (A, B and D–F) Confocal sections of fixed fat bodies stained with GFP (green), Alexa 546-phalloidin (red) and DAPI (blue) are shown. G, H: Bracketed numbers represent the number of individual cells examined. (** = p < 0.001, independence chi-square test). Genotypes: (A) y,w,hsFLP/+; UAS-GFP-LC3/+, Ac > CD2 > Gal4/+ (B) y,w,hsFLP/+; UAS-GFP-LC3/UAS-dUsp36-IR, Ac > CD2 > Gal4/+ (D) y,w,hsFLP/+; UAS-GFP-LC3,UAS-dUsp36-IR/+, Ac > CD2 > Gal4/+ (E) y,w,hsFLP/UAS-Atg5-IR; UAS-GFP-LC3,UAS-dUsp36-IR/+, Ac > CD2 > Gal4/+ (F) y,w,hsFLP/+; UAS-GFP-LC3,UAS-dUsp36-IR/UAS-ref(2)P-IR, Ac > CD2 > Gal4/+. Scale bar: 10 µm.
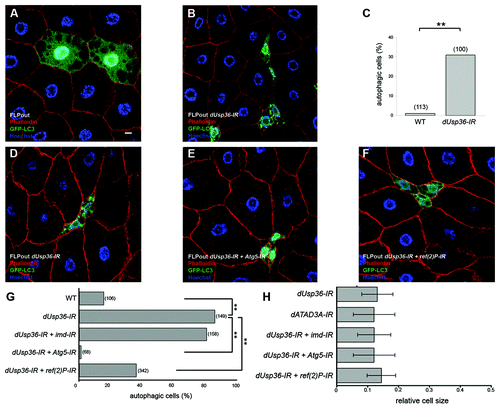
Co-expression of the dUsp36-IR transgene and of an imd-IR transgene, which had been previously shown to efficiently inhibit imd gene expression,Citation23 did not significantly modify the level of autophagy induction () or the cell size reduction (), compared with cells expressing only the dUsp36-IR transgene. These results indicate that autophagy activation and cell growth inhibition induced by dUsp36 silencing are not caused by the activation of the IMD pathway or the connected JNK pathway.
Cell growth inhibition and autophagy activation
Overexpression of the autophagy regulator ATG1 in fat body cells has been shown to strongly activate autophagy and cause cell growth inhibition.Citation26 Thus, we considered the possibility that autophagy activation in dUsp36 mutant fat body cells might lead to the observed growth defect. To test this hypothesis, autophagy was inhibited in dUsp36-IR expressing cells by co-expressing an RNAi transgene targeting Atg5,Citation13 an obligatory component of the autophagy machinery. We observed that cells expressing dUsp36-IR and Atg5-IR showed a dramatic reduction of autophagy activity (). This result indicates that autophagy induced by dUsp36 silencing requires Atg5 function. We then looked at the relative size of cells expressing dUsp36-IR alone or in combination with Atg5-IR. We found no significant differences in the size of cells (). Thus, we conclude that autophagy inhibition does not significantly rescue the size of dUsp36 silenced cells.
We also expressed the dUsp36-IR transgene in the fat body of wild-type or Atg1 null mutant larvae which further confirmed that prevention of autophagy induction in dUsp36-IR expressing cells does not modify the cell growth defect (Fig. S3). Altogether, our findings show that the cell growth inhibition induced by dUsp36 loss of function is not a consequence of autophagy activation.
Conversely, we asked whether cell growth inhibition per se could trigger autophagy. To this end, we inactivated the dATAD3A/belphegor (bor) geneCitation35 which encodes a mitochondrial protein required for cell growth.Citation31 FLPout mediated expression of an RNAi construct targeting dATAD3A resulted in cell size inhibition in the same range as dUsp36 inactivation (). However, none of the dATAD3A knockout cells showed GFP-LC3 recruitment to autophagosomes (n = 38, data not shown). This result indicates that cell growth inhibition is not sufficient to activate autophagy and suggests that dUSP36 plays a specific role in preventing autophagy activation.
Autophagy induced by dUsp36 inactivation requires the p62/Ref(2)P protein
The p62 adaptor protein has been identified as a key component of selective autophagy.Citation16 The Drosophila p62 homolog, refractory to Sigma P (ref(2)P), shares similar functional motifsCitation36,Citation37 and has been shown to play a major role in the formation of ubiquitin-positive protein aggregates in the Drosophila adult brain.Citation38 To address the question of genetic interactions between dUsp36 and ref(2)P, we used two ref(2)P loss-of-function alleles: ref(2)Pod2, which lacks the PB1 domain required for multimerization and formation of protein aggregates and ref(2)Pod3, which lacks the UBA (ubiquitin-associated) domain required for binding to ubiquitinated proteins.Citation39 ref(2)P homozygous mutants are viable and do not show any developmental defects except male sterility.Citation40 Compared with wild-type larvae, dUsp36 null larvae are developmentally delayed and, at 96 h AEL, they are in 2nd larval instar whereas wild-type larvae are in 3rd larval instar (Fig. S4). However, they do not exhibit significant larval lethality before this time point (). In contrast, most of the ref(2)P, dUsp36 double mutant larvae have died by 96 h AEL (). This strong genetic interaction is observed with the two ref(2)P alleles and prompted us to investigate the role of ref(2)P in autophagy induced by dUsp36 inactivation.
Figure 4. Genetic interaction between dUsp36 and ref(2)P. (A) Survival rate of dUsp36 null mutant larvae or of ref(2)P; dUsp36 double mutant larvae at 4 d AEL (**p < 0.001, independence chi-square test, n = 50 for each genotype). Fluorescent microscope imaging of unfixed fat bodies from dUsp36 null mutant larvae (B) or ref(2)Pod2; dUsp36 double mutant larvae (C) stained with Lysotracker Red (red) and Hoechst 33342 (blue). Confocal sections of fixed fat body cells stained with Lysotracker Red (red), GFP (green), and DAPI (blue) from dUsp36 null mutant clone generated either in a wild-type (D) or in a ref(2)Pod2 mutant background (E). Genotypes: (B) dUsp36Δ43/dUsp36Δ43 (C) ref(2)Pod2/ref(2)Pod2; dUsp36Δ43/dUsp36Δ43 (D) y,w,hsFLP/+; FRT79 dUsp36Δ43/FRT79UbiGFP (E) y,w,hsFLP/+; ref(2)Pod2/ref(2)Pod2; FRT79 dUsp36Δ43/FRT79UbiGFP. Scale bar: 10 µm.
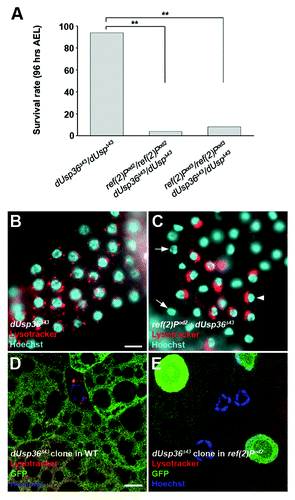
We first performed Lysotracker staining. The pattern of the few surviving double mutants revealed a striking difference compared with that of dUsp36 mutant larvae (). Double mutant fat body cells showed either a complete absence of lysosomes (, arrow) or a juxta-nuclear accumulation of Lysotracker-positive structures (, arrowhead). To confirm this result at the cellular level, we performed Lysotracker staining on clones of dUsp36 null mutant cells generated either in a wild-type or in a ref(2)Pod2 mutant background (). The autolysosomes observed in dUsp36 mutant cells were not present in the ref(2)P; dUsp36 double mutant cells. This result suggests that the p62/Ref(2)P protein is required for autophagy in dUsp36 mutant cells.
We then directly looked at autophagy by using the FLPout method to clonally activate expression of the GFP-LC3 reporter and of RNAi transgenes targeting dUsp36 and/or ref(2)P. We observed that clones of cells co-expressing the dUsp36-IR and ref(2)P-IR transgenes showed a significant reduction of autophagy activity compared with cells expressing dUsp36-IR alone (). We have also quantified autophagy in white pre-pupae, a stage during which nonselective autophagy is strongly activated in fat body cells (Fig. S5). As expected, compared with wild-type (WT) cells, Atg5-IR expressing cells have reduced autophagy activity. This was not the case with ref(2)P-IR expressing cells in which the autophagy levels are not modified. This indicates that Ref(2)P is not required for nonselective autophagy but specifically contributes to autophagy induced by dUsp36 loss of function. Lastly the relative cell size is not significantly modified by ref(2)P inactivation () further indicating that cell growth defects are independent of autophagy induction in dUsp36 mutant cells.
The role of ref(2)P was also confirmed in a dUsp36 null mutant background. For this, the MARCM system was used to drive expression of GFP-LC3 and ref(2)P-IR in dUsp36 null mutant cells. We observed that ref(2)P silencing significantly inhibited autophagy induced in dUsp36 null mutant cells (Fig. S6). Taken together, these results show that the p62/Ref(2)P protein contributes to autophagy induced by dUsp36 inactivation.
dUsp36 loss of function leads to the accumulation of ubiquitinated proteins in both the nucleus and the cytoplasm
We have shown that autophagy in dUsp36 mutant cells is dependent on the p62/Ref(2)P protein, a key component of ubiquitin-targeted autophagy. This suggests that, in the absence of the deubiquitinating enzyme dUSP36, ubiquitinated substrates accumulate and induce selective autophagy. We found that ubiquitinated proteins were readily detected in the nucleus of the dUsp36 mutant cells (). They formed dense dots in close association, but not colocalized with DNA. Importantly, MARCM induced expression of the dUSP36 protein in dUsp36 mutant cells prevented the apparition of these nuclear aggregates (). This indicates that ubiquitinated proteins accumulation results from dUsp36 loss of function. The dUSP36 deubiquitinating enzyme has been shown to deubiquitinate the histone H2B in both Drosophila embryo and stem cells.Citation25 In accordance with a role in H2B ubiquitination, dUsp36 mutant fat body cells accumulated ubiquitinated H2B in their nuclei (Fig. S7). These results show that dUSP36 is required to prevent nuclear accumulation of ubiquitinated proteins, among which the histone H2B was identified.
Figure 5. Ubiquitinated proteins accumulate in dUsp36 mutant cells and are eliminated by autophagy. Confocal sections of fat body cells stained with nuclear GFP (green), the FK2 monoclonal antibody detecting ubiquitinated proteins (red), and DAPI (blue). dUsp36 null mutant cells generated by MARCM (A, A’) and dUsp36 RNAi silenced cells generated by FLPout (C, C’) accumulate nuclear ubiquitinated protein aggregates. These aggregates are not observed in rescued cells (B, B’). Cytoplasmic ubiquitinated protein aggregates are observed upon autophagy inhibition in Atg5; dUsp36 double knockout cells. Genotypes: (A, A’) y,w,hsFLP/+; Cg-Gal4, UAS-GFPnls/+; FRT80 dUsp36Δ43/FRT80 TubGal80 (B, B’) y,w,hsFLP/UAS-dUsp36; Cg-Gal4, UAS-GFPnls/+; FRT80 dUsp36Δ43/FRT80 TubGal80 (C, C’) y,w,hsFLP/+; UAS-GFPnls,UAS-dUsp36-IR/+, Ac > CD2 > Gal4/+ (D, D’) y,w,hsFLP/UAS-Atg5-IR; UAS-GFPnls,UAS-dUsp36-IR/+, Ac > CD2 > Gal4/+. Scale bar: 10 µm.
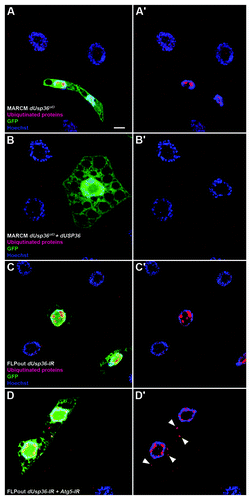
By contrast with the nucleus, ubiquitinated proteins were not readily observed in the cytoplasm of dUsp36 knockout cells (). We hypothesized that their elimination by autophagy may have prevented their detection. Indeed, autophagy inhibition using the Atg5-IR transgene revealed the presence of numerous ubiquitinated proteins in the cytoplasm of dUsp36 silenced cells (). Previous work showed that autophagy inhibition induced accumulation of ubiquitinated aggregates in the DrosophilaCitation38 and mouseCitation41,Citation42 central nervous system. In contrast, this was not observed in Atg5 silenced larval fat body cells (data not shown). This indicates that the observed ubiquitinated proteins are not due to autophagy inhibition by itself, but rather to dUsp36 loss of function. Our observations thus show that dUsp36 loss of function results in the accumulation of ubiquitinated proteins, in both the nucleus and the cytoplasm where they are permanently eliminated by autophagy.
As we had shown that the Ref(2)P/p62 protein contributes to autophagy induced by dUsp36 inactivation, we asked whether it accumulates in dUsp36 mutant cells. We first addressed this point by overexpressing a Ref(2)P-GFP fusion protein in WT and dUsp36-IR expressing cells (). We observed that Ref(2)P-GFP accumulates as dots in WT cells and that the size and intensity of these dots were greatly enhanced in dUsp36 knockdown cells. To rule out the possibility that this result might be due to Ref(2)P-GFP overexpression, we then used an anti-Ref(2)P antibody to study expression of the endogenous Ref(2)P protein. In WT and dUsp36-IR expressing fat body cells, the Ref(2)P protein was detected at very low levels (). In contrast, in cells co-expressing dUsp36-IR and Atg5-IR, larger Ref(2)P-positive dots were observed (arrows, ). This indicates that accumulation of ubiquitinated proteins in dUsp36 mutant cells is correlated with accumulation of Ref(2)P and supports our conclusion that the p62/Ref(2)P protein is required to target ubiquitinated proteins induced upon dUsp36 inactivation to autophagy degradation.
Figure 6. The Ref(2)P/p62 protein accumulates in dUsp36 silenced cells. Confocal sections of fat body cells after FLPout induced expression of the Ref(2)P-GFP fusion protein in wild-type (A) or dUsp36 knock-down cells (B). The size and intensity of Ref(2)P-GFP dots are enhanced by dUsp36 inactivation. Expression of the endogenous Ref(2)P protein in fat body cells expressing the dUsp36-IR transgene alone (C) or in combination with Atg5-IR (D) shows that, as ubiquitinated proteins, the endogenous Ref(2)P protein accumulates in Atg5; dUsp36 double knockout cells. Genotypes: (A) y,w,hsFLP/+; UAS-Ref(2)P-GFP/+, Ac > CD2 > Gal4/+ (B) y,w,hsFLP/+; UAS-Ref(2)P-GFP,UAS-dUsp36-IR/+, Ac > CD2 > Gal4/+ (C) y,w,hsFLP/+; UAS-GFPnls,UAS-dUsp36-IR/+, Ac > CD2 > Gal4/+ (D) y,w,hsFLP/UAS-Atg5-IR; UAS-GFPnls,UAS-dUsp36-IR/+, Ac > CD2 > Gal4/+. Scale bar: 10 µm.
Human USP36 is required for p62-dependent autophagy inhibition
Human and Drosophila USP36 show 37% identity in their catalytic domain and share two conserved domains at their N and C-terminal parts.Citation23,Citation24 The human USP36 protein was described to function in the nucleolus where it deubiquitinates fibrillarin,Citation43 but its role in autophagy is unknown. We asked whether human USP36 would also be required to prevent autophagy activation in cultured cells. We used specific short interference (si)-RNA to reduce hUSP36 expression in GFP-LC3 HeLa cells.Citation7 We observed a drastic increase of GFP-LC3 dots, corresponding to GFP-LC3-containing autophagosomes in siUSP36-treated cells compared with siControl-treated cells (). As observed in Drosophila, hUSP36 inactivation does not inhibit TOR pathway activity as judged by levels of P-S6K and P-4EBP (Fig. S2B). An increase in the lipidated, autophagosome-associated form of LC3 (LC3-II) was also observed in siUSP36-treated cells (). Reduction of ATG5 expression was used as a control of autophagy induction and strongly prevented autophagosome formation (). Furthermore, as observed in Drosophila, reduction of ATG5 expression strongly prevented autophagosome formation in hUSP36 silenced cells (data not shown). We next determined if autophagosome detection subsequent to siUSP36 treatment was the result of an increased autophagic flux or an accumulation of basal autophagosomes due to the inhibition of their degradation. First, we found that, in contrast to siATG5, siUSP36 treatment did not lead to accumulation of endogenous p62/SQSTM1 (). Next, we analyzed autophagy in the presence of bafilomycin A1 (BAF), an inhibitor of autolysosome acidification. We found that BAF treatment further increased the total number of GFP-LC3 puncta observed in siUSP36-treated cells as compared with siControl (). We also observed a clear colocalization of GFP-LC3 and p62 indicating that USP36 dependent autophagy results in p62 recruitment to autophagosomes (Fig. S8A and S8B). Finally, we observed a partial colocalization of the endogenous LC3B protein with the lysosomal marker LAMP-1 (Fig. S8C and S8D) indicating that autophagosomes induced upon hUSP36 inactivation are bona fide autophagosomes capable of fusing with lysosomes. This result strongly supports that reduced expression of USP36 induces de novo autophagosome formation.
Figure 7. The reduced expression of USP36 induces p62-dependent autophagy in human cells. Confocal section of GFP-LC3-HeLa cells transfected with control siRNA (A), or siRNA targeting either ATG5 (B) or USP36 (C). Western blot analysis of LC3 (D) and p62 (E) expressions in total cell lysates. (F–H) Quantification of GFP+ vesicles per cell profile. Comparison of siControl, siATG5 and siUSP36-transfected cells (F), of siControl and siUSP36-transfected cells treated with bafilomycin A1 (BAF, G) and of siControl, siUSP36, sip62 and siUSP36+sip62-transfected cells (H). Results are expressed as the relative fold induction of dots number in siATG5 or siUSP36, sip62 or siUSP36+sip62-treated cells compared with control cells for 3 individual experiments (**p < 0.01, *p < 0.05, Student’s test).
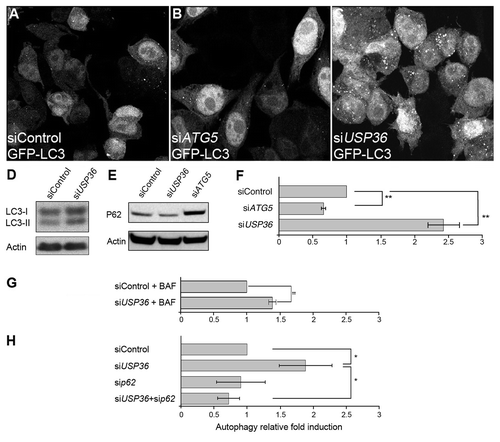
We next tested whether autophagy induced by silencing hUSP36 depends on p62. We found that GFP-LC3 HeLa cells cotransfected with siRNAs targeting hUSP36 and p62 displayed reduced number of autophagosome compared with single siUSP36-transfected cells (). Together, these results show that autophagy induction upon hUSP36 silencing depends on the p62 protein. Thus, the function of hUSP36 in autophagy is conserved between Drosophila and human cells and relies on the expression of p62.
Discussion
Our work identifies USP36 as a deubiquitinating enzyme that inhibits selective, p62-dependent autophagy in Drosophila and human cells and which is further required for cell growth in Drosophila.
We found very reminiscent phenotypes between dUsp36 and dTOR mutants. The TOR pathway regulates autophagy induction upon starvation in human and in Drosophila cells.Citation13 This pathway has also been shown to control cell growth in Drosophila.Citation30 These similarities raised the possibility that USP36 is required for activation of the TOR pathway. However, monitoring TOR pathway activity in Drosophila larvae and in human cells did not convincingly implicate USP36 in this pathway. Moreover, in Drosophila epistastasis experiments indicate that dUSP36 is not part of the TOR pathway. Thus, our results strongly suggest that USP36 does not function in the TOR pathway.
We next considered different hypotheses to account for the growth defect of dUsp36 mutant cells. First, dUSP36 is a negative regulator of the IMD pathway,Citation23 suggesting that the constitutive activation of this pathway may impair Drosophila normal development as previously shown in other tissues.Citation44 Second, constitutive autophagy activation following dUsp36 inactivation may directly inhibit cell growth.Citation26 However, silencing either Imd or Atg5 does not rescue the growth defect of dUsp36 mutant cells, which rules out these two hypotheses. This led us to consider other mechanisms to explain the cell-growth defects induced by dUsp36 loss of function. It has been shown that hUSP36 regulates nucleolar structure and function in human cells.Citation43 If this function is conserved in Drosophila, it could account for the growth defect of dUsp36 mutant cells as a result of a general diminution of protein synthesis and metabolism. However, when we examined fibrillarin nucleolar protein expression by immunofluorescence as well as the levels of rRNA transcription by quantitative RT PCR, we did not observe any evidence of nucleolar dysfunction in dUsp36 mutants (data not shown). In contrast, we observed the accumulation of ubiquitinated proteins and in particular of the ubiquitinated form of the known dUSP36 substrate H2B in the nucleus of the mutant cells. This likely reflects profound defects in the chromatin structure of dUsp36 mutant cells. These defects may interfere with gene expression and potentially inhibit cell growth. Further investigations are needed to determine whether the aberrant ubiquitination of histone H2B contributes to defective cell growth of dUsp36 mutant cells.
Next, we characterized the role of the deubiquitinating enzyme USP36 in autophagy. In both Drosophila and human cell lines we observed accumulation of GFP-LC3 positive vesicles when Usp36 was knocked down. These vesicles were dependent on the presence of ATG1 and ATG5 indicating that they are bona fide autophagosomes. This was confirmed using an anti-LC3B antibody to detect the endogenous LC3 protein: we observed an increase of the lipidated form of LC3 and of the number of autophagosomes following USP36 inactivation in human cell lines. Accumulation of autophagosomes could be due either to de novo autophagosomes formation or to inhibition of autophagosomes maturation. In Drosophila, the increase in autophagosomes number was correlated with an increase of lysosomes. In human cells, normal degradation of the autophagy substrate p62 and partial colocalization of autophagosomes with the lysosomal marker LAMP-1 were observed. Moreover, bafilomycin A1, an inhibitor of autolysosome acidification, did not block the effects of hUSP36 inactivation on autophagy. These results indicate that the whole autophagy flux is activated in the mutant cells. Thus, USP36 inhibits upstream events of the autophagosome formation complex. In order to depict the molecular involvement of USP36 in the autophagy flux regulation, we tested the ability of USP36 to interact with several autophagy-related proteins involved in this process. Through a yeast-2-hybrid assay, we did not identify interactions between hUSP36 and either ATG3/4/9/10/12, LC3, BECLIN 1, or VPS34 (data not shown). However, we showed that autophagy induced by Usp36 knockdown required the p62/SQSTM1 adaptor protein in both Drosophila and human cells. This result implies that Usp36 knockdown induces selective ubiquitin-mediated autophagy. The involvement of p62 suggests that ubiquitinated substrates accumulate following Usp36 inactivation. These ubiquitinated proteins may then be selectively recognized and aggregated by p62, and ultimately targeted to autophagosomes. This hypothesis is supported since cytoplasmic ubiquitinated proteins and p62 itself accumulated in dUsp36 mutant cells in which autophagy is inhibited. Therefore, autophagy is required to degrade ubiquitinated proteins accumulating upon dUsp36 loss of function. Proteasome inhibition is known to promote the accumulation of ubiquitinated proteins and activate autophagy.Citation45 However, USP36 is unlikely to directly interfere with proteosomal function. Indeed, despite extensive proteomic analysis of the proteasome, USP36 has never been described as a proteosome associated deubiquitinating enzyme. In metazoans, the three proteasomal deubiquitinating enzymes are RPN11 (also known as POH1), USP14 (the S. cerevisiae ortholog of which is known as Ubp6), and UCH37 (also known as UCH-L5).Citation46 Alternatively, we propose that USP36 is required for the deubiquitination of specific, as yet unknown, autophagy substrates thereby regulating their targeting to autophagosomes (Fig. S9). Identification of these substrates will provide a better understanding of the molecular mechanisms involved in the selective elimination of autophagy substrates mediated by p62.
Our study demonstrates that by preventing accumulation of ubiquitinated proteins, USP36 inhibits selective autophagy and thus identifies for the first time a deubiquitinating enzyme controlling activation of selective autophagy. Interestingly, a genome-wide siRNA screen in human cells has recently identified two deubiquitinating enzymes, USP19 and USP24, as negative regulators of autophagy under normal nutritional conditions.Citation47 The mechanisms by which these two deubiquitinating enzymes negatively regulate autophagy remain to be elucidated. This suggests that USP36 may not be the only deubiquitinating enzyme acting as an inhibitor of selective autophagy and that the interplay between ubiquitination and deubiquitination participates in selective autophagy regulation.
Materials and Methods
Drosophila stocks and clonal analysis
Flies were reared at 25°C on standard cornmeal–yeast medium. dUsp36Δ43, a null dUsp36 allele, and the UAS-dUsp36 transgene are described in Thevenon et al.Citation23 The strains required for clonal analysis and the UAS-InR, UAS-Akt1 and UAS-S6K transgenic lines are from the Bloomington Stock Center. The strains required for FLPout and the UAS-Rheb transgenic line were from Dr. Leopold’s laboratory. The RNAi transgenes, except UAS-Atg5-IR, were obtained from the Vienna Drosophila RNAi Center. The UAS-GFP-LC3 transgene was provided by Dr. H. Stenmark. The UAS-Atg5-IR and UAS-Ref(2)P-GFP transgenes and the null atg1 allele atg1Δ3D were obtained from Dr. T. Neufeld. The Ref(2)Pod2 and Ref(2)Pod3 mutant alleles were obtained from Dr. S. Gaumer.
For clonal analysis in the fat body, 0–6 h embryos were subjected to heat-shock for 1 h at 37°C and resulting female L3 larvae were dissected at the indicated time point. For the FLPout GAL4/UAS method, spontaneous activation of the GAL4 transcription factor has been reportedCitation48 and allows for the induction of GAL4 expressing cells without heat-shock. Female L3 larvae were dissected at the indicated time point.
Immunocytochemistry and microscopy
Lysotracker staining was performed on unfixed larval fat bodies as described in Scott et al.Citation13 Images were obtained with a Zeiss Axiovert 200M microscope.
For immunocytochemistry, lateral lobes of larval fat bodies were dissected in PBS and fixed for 30 min in 4% paraformaldehyde. For quantification of cell size and autophagy levels, after rinsing in PBS, samples were incubated overnight at 4°C in 2 U/ml Alexa Fluor 546 Phalloidin (Invitrogen, A22283). For antibody staining, samples were blocked for 1 h in PBS, 0.1% Triton X-100, 5% normal goat serum and then incubated overnight at 4°C with the FK2 monoclonal antibody (Millipore, 04–263) diluted 1:10,000, with the anti-ubiquitinated Histone H2B antibody (Millipore, clone 56) diluted 1:500 or with the anti-Ref(2)P antibodyCitation39 diluted 1:500. After mounting in DAPI containing Vectashield (Vector Laboratories, H-1200), the samples were imaged with a 40x or 63x magnification (oil immersion) using a Leica TCS SP2 confocal microscope and the LCS software. Cell size was quantified as the ratio between the areas of a GFP+ cell and of neighboring GFP- cells.
GFP-LC3-HeLa cells were fixed with 4% paraformaldehyde and analyzed using a Zeiss Axiovert 100M microscope equipped with the LSM 510 system and observed with a magnification of 63× (oil immersion). The number of GFP+ vesicles per cell profile was enumerated from one single plan section per cell. The number of GFP+ vesicles was counted using 100 to 200 cells for each independent experiment.
Bafilomycin A1 for flux experiments was purchased from Sigma (Sigma, B1793). GFP-LC3 HeLa cells were treated for 5hrs with 100 nM of bafilomycin.
Human cells and antibodies
HeLa and GFP-LC3-HeLa cells were maintained in RPMI 1640 supplemented with 0.5mg/mL gentamicin, 2mM L-glutamine and 10% fetal calf serum. Anti-actin (A2066), anti-LC3B (L7543) and anti-ATG5 (A0856) were purchased from Sigma. Anti-p62 (sc-28359) was purchased from Santa Cruz biotechnology. Anti-rabbit HRP (NA 934) was purchased from Amersham Biosciences. Anti-LAMP1 (555798) was purchased from BD Pharmigen.
Small interfering RNA experiments
Smartpool si-ATG5, si-p62, si-USP36 and control si-RNA were from Dharmacon. 2.10Citation5 HeLa or HeLa-GFP-LC3 were plated in 6-well plates 24 h prior to transfection with 100 pmol si-RNA using Lipofectamine RNAiMAX (Invitrogen, 13778-100) according to manufacturer’s instructions.
Additional material
Download Zip (760.5 KB)Acknowledgments
We thank J. Jackson and D. Schneider for critical reading of the manuscript. We are grateful to R. Delanoue, S. Gaumer, P. Leopold, T. Neufeld and H. Stenmark for providing mutant and transgenic flies. We also thank S. Gaumer and D. Contamine for the Ref(2)P antibody, D. Grunwald and O. Azocar for help with confocal microscopy, E. Engel and P.E. Joubert for testing and validating siUSP36, and C. Bama and I. Maréchal for fly food preparation. The Bloomington Drosophila Stock Center and Vienna Drosophila RNAi Center contributed to this work by providing mutant and transgenic fly strains. This work was supported by grant from Cluster 10 Infectiologie, Région Rhône Alpes and ANR-08-JCJC-0064-01.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- García-Arencibia M, Hochfeld WE, Toh PP, Rubinsztein DC. Autophagy, a guardian against neurodegeneration. Semin Cell Dev Biol 2010; 21:691 - 8; http://dx.doi.org/10.1016/j.semcdb.2010.02.008; PMID: 20188203
- Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res 2010; 70:3431 - 4; http://dx.doi.org/10.1158/0008-5472.CAN-09-4027; PMID: 20424122
- Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009; 30:588 - 98; http://dx.doi.org/10.1016/j.immuni.2009.02.009; PMID: 19362021
- Deretic V. Autophagy in infection. Curr Opin Cell Biol 2010; 22:252 - 62; http://dx.doi.org/10.1016/j.ceb.2009.12.009; PMID: 20116986
- Yano T, Mita S, Ohmori H, Oshima Y, Fujimoto Y, Ueda R, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat Immunol 2008; 9:908 - 16; http://dx.doi.org/10.1038/ni.1634; PMID: 18604211
- Kanki T, Klionsky DJ. The molecular mechanism of mitochondria autophagy in yeast. Mol Microbiol 2010; 75:795 - 800; http://dx.doi.org/10.1111/j.1365-2958.2009.07035.x; PMID: 20487284
- Joubert PE, Meiffren G, Grégoire IP, Pontini G, Richetta C, Flacher M, et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009; 6:354 - 66; http://dx.doi.org/10.1016/j.chom.2009.09.006; PMID: 19837375
- Inoue Y, Klionsky DJ. Regulation of macroautophagy in Saccharomyces cerevisiae. Semin Cell Dev Biol 2010; 21:664 - 70; http://dx.doi.org/10.1016/j.semcdb.2010.03.009; PMID: 20359542
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102 - 9; http://dx.doi.org/10.1038/ncb1007-1102; PMID: 17909521
- Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 2005; 1:46 - 52; http://dx.doi.org/10.4161/auto.1.1.1542; PMID: 16874027
- Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci 2006; 119:259 - 70; http://dx.doi.org/10.1242/jcs.02735; PMID: 16390869
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124 - 31; http://dx.doi.org/10.1016/j.ceb.2009.11.014; PMID: 20034776
- Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell 2004; 7:167 - 78; http://dx.doi.org/10.1016/j.devcel.2004.07.009; PMID: 15296714
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259 - 69; http://dx.doi.org/10.1016/j.molcel.2009.04.026; PMID: 19450525
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Kirkin V, Lamark T, Johansen T, Dikic I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 2009; 5:732 - 3; http://dx.doi.org/10.4161/auto.5.5.8566; PMID: 19398892
- Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy 2011; 7:341 - 5; http://dx.doi.org/10.4161/auto.7.3.14046; PMID: 21079414
- Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010; 6:330 - 44; http://dx.doi.org/10.4161/auto.6.3.11226; PMID: 20168092
- Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 2010; 38:265 - 79; http://dx.doi.org/10.1016/j.molcel.2010.04.007; PMID: 20417604
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119 - 31; http://dx.doi.org/10.1038/ncb2012; PMID: 20098416
- Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005; 123:773 - 86; http://dx.doi.org/10.1016/j.cell.2005.11.007; PMID: 16325574
- Thevenon D, Engel E, Avet-Rochex A, Gottar M, Bergeret E, Tricoire H, et al. The Drosophila ubiquitin-specific protease dUSP36/Scny targets IMD to prevent constitutive immune signaling. Cell Host Microbe 2009; 6:309 - 20; http://dx.doi.org/10.1016/j.chom.2009.09.007; PMID: 19837371
- Ribaya JP, Ranmuthu M, Copeland J, Boyarskiy S, Blair AP, Hay B, et al. The deubiquitinase emperor’s thumb is a regulator of apoptosis in Drosophila. Dev Biol 2009; 329:25 - 35; http://dx.doi.org/10.1016/j.ydbio.2009.02.005; PMID: 19217892
- Buszczak M, Paterno S, Spradling AC. Drosophila stem cells share a common requirement for the histone H2B ubiquitin protease scrawny. Science 2009; 323:248 - 51; http://dx.doi.org/10.1126/science.1165678; PMID: 19039105
- Scott RC, Juhász G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol 2007; 17:1 - 11; http://dx.doi.org/10.1016/j.cub.2006.10.053; PMID: 17208179
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci 2001; 24:251 - 4; http://dx.doi.org/10.1016/S0166-2236(00)01791-4; PMID: 11311363
- Rusten TE, Lindmo K, Juhász G, Sass M, Seglen PO, Brech A, et al. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell 2004; 7:179 - 92; http://dx.doi.org/10.1016/j.devcel.2004.07.005; PMID: 15296715
- Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol 2009; 452:1 - 12; http://dx.doi.org/10.1016/S0076-6879(08)03601-X; PMID: 19200872
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev 2000; 14:2712 - 24; http://dx.doi.org/10.1101/gad.835000; PMID: 11069888
- Gilquin B, Taillebourg E, Cherradi N, Hubstenberger A, Gay O, Merle N, et al. The AAA+ ATPase ATAD3A controls mitochondrial dynamics at the interface of the inner and outer membranes. Mol Cell Biol 2010; 30:1984 - 96; http://dx.doi.org/10.1128/MCB.00007-10; PMID: 20154147
- Juhász G, Hill JH, Yan Y, Sass M, Baehrecke EH, Backer JM, et al. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol 2008; 181:655 - 66; http://dx.doi.org/10.1083/jcb.200712051; PMID: 18474623
- Hennig KM, Neufeld TP. Inhibition of cellular growth and proliferation by dTOR overexpression in Drosophila. Genesis 2002; 34:107 - 10; http://dx.doi.org/10.1002/gene.10139; PMID: 12324961
- Pignoni F, Zipursky SL. Induction of Drosophila eye development by decapentaplegic. Development 1997; 124:271 - 8; PMID: 9053304
- Calgaro S, Boube M, Cribbs DL, Bourbon HM. The Drosophila gene taranis encodes a novel trithorax group member potentially linked to the cell cycle regulatory apparatus. Genetics 2002; 160:547 - 60; PMID: 11861561
- Avila A, Silverman N, Diaz-Meco MT, Moscat J. The Drosophila atypical protein kinase C-ref(2)p complex constitutes a conserved module for signaling in the toll pathway. Mol Cell Biol 2002; 22:8787 - 95; http://dx.doi.org/10.1128/MCB.22.24.8787-8795.2002; PMID: 12446795
- Carré-Mlouka A, Gaumer S, Gay P, Petitjean AM, Coulondre C, Dru P, et al. Control of sigma virus multiplication by the ref(2)P gene of Drosophila melanogaster: an in vivo study of the PB1 domain of Ref(2)P. Genetics 2007; 176:409 - 19; http://dx.doi.org/10.1534/genetics.106.063826; PMID: 17409092
- Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, et al. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol 2008; 180:1065 - 71; http://dx.doi.org/10.1083/jcb.200711108; PMID: 18347073
- Wyers F, Petitjean AM, Dru P, Gay P, Contamine D. Localization of domains within the Drosophila Ref(2)P protein involved in the intracellular control of sigma rhabdovirus multiplication. J Virol 1995; 69:4463 - 70; PMID: 7769706
- Dezelee S, Bras F, Contamine D, Lopez-Ferber M, Segretain D, Teninges D. Molecular analysis of ref(2)P, a Drosophila gene implicated in sigma rhabdovirus multiplication and necessary for male fertility. EMBO J 1989; 8:3437 - 46; PMID: 2510997
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/10.1038/nature04723; PMID: 16625205
- Endo A, Matsumoto M, Inada T, Yamamoto A, Nakayama KI, Kitamura N, et al. Nucleolar structure and function are regulated by the deubiquitylating enzyme USP36. J Cell Sci 2009; 122:678 - 86; http://dx.doi.org/10.1242/jcs.044461; PMID: 19208757
- Maillet F, Bischoff V, Vignal C, Hoffmann J, Royet J. The Drosophila peptidoglycan recognition protein PGRP-LF blocks PGRP-LC and IMD/JNK pathway activation. Cell Host Microbe 2008; 3:293 - 303; http://dx.doi.org/10.1016/j.chom.2008.04.002; PMID: 18474356
- Jänen SB, Chaachouay H, Richter-Landsberg C. Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia 2010; 58:1766 - 74; http://dx.doi.org/10.1002/glia.21047; PMID: 20645412
- Lee MJ, Lee BH, Hanna J, King RW, Finley D. Trimming of ubiquitin chains by proteasome-associated deubiquitinating enzymes. Mol Cell Proteomics 2011; 10:R110.003871; http://dx.doi.org/10.1074/mcp.R110.003871; PMID: 20823120
- Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell 2010; 18:1041 - 52; http://dx.doi.org/10.1016/j.devcel.2010.05.005; PMID: 20627085
- Hennig KM, Colombani J, Neufeld TP. TOR coordinates bulk and targeted endocytosis in the Drosophila melanogaster fat body to regulate cell growth. J Cell Biol 2006; 173:963 - 74; http://dx.doi.org/10.1083/jcb.200511140; PMID: 16785324