Abstract
Direct intercellular communication mediated by gap junctions (GJs) is a hallmark of normal cell and tissue physiology. In addition, GJs significantly contribute to physical cell-cell adhesion. Clearly, these cellular functions require precise modulation. Typically, GJs represent arrays of hundreds to thousands of densely packed channels, each one assembled from two half-channels (connexons), that dock head-on in the extracellular space to form the channel arrays that link neighboring cells together. Interestingly, docked GJ channels cannot be separated into connexons under physiological conditions, posing potential challenges to GJ channel renewal and physical cell-cell separation. We described previously that cells continuously—and effectively after treatment with natural inflammatory mediators—internalize their GJs in an endo-/exocytosis process that utilizes clathrin-mediated endocytosis components, thus enabling these critical cellular functions. GJ internalization generates characteristic cytoplasmic double-membrane vesicles, described and termed earlier annular GJs (AGJs) or connexosomes. Here, using expression of the major fluorescent-tagged GJ protein, connexin 43 (Cx43-GFP/YFP/mApple) in HeLa cells, analysis of endogenously expressed Cx43, ultrastructural analyses, confocal colocalization microscopy, pharmacological and molecular biological RNAi approaches depleting cells of key-autophagic proteins, we provide compelling evidence that GJs, following internalization, are degraded by autophagy. The ubiquitin-binding protein p62/sequestosome 1 was identified in targeting internalized GJs to autophagic degradation. While previous studies identified proteasomal and endo-/lysosomal pathways in Cx43 and GJ degradation, our study provides novel molecular and mechanistic insights into an alternative GJ degradation pathway. Its recent link to health and disease lends additional importance to this GJ degradation mechanism and to autophagy in general.
Introduction
Direct cell-to-cell communication is a pivotal cellular function of multicellular organisms. It is established by gap junction (GJ) channels, that bridge apposing plasma membranes of neighboring cells. Typically, hundreds to thousands of GJ channels cluster into densely packed two-dimensional arrays, termed GJ plaques that can reach several square-micrometers in size. In addition to providing intercellular communication, GJs, based on their characteristic double-membrane configuration, significantly contribute to physical cell-to-cell adhesion. The ability to modulate (up- and downregulate) the level of GJ-mediated intercellular communication (GJIC), and of physical cell-cell adhesion is as vitally important as the basic ability of GJ formation itself; and is for example crucial for many physiological and pathological conditions, including cell migration during development and wound healing, mitosis, apoptosis, leukocyte extravasation, ischemia, hemorrhage, edema, and cancer metastasis.
GJ channels are assembled from a ubiquitously expressed class of four-pass trans-membrane proteins, termed connexins, with connexin 43 (Cx43) being the most abundantly expressed connexin type. Six connexin polypeptides oligomerize into a ring to form a hexameric structure with a central hydrophilic pore, called hemi-channel or connexon. Once trafficked to the plasma membrane, two connexons, one provided by each of two neighboring cells, dock head-on in the extracellular space to form the complete, tightly sealed to the outside, transmembrane GJ channel. Recruitment of additional GJ channels along the outer edge then enlarges the channel plaques, while simultaneous removal of older channels from plaque centers balances GJ channel turnover.Citation1-Citation3
While GJ channels can open and close (gate) to regulate electrical and chemical cell-cell coupling, GJ channel gating does not provide a means for modulating cell-to-cell adhesion, or for plasma membrane GJ channel renewal. Moreover, docked GJ connexons were found to be inseparable under physiological conditions,Citation4 posing potential challenges to these cellular functions. How is then the removal of GJ channels from the plasma membrane achieved?
We reported previously that cells appear to continuously internalize and turn over their GJs via a combined endo-/exocytic process that utilizes clathrin-mediated endocytosis components.Citation5-Citation8 GJ internalization generates characteristic, cytoplasmic double-membrane GJ vesicles, termed earlier annular GJs (AGJs) or connexosomes, preferentially in one of two coupled cells. We and others further found that internalization can occur highly efficient and regulated, for example in response to natural inflammatory mediators such as thrombin and endothelin,Citation5 well-known inhibitors of GJIC;Citation9-Citation12 and under pathological conditions as e.g., in the failing canine ventricular myocardium.Citation13 Continuous, as well as spontaneous internalization of GJ channels as complete, double-membrane spanning protein structures is supported by the fundamental observation that connexons, once docked, are inseparable under physiological conditions,Citation4 and by a general, surprisingly short half-life of Cxs and GJ channels of only 1–5 h.Citation2,Citation3,Citation14-Citation16 A similar internalization of GJs into cytoplasmic AGJ vesicles has been observed by others in cells in culture, as well as in situ in tissues.Citation13,Citation17-Citation22
Previous studies identified proteasomal, endo-/lysosomal, and to a lesser extent phago-/lysosomal degradation pathways in the regulation of GJ stability and connexin protein degradation.Citation13,Citation20,Citation23-Citation27 We thus embarked on investigating, on a molecular level, the fate of internalized AGJ vesicles. Following internalization, AGJ vesicles were observed to move away from the plasma membrane and to translocate deeper into the cytoplasm in a process that involves actin-filaments and the retrograde actin motor protein myosin VI (myo6).Citation8 Here we report that GJs, following internalization into cytoplasmic AGJ vesicles, are targeted to autophagosomes via the ubiquitin-binding protein, p62/SQSTM1, and are degraded by autophagy.
Results
GJs lose fluorescence subsequent to internalization into AGJ vesicles suggesting degradation
Using expression of C-terminal fluorescent protein-tagged Cx43 (Cx43-GFP/YFP) in HeLa cells, combined with time-lapse live-cell imaging, others and we found that entire GJs, or large portions thereof, continuously appear to be internalized into the cytoplasm.Citation8,Citation18 Characteristic GJ plaques, visible as straight green-fluorescent lines and puncta, assemble in the plasma membranes between expressing cells (, panel 1; , time points 0:00 to 4 h:44 min; Movie 1). Over time, plaques were observed to bulge inward, detach from the plasma membrane and form spherical cytoplasmic GJ vesicles, termed previously annular gap junctions (AGJs) or connexosomes, within 10–60 min (, panels 2 and 3; , time points 4 h:48 min to 5 h:28 min; Movie 1).Citation8,Citation18 Note that the outer membrane of the AGJ vesicle corresponds to plasma membrane of the host-cell, while inner membrane and vesicle lumen correspond to plasma membrane and cytoplasm of the neighboring donor cell ().Citation8 We also characterized similar stages of AGJ vesicle formation by electron microscopic ultrastructural analyses.Citation8 Internalization was found to occur preferentially into one of two coupled cells, indicating a highly regulated process.Citation8 The scaffolding protein, ZO-1, a well-known binding protein of Cx43Citation28-Citation30 was found to be displaced from GJ plaques on the side to which the plaques invaginate, potentially regulating directionality of internalization.Citation5,Citation6 Further analyses indicated that GJ internalization involves the classical endocytic coat protein, clathrin, the clathrin-adaptors AP-2 and Dab2, the GTPase dynamin2, actin polymerization, and the retrograde actin motor, myosin VI (myo6).Citation7,Citation8 Furthermore, we confirmed that efficient GJ internalization can occur under normal conditions in vivo, for example in primary porcine pulmonary artery endothelial cells (PAECs) in response to natural inflammatory mediators such as thrombin and endothelin,Citation5 or in Sertoli cells in response to the nongenomic carcinogen lindane.Citation6 Similar cytoplasmic AGJ vesicles were previously observed in ultrastructural analyses in cells in situ, especially in differentiating tissues, sometimes in association with lysosomes;Citation13,Citation17,Citation19-Citation21,Citation27,Citation31 and were seen to form in living Cx43-GFP-transfected NRK cells.Citation18 We therefore wanted to investigate what will happen to AGJ vesicles after their generation. Time-lapse recordings of GFP-, or YFP-tagged Cx43-expressing HeLa cells showed that AGJ vesicles moved about in the cytoplasm, sometimes fragmented into smaller vesicles in a process that appeared similar to vesicle budding,Citation8 and finally their fluorescence disappeared, suggesting degradation (, time point 19 h:16 min; Movie 1).
Figure 1. Plasma membrane GJs internalize to form cytoplasmic double-membrane GJ vesicles [termed annular gap junctions (AGJs) or connexosomes] that subsequently are degraded. (A) HeLa cells transfected with Cx43-GFP efficiently express and assemble GJs in the adjacent plasma membranes of transfected cells after overnight expression (visible as green fluorescent lines and puncta, such as the one shown in insert 1). Over time, GJs bulge inward (insert 2), detach from the plasma membrane and form cytoplasmic AGJ vesicles or connexosomes (insert 3). (B) Selected still-images of a time-lapse recording of stably transfected Cx43-YFP expressing HeLa cells covering over 19 h (2 min image intervals), showing the formation of a GJ plaque, its endocytic internalization into the cytoplasm of one of the previously coupled cells, and final degradation of the generated AGJ vesicle, indicated by the loss of its fluorescence (marked with arrows). Combined phase contrast and fluorescence images are shown in (A and B) and in Movie 1. (C) Schematic representation depicting the progressive stages of GJ internalization. For ultrastructural and additional analyses of internalizing GJs see reference Citation8. (D) western blot analyses of total Cx43-GFP, or Cx43-YFP protein in transiently and stably expressing HeLa cells 72 h after BECN1 protein was depleted by RNAi-oligonucleotide transfection. SI-control cells were transfected with a scrambled non-targeting RISC-activating control oligonucleotide. Cx43-GFP/YFP was detected by probing with polyclonal anti-Cx43 antibodies. After stripping, blots were re-probed with anti-α-tubulin antibodies as a loading control. Normalized quantitative analyses revealed a two to 3-fold increase of Cx43-GFP/YFP in the BECN1-KD over SI-control cells.
![Figure 1. Plasma membrane GJs internalize to form cytoplasmic double-membrane GJ vesicles [termed annular gap junctions (AGJs) or connexosomes] that subsequently are degraded. (A) HeLa cells transfected with Cx43-GFP efficiently express and assemble GJs in the adjacent plasma membranes of transfected cells after overnight expression (visible as green fluorescent lines and puncta, such as the one shown in insert 1). Over time, GJs bulge inward (insert 2), detach from the plasma membrane and form cytoplasmic AGJ vesicles or connexosomes (insert 3). (B) Selected still-images of a time-lapse recording of stably transfected Cx43-YFP expressing HeLa cells covering over 19 h (2 min image intervals), showing the formation of a GJ plaque, its endocytic internalization into the cytoplasm of one of the previously coupled cells, and final degradation of the generated AGJ vesicle, indicated by the loss of its fluorescence (marked with arrows). Combined phase contrast and fluorescence images are shown in (A and B) and in Movie 1. (C) Schematic representation depicting the progressive stages of GJ internalization. For ultrastructural and additional analyses of internalizing GJs see reference Citation8. (D) western blot analyses of total Cx43-GFP, or Cx43-YFP protein in transiently and stably expressing HeLa cells 72 h after BECN1 protein was depleted by RNAi-oligonucleotide transfection. SI-control cells were transfected with a scrambled non-targeting RISC-activating control oligonucleotide. Cx43-GFP/YFP was detected by probing with polyclonal anti-Cx43 antibodies. After stripping, blots were re-probed with anti-α-tubulin antibodies as a loading control. Normalized quantitative analyses revealed a two to 3-fold increase of Cx43-GFP/YFP in the BECN1-KD over SI-control cells.](/cms/asset/d557a1b5-28a2-4ad9-8abf-40ebe16bf4b9/kaup_a_10919390_f0001.gif)
Total Cx43 protein accumulates in cells in which autophagosomal degradation is inhibited
Cells have developed proteasomal, endo-/lysosomal and phago-/lysosomal (autophagic) degradation machineries, and all three have been linked to connexin protein and GJ degradation.Citation13,Citation20,Citation23-Citation27 While proteasomal degradation in general degrades short-lived, misfolded or otherwise unwanted polypeptides, endo-/lysosomal and phago-/lysosomal pathways allow the degradation of more complex protein structures that either are endocytosed at the plasma membrane, or are present in the cytoplasm. In both, endo-/lysosomal as well as autophagic degradation pathways, the lysosome plays a key role in mediating final degradation. Internalized, AGJ vesicles are cytoplasmic, complex multiprotein structures and thus might be degraded via autophagy (macroautophagy). To test this hypothesis, we depleted HeLa cells, either transiently, or stably expressing Cx43-GFP or Cx43-YFP, of a key-autophagic protein, BECN1/(Atg6) (see below for a more detailed description), using RNAi-technology, and analyzed total Cx43-GFP/YFP protein by western blot analyses (). Qualitative, as well as quantitative analyses indicated a two- to three-fold increase of FP-tagged Cx43 protein after 72 h in BECN1 knockdown (KD) cells compared with SI-control cells, suggesting that GJs, following their internalization into AGJ vesicles are degraded by autophagy. This hypothesis was explored further in the experiments described below.
AGJ vesicles robustly colocalize with lysosomal and autophagosomal marker proteins but only insignificantly with endosomal markers
To further investigate how AGJ vesicles are degraded, we performed confocal colocalization studies of AGJ vesicles formed in Cx43-GFP expressing HeLa cells with fluorescent probes and antibodies specific for endosomal, lysosomal, and autophagosomal compartments (, marked in blue). AGJ vesicles were recognized by their large size (0.5–2 μm in diameter) that is much larger than the diameter of secretory GJ vesicles (in general ≤ 0.2 μm in diameter), their bright fluorescence, and their spherical structure (see , panel 3; , time point 5 h:28 min; and refs.Citation1, Citation2, Citation7 and Citation8). For proteins directly involved in the endosomal degradation pathway, such as the early endosomal antigen 1 (EEA1), and the late endosomal marker Rab7, only weak and infrequent colocalization with small GJ vesicles was detected (, panels b and c). In contrast, a more robust colocalization of GJ vesicles was observed with the lysosomal-associated membrane proteins, LAMP-1 (, panels d and e) and LAMP-2 (, panel 3), and with the acidophilic compartment-specific live cell probe, LysoTracker (, panel f). A notable amount of internalized GJ vesicles also colocalized with Rab5 (, panel a). This endocytic GTPase has been found essential for the uncoating of AP-2-mediated clathrin coats,Citation32 however has been linked also to the targeting of internalized cytoplasmic cargo to the autophagic degradation pathway.Citation33
Figure 2. AGJ vesicles robustly colocalize with lysosomal and autophagosomal markers but only insignificantly with endosomal markers. (A) Schematic representation of phago-/lysosomal and endo-/lysosomal cellular degradation pathways and respective marker proteins (highlighted with blue shading) that have been investigated in this study. (B) Cx43-GFP or Cx43-mApple expressing HeLa cells were either labeled with antibodies specific for endosomal and lysosomal marker proteins (a–d), cotransfected with LAMP-1-cerulean (e), or stained with the acidophilic fluorescent probe, LysoTracker Red (f). Representative merged confocal fluorescence images obtained 20–24 h after transfection are shown. Individual and merged fluorescence signals of the boxed areas are shown at higher magnification on the right. Robust colocalization of vesicular Cx43 was observed with lysosomal markers (LAMP-1, LAMP-1-cerulean, and LysoTracker, panels d-f, as indicated by yellow, the resulting color of overlaying red and green emission signals), but only insignificantly with endosomal markers (EEA1 and Rab7, panels b and c). A notable colocalization of Cx43-GFP was also observed with the GTP-binding protein Rab5 (panel a), described to be involved in early steps of endocytosis (uncoating of clathrin-coated vesicles), and in targeting cytoplasmic cargo to autophagic degradation (see text). Representative colocalizing vesicles are marked with arrows; GJs are marked with arrowheads.
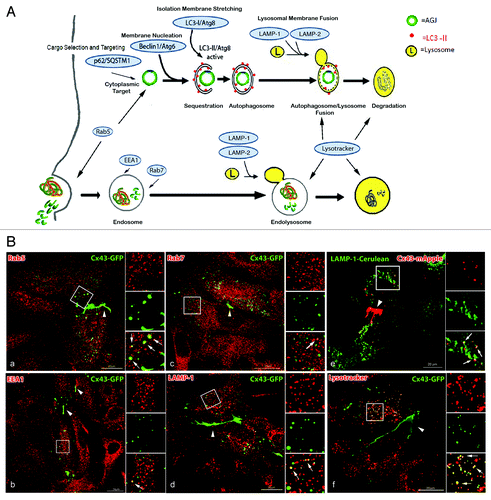
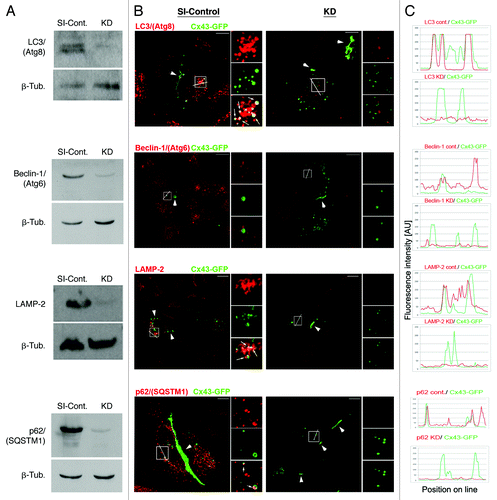
Figure 5. Efficient RNAi-mediated depletion of autophagy-relevant proteins monitored by western blot and quantitative fluorescence intensity measurements. HeLa cells were transfected with SI-control RNA-oligonucleotides, or with RNA-oligos targeting the autophagy-relevant proteins, LC3/(Atg8), BECN1/(Atg6), LAMP-2 and p62/SQSTM1 (KD). 48 h later cells were transfected with Cx43-GFP cDNA and analyzed for KD efficiency 72 h after oligo-transfection. (A) Target protein-depletion assayed by western blot analyses in SI-control and RNAi-KD cells. (B) Representative merged confocal fluorescence images of SI-control and KD cells acquired at identical exposure settings are shown. Individual and merged fluorescence signals of the boxed areas are shown at higher magnification on the right. Robust colocalization of Cx43-GFP containing cytoplasmic vesicles was observed with LC3/(Atg8), LAMP-2, and p62/SQSTM1 in mock treated, but not in KD cells (marked with arrows). GJ plaques are marked with arrowheads. Bars = 10 μm. (C) Fluorescence intensities (in arbitrary units) of Cx43-GFP (green) and RNAi-depleted protein (red) for both, SI-control and RNAi-treated cells, measured along lines depicted in (B). Efficient KD of all targeted proteins (> 90%) was confirmed by western blot and quantitative fluorescence line-scan analyses. BECN1/(Atg6) is known not to efficiently localize to autophagosomes and was not detected to colocalize with AGJ vesicles (B, panel 2). However, a significant cytoplasmic accumulation of AGJ vesicles and reduced AGJ/phagosome colocalization was observed in BECN1/(Atg6) depleted cells ().
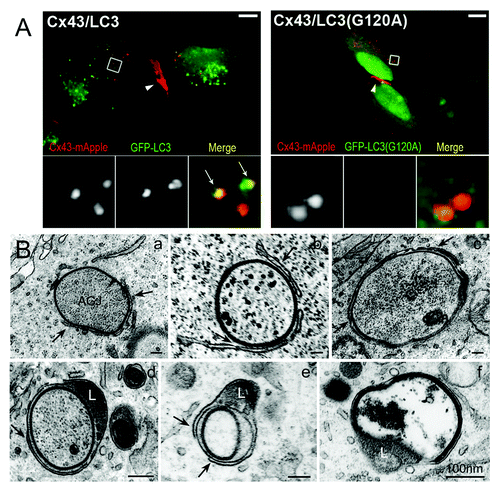
We further observed a robust colocalization of cytoplasmic AGJ vesicles formed in Cx43-mApple-expressing HeLa cells with the generic GFP-tagged autophagy-marker protein LC3 (GFP-LC3, the mammalian homolog of the yeast autophagic protein Atg8), but not with the LC3-dead mutant GFP-LC3(G120A) that is incapable of activation and autophagosomal membrane conjugation (). Microtubule-associated protein 1 light chain 3 (LC3) is a soluble cytoplasmic protein (LC3-I). It is recruited to developing phagophores, is covalently bound to phosphatidyl-ethanolamine (PE) of the phagophore membrane (LC3-II), and remains on autophagosomes for most of their lifetime.Citation34,Citation35 Thus, LC3 is a useful generic marker-protein for the characterization of autophagosomes ().Citation34
Figure 3. Fluorescence and ultrastructural evidence for autophagic AGJ vesicle degradation. (A) HeLa cells were cotransfected with Cx43-mApple and the mammalian autophagy marker protein GFP-LC3 (panel 1), or the activation-deficient LC3-mutant GFP-LC3(G120A) (panel 2). In cells, a fraction of cytoplasmic LC3 (LC3-I) is conjugated to phagophore-membranes (LC3-II) that localizes to autophagosomes; LC3(G120A) cannot be conjugated and remains cytoplasmic. Representative merged fluorescence images acquired 24 h post transfections are shown. Individual and merged fluorescence signals of the boxed areas are shown below at higher magnification. Robust colocalization of cytoplasmic AGJ vesicles present in Cx43-mApple expressing cells (red puncta) with GFP-LC3-II (green puncta) was observed in GFP-LC3 expressing cells, but not in GFP-LC3(G120A) expressing cells. Representative colocalizing AGJ vesicles are marked with arrows; GJs are marked with arrowheads. Bars = 10 μm. (B) Multiple stages characteristic of progressive autophagosome formation and maturation that formed around AGJ vesicles were revealed by ultrastructural analyses of Cx43-GFP expressing HeLa cell preparations. Double-membrane cisternae (phagophores, marked with arrows) progressively encircled AGJ vesicles (panels a–c), coalesced into phagophores (panels c–e) and fused with lysosomes (L, panels d and e), resulting in AGJ degradation inside the phagosome (panel f).
We also observed a robust colocalization of Cx43-GFP-labeled cytoplasmic GJ vesicles with p62/sequestosome1 (SQSTM1) (, panel 4). This ubiquitin-binding protein was found essential for sequestering ubiquitinated, cytoplasmic protein complexes to autophagic degradation (see below).Citation36 Taken together these results suggest that under normal steady-state conditions, internalized sufficiently matured AGJ vesicles are targeted for autophagic, rather than endo-/lysosomal degradation.
Ultrastructural analyses revealed multiple stages of phagophore formation around AGJ vesicles
Autophagosomes have a characteristic appearance on ultrathin-sectioned preparations of cells, making electron microscopy a reliable tool to characterize autophagic structures. Ultrastructural analyses of Cx43-GFP expressing HeLa cells revealed numerous cytoplasmic double-membrane GJ vesicles at various stages of apparent degradation. Numerous evidently maturing AGJ/autophagosome-intermediates, with double-membrane cisternae (marked with arrows) progressively encircling AGJs, were clearly identifiable (, panels a–c). These membrane cisternae (termed phagophores) appeared to coalesce and form characteristic double-membrane phagophores around the AGJs (, panels c–e), and to merge with a lysosome on one side (marked with an “L,” , panels d and e), resulting in the complete degradation of the AGJ inside the phagophore lumen (, panel f). These partially and completely double-membrane-encircled AGJ vesicles appeared structurally identical to the known stages that lead to the progressive formation of autophagosomes: formation of (1) the phagophore, (2) autophagophore, and (3) autophagolysosomeCitation37,Citation38 (see , top portion). Our ultrastructural analyses are consistent with our co-immunofluorescence analyses described above and further support the hypothesis that AGJ vesicles are degraded by autophagy. Strikingly similar ultrastructural images of AGJ/autophagosome-intermediates were also observed in situ, in tissues and cells expressing endogenous Cxs,Citation20,Citation27 and more recently in failing canine ventricular myocardium known to loose GJs from intercalated disc structures.Citation13
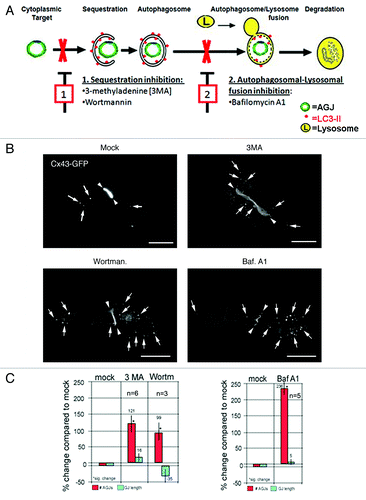
Pharmacological inhibition of autophagy prevents degradation of internalized AGJ vesicles
Numerous studies describe the use of pharmacological drugs that inhibit lysosomal and proteasomal function, to demonstrate the involvement of either or both organelles in the degradation of connexins and GJs.Citation15,Citation23,Citation25,Citation26 While drugs previously used in conjunction with connexin and GJ degradation analyses, such as MG132, ALLN and lactacystin, are proteasomal pathway specific, drugs such as leupeptin, chloroquine, pepstatin A, NH4Cl and E-64 act on lysosmal function, and thus will inhibit both, endo-/lysosomal as well as phago-/lysosomal degradation pathways. Therefore, in this study, we considered drugs that others have used previously to target and inhibit individual steps of the autophagic degradation pathway: autophagosome formation, phagophore/lysosome fusion, and ultimately phago-/lysosomal degradation (, steps 1 and 2). Treating Cx43-GFP expressing HeLa cells for 5 h with the purine, 3-methyladenine (3MA), or with the specific PI3-kinase blocker, wortmannin, that were found to inhibit autophagosomal cargo-sequestration as well as phagophore-maturation,Citation39,Citation40 we observed a significant increase of AGJ vesicles in the cytoplasm of treated cells by 121 ± 28% (n = 6), and 99 ± 23% (n = 3), compared with the number of AGJ vesicles in the cytoplasm of untreated (mock) cells (, panel 1, red bars). Treating Cx43-GFP expressing HeLa cells with Bafilomycin A1, a known lysosomal inhibitor and a potent and specific inhibitor of vacuolar H+ ATPase (V-ATPase) that more recently has also been described to prevent autophagosome/lysosome fusion, and therefore will also prevent autophagic degradation,Citation41 we observed an even higher significant increase of AGJ vesicles by 236 ± 21% (n = 5) (, panel 2). In none of these treatments a significant change of overall GJ plaque number and length was observed (, blue bars). Representative images of treated and untreated Cx43-GFP expressing HeLa cells are shown in . Taken together these results further support the hypothesis that GJs, following internalization into AGJ vesicles, are degraded by autophagy.
Figure 4. Pharmacological inhibition of autophagic degradation results in the accumulation of AGJ vesicles in the cytoplasm of drug-treated cells. (A) Schematic representation of autophagosome-maturation, and of representative steps blocked by drugs used in this study: (1) inhibition of cargo sequestration by 3MA and wortmannin; (2) inhibition of autophagosome/lysosome fusion by Bafilomycin A1. (B) Fluorescence images of drug-treated and control cells (mock) acquired approximately 21–23 h post transfection and 5 h into drug treatments are shown. Representative AGJ vesicles are marked with arrows. GJs are marked with arrowheads. Bars = 20 μm. (C) Quantitative analyses of total number of cytoplasmic AGJ vesicles (red columns) and GJ length (blue columns), and compared in percent to mock-treated cells are shown. In all drug treatments, a significant increase in the number of cytoplasmic AGJ vesicles was observed (99, 121 and 236%, marked with asterisks). GJ plaque length was not changed significantly in any of the experiments.
RNAi-mediated knockdown of autophagy-relevant proteins inhibits degradation of internalized AGJ vesicles
To further support our assumption that internalized AGJ vesicles are degraded by autophagy, we depleted cells of autophagy-relevant proteins using RNA-interference (RNAi) technology. We depleted cells of LC3B/(Atg8), BECN1/(Atg6), and LAMP-2 (see for function), and investigated its impact on AGJ vesicle degradation and AGJ vesicle/autophagosome colocalization. As part of a class III PI3-kinase complex, BECN1 (the mammalian homolog of the yeast protein Atg6) is crucial for mediating the localization of autophagic machinery-proteins to phagophore assembly sites, and BECN1 has been identified as a key-protein required for isolation-membrane nucleation.Citation42 As described above, LC3 has been identified as a reliable autophagy marker, due to its extended association with autophagic membranes throughout most stages of autophagosome formation and autophagic degradation. The lysosome-associated membrane protein 2 (LAMP-2) is required for the fusion of lysosomes with vacuolar target-membranes including autophagosomes,Citation43 enabling final phago-/lysosomal cargo degradation. We performed three independent sets (n = 3) of knockdown (KD) experiments in which the levels of each of these proteins were significantly depleted as verified by western blot and quantitative fluorescence line-scan analyses. Both assays demonstrated a significant reduction of all three proteins in the HeLa KD cells (, panels 1–3). RNAi-oligonucleotide transfection-efficiency was confirmed in control experiments to be above 90%, using a fluorescently labeled siGLO RISC-free oligonucleotide (not shown).Citation7 Following efficient depletion of endogenous BECN1 and LAMP-2, we observed a statistically significant increase in the number of Cx43-GFP AGJ vesicles in the cytoplasm of the treated cells when compared with SI-control treated cells by 50 ± 7% (n = 3), and 50 ± 4% (n = 3) (). LC3 knockdown (KD) also resulted in an increased, although insignificant accumulation of un-degraded GJ vesicles by 32 ± 9% (n = 4) (). As described above, the amount of LC3-protein that is covalently bound to phagophore membranes (LC3-II) is only a small fraction of the total cytoplasmic LC3-protein pool (LC3-I). Thus, despite an over 90% efficient depletion of total cellular LC3 in our KD experiments (, panel 1), enough cyoplasmic LC3-I may have remained in the cytoplasm of the KD cells and to be linked to phagophore membranes (LC3-II), potentially diminishing the LC3 KD effect. No significant change in the number of cytoplasmic AGJ vesicles or plasma membrane GJ plaques was observed in control cells that were treated with a scrambled nontargeting oligonucleotide (not shown).Citation7
Figure 6. RNAi-mediated depletion of key-autophagic proteins inhibits degradation of AGJ vesicles and decreases the number of AGJ-containing LC3-labled phagosomes. HeLa cells were transfected with SI-control RNA-oligonucleotides, or with RNA-oligos targeting the autophagy-relevant proteins, LC3/(Atg8), BECN1/(Atg6), LAMP-2, and p62/SQSTM1 (KD) as in . 48 h post oligo transfection cells were transfected with Cx43-GFP cDNA and 24 h later, total number of AGJs (in C), and colocalization with LC3-positive phagosomes (in A and B) was analyzed. (A) BECN1 and p62 KD and SI-control cells were stained with anti-LC3 antibodies 72 h post oligo, and 24 h post Cx43-GFP cDNA transfections. Representative immuno-fluorescence images are shown. Individual and merged fluorescence signals of the boxed areas are shown at higher magnification on the right. Colocalization of Cx43-GFP AGJ vesicles and LC3-positive phagosomes was observed preferentially in the SI-control cells (marked with arrows). Bars = 20 μm. (B) Quantitative analyses of AGJ vesicles (panel 1), LC3-positive autophagosomes (panel 2), and colocalizing AGJ/autophagosomes (panel 3) revealed a significant increase of AGJs, a significant decrease of autophagosomes, and significantly reduced AGJ/autophagosome colocalization in the BECN1 and p62 KD knockdown cells in all three independent experiments (**p < 0.01; ***p < 0.001) (see , and for total counts). (C) Quantitative AGJ vesicle analyses performed 72 h post oligo-transfection indicated a significant cytoplasmic AGJ accumulation in BECN1/(Atg6), LAMP-2, and p62/SQSTM1 depleted cells (≥ 50%, marked with asterisks) compared with SI-control cells. Less pronounced AGJ vesicle-accumulation was observed in LC3-depleted cells (32%), and this was attributed to sufficient inactive LC3-I that may remained in the LC3-KD cells and may have been converted into active LC3-II.
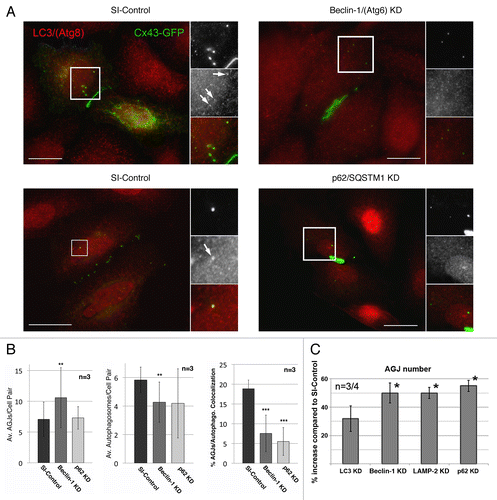
RNAi-mediated knockdown of the autophagy-mediator protein p62/SQSTM1 inhibits degradation of internalized GJ vesicles
The Ub-binding protein p62, also named sequestosome 1 (SQSTM1), has been found instrumental in recognizing and targeting ubiquitinated cytoplasmic protein complexes to autophagic degradation,Citation36,Citation44,Citation45 and to interact directly with LC3/(Atg8).Citation46 As cytoplasmic GJ vesicles robustly colocalize with p62/SQSTM1-specific antibodies in Cx43-GFP expressing Hela cells (, panel 4), internalized GJs have been described to be ubiquitinated,Citation47-Citation49 and AGJs consist of densely packed GJ channel aggregates, it is tempting to speculate that p62/SQSTM1 might also target AGJ vesicles to autophagic degradation. To further test this hypothesis, we depleted Cx43-GFP expressing HeLa cells of endogenous p62/SQSTM1-protein using RNAi technology. Efficient knockdown of p62/SQSTM1-protein was confirmed by western blotting (> 90%, , panel 4), and by quantitative fluorescence line-scan analyses, that demonstrated a significant reduction of p62/SQSTM1-immunofluorescence intensity in p62/SQSTM1-depleted cells (, panel 4). Downregulation of p62/SQSTM1 expression resulted in the highest, statistically significant increase of the number of cytoplasmic GJ vesicles in our study by 55 ± 4% (n = 4) (), further supporting our hypothesis that p62/SQSTM1 targets internalized, ubiquitinated AGJ vesicles to autophagic degradation.
RNAi-mediated knockdown of BECN1 and p62/SQSTM1-protein significantly reduced AGJ/phagosome colocalization
Staining BECN1/(Atg6) KD and SI-control cells with LC3-specific antibodies followed by quantitative analyses revealed that a significantly smaller portion of AGJ vesicles (60.8 ± 21.8%) colocalized with LC3-positive phagosomes in the BECN1 KD cells (7.6 ± 4.6%, n = 3, p < 0.001) when compared with SI-control cells (18.9 ± 2.2%, n = 3) (, panel 3 and , and ). Similar results (69.5 ± 21.1% reduction) were obtained for p62/SQSTM1 KD cells (5.5 ± 3.5%, n = 3, p < 0.001) (, panel 3 and , and ). As expected, an increased number of AGJs (, panel 1 and , and ), and a decreased number of autophagosomes (, panel 2 and , and ) were detected in all of these KD experiments as well. Representative LC3-immunofluorescence/Cx43-GFP images of BECN1 and p62/SQSTM1 KD, and SI-control cells, are shown in (, panels 1 and 2). LC3-positive Cx43-GFP AGJ vesicles are marked with arrows in the inserts. Total cell, autophagosome, and AGJ vesicle counts for all three independent KD experiments are shown in , and . Taken together, these results further support the assumption that under normal conditions (no induction or inhibition of autophagy or of any other cellular protein degradation pathway) internalized GJs are degraded by autophagy.
Table 1. AGJ/autophagosome colocalization in SI-Control, Beclin1 and p62 KD cells
Table 2. AGJ/autophagosome colocalization in SI-Control, Beclin1 and p62 KD cells
Table 3. AGJ/autophagosome colocalization in SI-Control, Beclin1 and p62 KD cells
LC3 and p62/SQSTM1 colocalize with Cx43 in primary PAE cells indicating autophagosomal degradation of AGJs in endogenously Cx43-expressing cells
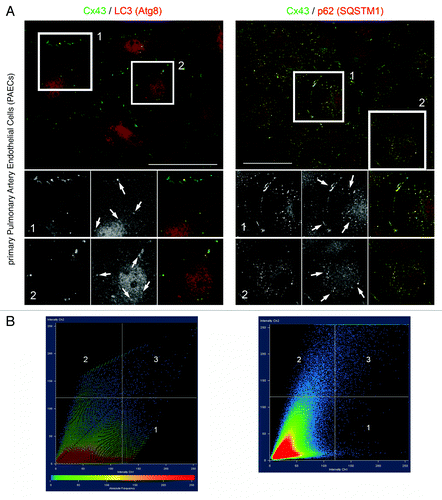
Given that all analyses described above were either done in transiently or stably Cx43-GFP/YFP expressing HeLa cells, we wanted to know whether autophagy-specific proteins would also co-localize with endogenously expressed Cx43. Cytoplasmic, AGJ vesicles had been seen before in situ under normal conditions inside autophagosomes,Citation20,Citation27 suggesting that autophagy might be the generic degradation pathway for internalized GJs. We stained endogenously Cx43 expressing primary pulmonary artery endothelial cells (PAECs), that we had characterized before for thrombin/endothelin-mediated GJ internalization for Cx43, LC3 and p62/SQSTM1 using specific antibodies and qualitatively and quantitatively analyzed potential Cx43-LC3/p62 colocalization. Representative images are shown in . Significant colocalization of both autophagic proteins with Cx43 was observed (, panels 1 and 2), even at stringent threshold settings (set to 120 of a maximum intensity of 250 arbitrary units; , panels 1 and 2). Interestingly, while LC3 appeared to exclusively colocalize with Cx43-AGJ vesicles located deep in the cytoplasm and occasionally juxtaposed to plasma membrane GJs, p62/SQSTM1 protein appeared to localize with both, particular plasma membrane GJs, as well as cytoplasmic AGJ vesicles; consistent with a role of p62 in sequestering ubiquitinated Cx43 to autophagosomes, and LC3 as a specific autophagy marker (, panels 1 and 2, marked with arrows in inserts 1 and 2).
Figure 7. LC3 and p62/SQSTM1 protein colocalize with endogenously expressed Cx43-based GJs and AGJ vesicles. Endogenously Cx43 expressing porcine primary pulmonary artery endothelial cells (PAECs) were stained for Cx43, LC3, and p62/SQSTM1 using specific mono- and polyclonal antibodies, and potential Cx43-LC3/p62 colocalization was qualitatively and quantitatively analyzed. (A) Representative merged fluorescence images of confluent PAECs are shown. Individual and merged fluorescence signals of the boxed areas are shown below at higher magnification. Significant colocalization of LC3 with AGJ vesicles, and of p62 with AGJ vesicles and individual plasma membrane GJs was observed, even at stringent threshold settings (marked with arrows). Bars = 20 μm. (B) Quantitative scatter-blot colocalization analyses of the images shown in (A) with applied maximum intensity threshold settings of 120 (of maximally 250 arbitrary units; white lines) indicated.
Discussion
In this manuscript we report multiple lines of independent evidence that suggest that GJs, following internalization into cytoplasmic AGJ vesicles, are cleared from the cytoplasm (Movie 1) and are degraded by autophagy. While previous studies identified preferentially proteasomal and endo-/lysosomal degradation pathways in the regulation of GJ stability and connexin degradation, our study provides novel molecular and mechanistic insights into the autophagic degradation of endocytosed GJs. (1) Confocal colocalization studies revealed a robust colocalization of internalized GJs (cytoplasmic AGJ vesicles) with proteins and markers indicative of lysosomal degradation, but only insignificantly with markers indicative of endosomal degradation (, and ). Furthermore, we observed significant colocalization of AGJ vesicles with marker proteins specific for autophagosomal degradation (LC3/Atg8), or autophagosomal cargo sequestration (Rab5, p62/SQSTM1) (, and –). (2) Ultrastructural analyses of these cells identified the progressive formation of phagophores, typical of phagophore formation, around AGJ vesicles, and the progressive lysosomal degradation of AGJs inside phagophores (). (3) Functional studies using pharmacological agents known to block specific steps of autophagosomal degradation; or (4) RNAi-mediated downregulation of key-autophagic proteins (LC3, BECN1, LAMP-2), or (5) of p62/SQSTM1, a protein known to sequester ubiquitinated protein complexes to the autophagic degradation pathway,Citation36,Citation46 resulted in a significant accumulation of total Cx43-protein () and of cytoplasmic Cx43-GFP AGJ vesicles (, and ); and a significant reduction of LC3-positive AGJ vesicles in the KD cells (, , and ). (6) Finally, colocalization between autophagic proteins, GJs and AGJ vesicles was observed in primary PAE cells, endogenously expressing Cx43 protein (). Together, these findings strongly suggest that under normal physiological conditions, internalized AGJ vesicles, similarly to cellular organelles, cytoplasmic protein aggregates, and other superfluous macromolecular protein-complexes, such as the mitotic midbody ring, are cleared from the cytoplasm and degraded by autophagy.Citation36,Citation45,Citation50-Citation52
It should be noted that lysosomal inhibitors such as leupeptin, chloroquine, NH4Cl and E-64, that previously have been used to gain evidence for endo-/lysosomal degradation of GJs,Citation15,Citation23,Citation25,Citation26 will also inhibit autophagic GJ degradation, and thus obtained results may not have been interpreted entirely adequate. Future better-designed experiments that specifically target one or the other cellular degradation pathway may be required to clarify specific roles of both pathways in GJ degradation. Remarkably, although autophagic degradation of GJs had been described in several classical ultrastructural analyses of various cells and tissues in situ, including heart, dermis, and liver,Citation20,Citation21,Citation27,Citation31 not much attention was attributed to this GJ degradation pathway. The recent observation that GJs appear to be internalized and degraded by autophagy in the failing ventricular myocardiumCitation13 lends further importance to this GJ degradation pathway.
At first glance, autophagic degradation of a membrane vesicle, such as an AGJ, might not appear intuitive. Cells have developed three principle degradation systems, the proteasomal, the endo-/lysosomal, and the phago-/lysosomal system (termed macroautophagy or simply autophagy), and all three have been implicated previously at various steps in the regulation of GJ stability and connexin degradation.Citation13,Citation20,Citation23-Citation27 While the two latter ones are designed for the degradation of protein aggregates, multi-protein complexes, and organelles, the proteasomal system is designed for the degradation of single polypeptide chains that require unfolding in order to be inserted into the tubular proteasome core. Since AGJ vesicles are highly complex multifaceted-protein assemblies, their degradation by the proteasome appears unlikely, and we did not gain any evidence that would suggest proteasome-mediated degradation of AGJ vesicles (data not shown).
Substantial research over the past decade has indicated that autophagy, beside its well-known function in organelle degradation during starvation, represents a common lysosome-based cellular degradation pathway specifically designed to remove and degrade protein aggregates, multiprotein complexes, organelles and invading pathogens from the cytoplasm.Citation33,Citation36,Citation45,Citation53 Recent studies have further shown that protein aggregates, such as the ones formed by huntingtin and β-amyloid protein, and cellular structures such as the midbody ring, a cytokinesis left-over multi-protein complex, are all degraded by autophagy.Citation33,Citation36,Citation45,Citation53 Clearly, these cellular structures are degraded by autophagy independent of starvation. In addition, autophagosomal degradation of membranous/vesicular organelles, as for example malfunctioning mitochondria, is common. Considering the structural organization of AGJs, the dense packing of channels that occupy most or even the entire surface of AGJs in a double-membrane arrangement, and their cytoplasmic location, autophagic degradation of AGJs appears quite plausible. In addition, the two tightly bound membranes of AGJ vesicles may not easily allow for their fusion with single-membrane endosomes. Since lysosomal enzymes cannot be released into the cytosol, it is likely that the phagophore, formed during the initial steps of autophagy, sequesters and separates the AGJ vesicle from the cytoplasm, thus providing the sealed membrane compartment (the phagophore) that is required for subsequent lysosomal degradation. Finally, the unique structural composition of AGJ vesicles with lumen and inner membrane derived from the neighboring cell (being foreign to the AGJ-receiving host cell) may further direct AGJs to autophagic degradation. Cell-cell communication provided by GJ channels prior to the internalization of the GJ plaque can only accommodate the diffusion of small molecules, ions, and second messengers across the connected plasma membranes. In the instance of GJ internalization however, other potentially harmful substances such as signaling proteins, other larger hazardous molecules, or even cytoplasmic pathogens, may be entrapped in the AGJ vesicle lumen that may renders autophagosomal degradation the default pathway for AGJ degradation.
Conjugation of ubiquitin (Ub) moieties to proteins has been recognized as a signal for both, proteasomal targeting (addition of K48-linked poly-Ub chains), and more recently also as a sorting signal toward internal vesicles of the late endocytic pathway (addition of multiple mono-Ub moieties or of K63-linked poly-Ub chains) which ultimately leads to degradation by lysosomes.Citation54-Citation58 The latter includes Ub-conjugation that acts as an internalization signal for clathrin-mediated endocytosis.Citation59,Citation60 In this process, multiple mono-Ub moieties are attached to the target protein and are recognized by specific clathrin-mediated endocytic machinery protein-components that associate with a subset of Ub-binding proteins, specifically Epsin1 and Eps15.Citation61-Citation63 Further work has shown that the protein p62/SQSTM1 recognizes and interacts via its UBA-domain with poly-ubiquitinated proteinsCitation64-Citation66 and delivers polyubiquitinated (K63-linked) oligomeric protein complexes to the autophagic degradation pathway.Citation36,Citation46 Ubiquitination of Cx43-based GJs has been described previously,Citation47-Citation49,Citation67,Citation68 and is consistent with our own unpublished fluorescence analyses (data not shown). The finding that Cx43-based GJs can become ubiquitinated, the known affinity of p62/SQSTM1 for ubiquitinated protein complexes, its colocalization with plasma membrane GJs in PAECs (), and its apparent involvement in targeting AGJ vesicles for autophagic degradation (), suggests that ubiquitination of Cx43, besides serving as a likely signal for GJ internalization, also serves as a likely signal for targeting AGJ vesicles to autophagic degradation. Future research will be required to determine the probably multiple forms of Cx43 ubiquitination (see ref. Citation68 and Citation69 for recent reviews).
It is possible that a subpopulation of internalized GJ vesicles might be degraded by the endo-/lysosomal pathway, especially under conditions were autophagosomal degradation has been inhibited; and this might indeed be suggested by our experiments in which both, endo-/lysosomal as well as phago-/lysosomal degradation pathways were blocked by pharmacological means (i.e., bafilomycin A1-treatments). Under these conditions, a much more pronounced accumulation of AGJs was observed (> 200%) as compared with cells in which key-autophagic proteins were depleted by RNA-interference (> 50–70%). Interestingly, a recent paper by Leithe et al.Citation49 reports that in contrast to our observations made in untreated normal cells, in TPA-treated cells [a structural analog of the second messenger molecule diacylglycerol (DAG)], internalized GJs appear to be degraded by the endo-/lysosomal pathway. DAG is a known potent activator of protein kinase C (PKC), and PKC is known to hyper-phosphorylate and promote hyper-ubiquitination of Cx43.Citation10,Citation49,Citation67 Based on their and our own results, it is tempting to speculate that cells might be able to regulate by which pathway (endo-/lysosomal vs. phago-/lysosomal) specific cargo is sequestered and processed (e.g., endo-/lysosomal and phago-/lysosomal pathways might process internalized GJs in different ways), and that the level and type of cargo-phosphorylation, or ubiquitination might determine which pathway is ultimately chosen (see for a schematic representation).
Figure 8. Schematic representation of the proposed steps that lead to GJ internalization (1–3), AGJ vesicle formation and fragmentation (4 and 5), and AGJ vesicle degradation by phago-/lysosomal (6–10) and endo-/lysosomal (11–15) pathways based on the present and previous work by others and us (see text for details).
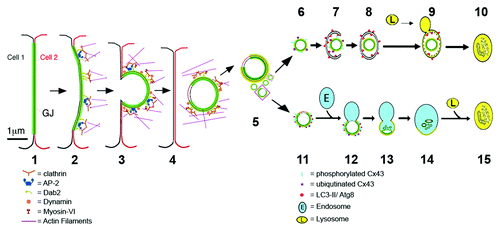
Endo-/lysosomal degradation of AGJs as observed in TPA-treated cells by Leithe et al.Citation49 of course raises the question of how, mechanistically, a double-membrane vesicle that consists of densely packed GJ channels and tightly bonded membrane layers can fuse with a single-membrane endosome. Interestingly, we found that AGJ vesicles examined by electron microscopy may include a small area where the two membranes are void of GJ channels and are not connected to each other (see Fig. 1H in ref. Citation8). Apparently, these nonjunctional membrane domains consist of plasma membrane that is derived from both neighboring cells, and we have postulated that these areas might originate from plasma membrane regions that were located immediately adjacent to the GJ plaques, and were internalized as well. It is tempting to speculate that this nonjunctional membrane domain could provide the unbonded membrane area that allows double-membrane AGJ vesicles to fuse with single-membrane endosomes (, steps 11–15).
In conclusion, our analyses provide molecular and mechanistic insights into a GJ degradation pathway that previously had not significantly been appreciated. Its recent link to health and diseaseCitation13 lends further importance to this GJ degradation pathway. Another recent study also reports that autophagy contributes to the degradation of endogenously and exogenously expressed wild type Cx43 protein, and of wild type and cataract-associated mutant Cx50 proteins in both, un-induced cells, and in cells in which autophagy was induced by starvation.Citation70 While this study was aimed more broadly at a potential role of autophagy contributing to connexin degradation in general, our study was aimed specifically at investigating the fate of internalized AGJ vesicles that we had characterized before.Citation5,Citation7,Citation8 Interestingly, while Lichtenstein et al.Citation70 knocked down the autophagy-related protein, Atg5 in both endogenously and exogenously Cx43 expressing cells, and used chloroquine to inhibit autophagy, we knocked down expression of the autophagy related proteins, BECN1 (Atg6), LC3 (Atg8), LAMP-2, and p62/SQSTM1, and used 3MA, wortmannin, and bafilomycin A1 in Cx43-GFP expressing HeLa cells; however taken together all three recent studies (refs. Citation13, Citation70 and our own) provide compelling evidence that endocytosed GJs are degraded by autophagy. In addition, all these works add to the growing consensus that autophagy plays an important role as a “normal” cellular degradation pathway, rather than representing solely a “rescue” pathway designed to supply critical nutrients during starvation.
Materials and Methods
Cell culture, cDNA constructs, transient and stable transfections
Human epitheloid cervix carcinoma cells (HeLa, American Type Culture Collection [ATCC], CCL2) were maintained under standard conditions as described.Citation8 Cells were cultured in a humidified atmosphere containing 5% CO2 at 37°C in low glucose (1000 mg/l) Dulbecco’s modified Eagle’s medium (DMEM, Thermo Scientific, SH30021.01). Medium was supplemented for a final concentration of 10% with fetal bovine serum (FBS, Atlanta Biologicals, S11050), 2 mM L-glutamine (Thermo Scientific, Hyclone, SH30034.01, stock 200 mM) and 50 I.U/ml penicillin and 50 µg/ml streptomycin (Cellgro, 30-001-CI). An inducible, stable-transfected HeLa tet-on cell line expressing Cx43-YFP was cultured and induced as described in reference Citation1. Porcine primary pulmonary artery endothelial cells (PAECs) were isolated and cultured as described in reference Citation5.
Fluorescent protein-tagged Cx43-GFP and Cx43-mApple constructs were described previously.Citation7,Citation71,Citation72 A LAMP-1-Cerulean fluorescent protein (FP) construct was generated by cloning the 1221 nucleotides (407 amino acid residues) of LAMP-1 (Lysosomal Associated Membrane Protein 1) into pEGFP-N1. GFP was replaced with the improved CFP-derivative, mCerulean,Citation73 resulting in a 60-nucleotide (20 amino acid) linker between the LAMP-1 and the mCerulean sequence. GFP-tagged wild-type and mutant LC3-constructs [GFP-LC3, GFP-LC3(G120A)] were generously provided by T. Yoshimori, National Institute of Genetics, Shizuoka, Japan and described previously.Citation34
For transfections cells were grown on 35mm collagen-coated glass bottom dishes (MatTek Corporation, P35GCol-1.5-14-C), or on 22 mm glass coverslips coated with poly-l-lysine (Sigma-Aldrich, P8920) for 24 h prior to transfection and allowed to reach 70–80% confluency. Cells were single and double-transfected with cDNAs using SuperFect® Transfection Reagent (Qiagen, 301307) according to manufacturer’s directions. Cells were observed 20–24 h post transfection, and transfection efficiencies between 15% and 30% were considered appropriate for qualitative and quantitative experiments.
siRNA duplexes and knockdown procedures
All RNAi oligonucleotides (oligos) were purchased from Dharmacon RNA Technologies and transfected into HeLa cells using Oligofectamine (Invitrogen, 12252011) according to manufacturer’s recommendations, and as described in reference Citation7. Oligonucleotides directed against human MAP1-LC3B/(Atg8) (siGENOME SMARTpool, M-012846-01-0005), BECN1/(Atg6) (siGENOME SMARTpool, M-010552-01), LAMP-2 (siGENOME SMARTpool, M-011715-00), p62/SQTM1 (siGENOME SMARTpool, M-010230-00-0005), as well as a nontargeting RISC-activating duplex control (siControl, D-001210-01-05) were used. RNAi-transfection efficiency was tested using a fluorescently labeled, non-targeting control oligonucleotide (siGLO RISC-Free, D-001600-01-05) and was established to be ≥ 90% efficient.Citation7 Forty-eight hours post oligo transfection cells were transfected with Cx43-GFP cDNA using SuperFect® Transfection Reagent (Qiagen, 301307), as recommended by the manufacturer. Cells were assayed 72 h post oligo transfection and 20–24 h into Cx43 expression for KD efficiency by western blot analyses, and quantitative fluorescence analyses (). To evaluate whether BECN1 and p62 KD would result in a reduced number of LC3-positive AGJ vesicles, KD and SI-control cells were stained with LC3-specific antibodies ().
Stains, antibodies and immunofluorescence analyses
Acidophilic LysoTracker Red stain (Molecular Probes/Invitrogen, L-7528; 1 mM stock in DMSO) was administered to the cell culture at 100 nM in regular growth medium and incubated at 37°C for 30 min, followed by a 15 min/37°C chase in fresh medium prior to imaging. Rabbit polyclonal anti-Rab5 (Cell Signaling Technology Inc., clone 2143), goat polyclonal anti-EEA1 (C-15) (Santa Cruz Biotechnology Inc., sc-6414), rabbit polyclonal anti-Rab7 (H-50) (Santa Cruz Biotechnology Inc., sc-10767), mouse monoclonal anti-LAMP-1 (Developmental Studies Hybridoma Bank, H4A3) and anti-LAMP-2 (Developmental Studies Hybridoma Bank, H4B4)-supernatant, mouse monoclonal anti-LC3 recognizing both, LC3-I and LC3-II of the human splice-variants LC3A, B, and C (MBL International Corp., M115-3), rabbit polyclonal BECN1(Cell Signaling, 3738), mouse monoclonal anti-3/p62 Lck ligand (BD Biosciences, 610832), and rabbit polyclonal anti-Cx43 (Sigma, 6219) antibodies were used at dilutions of 1:50–1:250 in 10% FBS/PBS. Secondary anti-mouse and anti-rabbit antibodies conjugated to Cy3 (Jackson Immunoresearch Laboratories, 305-165-003 and 115-165-003), Alexa Fluor 568 (Molecular Probes/Invitrogen, A11036 and A11004), or Alexa Fluor 488 (Molecular Probes/Invitrogen, A21206) respectively, were used at 1:100 –1:200 dilutions in 10% FBS/PBS.
Cells were fixed and permeabilized either in ice-cold MeOH for 5 min, or in 3.7% formaldehyde for 15 min followed by permeabilization in 0.1% Triton X-100 for 20 min at RT. Cells were washed three times in phosphate-buffered saline (PBS) between all steps. Following blocking with 10% FBS in PBS (20 min), cells were incubated with primary and secondary antibodies (diluted in blocking solution) for 60 min at RT each, then for 30 sec with DAPI (10 µg/ml), rinsed with PBS and diH2O, and mounted using Fluoromount G (Southern Biotechnology, 0100-01). Knockdown of proteins was evaluated by comparing quantitative fluorescence intensity signals measured along lines on images taken under identical exposure conditions from treated and mock-treated cells. Fluorescence colocalization signals in were quantified using the colocalization tool of the Zeiss LSM 510 ZEN 2008 (Version 5.0.0.267) software package.
Immunoblot analyses
Verification of protein knockdown (KD) efficiency, and total Cx43-GFP protein levels in stable and transient Cx43-GFP/YFP expressing HeLa cells after BECN1 KD, were assessed by western blotting 72 h post-RNAi transfection. Cells were scraped into PBS/protease inhibitor cocktail (Sigma-Aldrich, P8340). Cells were lysed in standard 4× SDS-PAGE sample buffer and boiled for 5 min. Biotinylated protein ladder (Cell Signaling Technology, 7727) was used to determine the molecular weight of relevant protein bands. Samples were resolved on 10–12% Bis/Acrylamide (1:29) gels and transferred onto nitrocellulose membranes (pore size 0.2µm, Whatman, Optitran BA-S83, 10439396). Following blocking in 5% dry milk in TBST, membranes were incubated in primary antibodies (1:1000 in 5% BSA in TBST) for 3hrs at RT or o/n at 4°C. Membranes were washed in TBST for 15min and incubated with horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit secondary antibodies (1:5000 in 5% BSA in PBST, Zymed Laboratories, 81-6520 and 81-6120) for 3 h at RT or o/n at 4°C. Immuno-reactive bands were detected using Amersham ECL Plus™ western Blotting Detection Reagents (GE Healthcare, RPN2132). Membranes were washed in TBST for 15 min, stripped in stripping buffer (Boston Bioproducts, BP-96) at 65°C for 30 min, followed by a stringent wash in TBST. Membranes were re-blocked with 5% dry milk in TBST and re-probed with mouse monoclonal antibodies directed against α-tubulin (Sigma, T9026) or β-tubulin (Developmental Studies Hybridoma Bank, clone E7) (1:5000 in 5% BSA in PBS) to serve as a loading control. Signals were quantified by scanning developed films using SCION software (NIH).
Drug treatments
To inhibit specific steps of the autophagosomal degradation pathway, HeLa cells expressing Cx43-GFP were treated with pharmacological inhibitors 16–18 h post transfection. Autophagic sequestration was blocked by using the purine derivative, 3-methyladenine (3MA) (Enzo Life Sciences/Biomol International, BML-AP502), or the specific phosphatidylinositol (PtdIns) 3-kinase inhibitor, wortmannin (Enzo Life Sciences/Biomol International, BML-ST415) as described.Citation39,Citation40 Cells were treated with 5 mM 3MA or 50 µM wortmannin in normal growth medium (a total of 5hrs each). To inhibit the phagosome-lysosome fusion step of autophagic degradation, cells were treated for 5 h in normal growth medium containing 200 nM Bafilomycin A1 (Enzo Life Sciences/Biomol International, BML-CM110, 10 mM stock in DMSO) as described.Citation41 Following treatments the number and size of cytoplasmic AGJ vesicles was counted and statistically evaluated ().
Microscopy and quantitative image analyses
Wide-field fluorescence microscopy was performed on a Nikon Eclipse TE 2000E inverted fluorescence microscope equipped with 40× Plan Fluor (numerical aperture [NA] 1.3), 60× and 100× Plan Apochromat (NA 1.4) oil immersion objectives; a forced-air-cooled Photometics CoolSnap HQ charge-coupled device camera (Roper Scientific), and a ProScan II motorized stage (Prior Scientific). Images were captured, analyzed, and processed using MetaVue software version 6.1r5 (Molecular Devices) and Adobe Photoshop (Adobe Systems). Fluorescence colocalization analyses were performed on a Zeiss Axiovert 200 M inverted fluorescence microscope (Carl Zeiss) equipped with an LSM510 META scan head and a 63x Apochromat oil-immersion objective (NA 1.4). Argon-ion and Helium-Neon lasers were used to generate the 488- and 543-nm excitation lines, and pinholes were typically set to 1 airy unit. Images were acquired using four-line mean averaging in separated channels to avoid bleed-through and LSM510 META 3.0 software. Extended live-cell recordings (phase contrast and fluorescence) shown in Movie 1 were performed on a Nikon BioStation IM Cell-S1 system using a 40× air-objective and glass-bottom dishes. Ultrastructural analyses of Cx43-GFP expressing HeLa cells were performed as described.Citation8
Statistical analyses
AGJ vesicles and GJ plaques were measured and counted on images taken of knockdown, drug treated and control cells in at least three independent experiments each. Only clearly recognizable GJs (a line of fluorescent puncta, or elongated plaques located between cell pairs), and AGJs (bright fluorescent spherical structures located in the cytoplasm and ≥ 0.5 µm in diameter) as defined in detail in Piehl et al.Citation8 and Gumpert et al.Citation7 were considered. Only spherical, clearly LC3-positive vesicular structures were considered as autophagosomes, and were counted as well. Participation of selected proteins and pathways in GJ internalization and subsequent degradation was evaluated by manually counting the number of Cx43-GFP expressing cells and by counting the number, and measuring the size of AGJ vesicles, GJ plaques, and LC3-positive autophagosomes. For statistical analyses, the total number of AGJ vesicles per experiment was divided by the number of cell pairs positive for Cx43-GFP expression and clearly coupled by GJs as described.Citation7,Citation8 Similarly, the total length of GJ plaques per experiment was determined and divided by the number of cell pairs. To compare the BECN1 and p62/SQSTM1 KD cells with the SI-control cells, the number of AGJs, autophagosomes, and AGJ/autophagosome colocalization was determined for each cell pair. For each experiment, the ratio of AGJ vesicles colocalizing with LC3-positive structures was then formed over the total number of AGJs and of autophagosomes that were counted per cell pair (, and ). For statistical analyses ratios of BECN1 and p62 KD cell pairs (99 and 40, see , and ) were compared with the ratios obtained in SI-control cells (99, see , and ). In all analyses, a p value ≤ 0.05 was considered statistically significant.” Statistical analyses were performed using Microsoft Excel’s “Students t-test: Two samples assuming equal or unequal variances” function of the data analysis package.
| Abbreviations: | ||
| AGJ | = | annular gap junction |
| Cx | = | connexin |
| GFP | = | green fluorescent protein |
| GJ | = | gap junction |
| GJIC | = | gap junction mediated intercellular communication |
| KD | = | knockdown |
Additional material
Download Zip (2.8 MB)Acknowledgments
We thank T. Yoshimori (National Institute of Genetics, Shizuoka, Japan) for providing fluorescent LC3 constructs; J.-P. Denizot (Unité de Neurosciences Intégratives et Computationnelles, CNRS, Gif sur Yvette, France) for the preparation of ultra-thin sections; D. Segretain (Institut National de la Santé et de la Recherche Médicale U895, Université Paris Descartes, 75006 Paris, France) for ultrastructural examination of our Cx43-GFP expressing HeLa cell samples; and L. Traub (University of Pittsburgh, Pittsburg, PA, USA), L. Cassimeris (Lehigh University), and Falk-lab members for critical advice. This work was supported by NIHs NIGMS (grant GM55725 to M.M.F.) and Lehigh University.
M.M.F. and A.M.G. designed the research; A.M.G., J.T.F, R.M.K. and M.M.F. performed the experiments; A.M.G., J.T.F., R.M.K., J.Y.M. and M.M.F. analyzed the data; M.W.D. provided essential research tools; A.M.G. and M.M.F. wrote the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials can be found at: www.landesbioscience.com/journals/autophagy/article/19390
References
- Lauf U, Giepmans BN, Lopez P, Braconnot S, Chen SC, Falk MM. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci U S A 2002; 99:10446 - 51; http://dx.doi.org/10.1073/pnas.162055899; PMID: 12149451
- Falk MM, Baker SM, Gumpert AM, Segretain D, Buckheit RW 3rd. Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol Biol Cell 2009; 20:3342 - 52; http://dx.doi.org/10.1091/mbc.E09-04-0288; PMID: 19458184
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, et al. Multicolor and electron microscopic imaging of connexin trafficking. Science 2002; 296:503 - 7; http://dx.doi.org/10.1126/science.1068793; PMID: 11964472
- Ghoshroy S, Goodenough DA, Sosinsky GE. Preparation, characterization, and structure of half gap junctional layers split with urea and EGTA. J Membr Biol 1995; 146:15 - 28; http://dx.doi.org/10.1007/BF00232677; PMID: 7563034
- Baker SM, Kim N, Gumpert AM, Segretain D, Falk MM. Acute internalization of gap junctions in vascular endothelial cells in response to inflammatory mediator-induced G-protein coupled receptor activation. FEBS Lett 2008; 582:4039 - 46; http://dx.doi.org/10.1016/j.febslet.2008.10.043; PMID: 18992245
- Gilleron J, Fiorini C, Carette D, Avondet C, Falk MM, Segretain D, et al. Molecular reorganization of Cx43, Zo-1 and Src complexes during the endocytosis of gap junction plaques in response to a non-genomic carcinogen. J Cell Sci 2008; 121:4069 - 78; http://dx.doi.org/10.1242/jcs.033373; PMID: 19033388
- Gumpert AM, Varco JS, Baker SM, Piehl M, Falk MM. Double-membrane gap junction internalization requires the clathrin-mediated endocytic machinery. FEBS Lett 2008; 582:2887 - 92; http://dx.doi.org/10.1016/j.febslet.2008.07.024; PMID: 18656476
- Piehl M, Lehmann C, Gumpert A, Denizot JP, Segretain D, Falk MM. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell 2007; 18:337 - 47; http://dx.doi.org/10.1091/mbc.E06-06-0487; PMID: 17108328
- Blomstrand F, Venance L, Sirén AL, Ezan P, Hanse E, Glowinski J, et al. Endothelins regulate astrocyte gap junctions in rat hippocampal slices. Eur J Neurosci 2004; 19:1005 - 15; http://dx.doi.org/10.1111/j.0953-816X.2004.03197.x; PMID: 15009148
- Postma FR, Hengeveld T, Alblas J, Giepmans BN, Zondag GC, Jalink K, et al. Acute loss of cell-cell communication caused by G protein-coupled receptors: a critical role for c-Src. J Cell Biol 1998; 140:1199 - 209; http://dx.doi.org/10.1083/jcb.140.5.1199; PMID: 9490732
- Spinella F, Rosanò L, Di Castro V, Nicotra MR, Natali PG, Bagnato A. Endothelin-1 decreases gap junctional intercellular communication by inducing phosphorylation of connexin 43 in human ovarian carcinoma cells. J Biol Chem 2003; 278:41294 - 301; http://dx.doi.org/10.1074/jbc.M304785200; PMID: 12907686
- van Zeijl L, Ponsioen B, Giepmans BN, Ariaens A, Postma FR, Várnai P, et al. Regulation of connexin43 gap junctional communication by phosphatidylinositol 4,5-bisphosphate. J Cell Biol 2007; 177:881 - 91; http://dx.doi.org/10.1083/jcb.200610144; PMID: 17535964
- Hesketh GG, Shah MH, Halperin VL, Cooke CA, Akar FG, Yen TE, et al. Ultrastructure and regulation of lateralized connexin43 in the failing heart. Circ Res 2010; 106:1153 - 63; http://dx.doi.org/10.1161/CIRCRESAHA.108.182147; PMID: 20167932
- Beardslee MA, Laing JG, Beyer EC, Saffitz JE. Rapid turnover of connexin43 in the adult rat heart. Circ Res 1998; 83:629 - 35; PMID: 9742058
- Berthoud VM, Minogue PJ, Laing JG, Beyer EC. Pathways for degradation of connexins and gap junctions. Cardiovasc Res 2004; 62:256 - 67; http://dx.doi.org/10.1016/j.cardiores.2003.12.021; PMID: 15094346
- Fallon RF, Goodenough DA. Five-hour half-life of mouse liver gap-junction protein. J Cell Biol 1981; 90:521 - 6; http://dx.doi.org/10.1083/jcb.90.2.521; PMID: 7287816
- Ginzberg RD, Gilula NB. Modulation of cell junctions during differentiation of the chicken otocyst sensory epithelium. Dev Biol 1979; 68:110 - 29; http://dx.doi.org/10.1016/0012-1606(79)90247-1; PMID: 437313
- Jordan K, Chodock R, Hand AR, Laird DW. The origin of annular junctions: a mechanism of gap junction internalization. J Cell Sci 2001; 114:763 - 73; PMID: 11171382
- Larsen WJ, Tung HN, Murray SA, Swenson CA. Evidence for the participation of actin microfilaments and bristle coats in the internalization of gap junction membrane. J Cell Biol 1979; 83:576 - 87; http://dx.doi.org/10.1083/jcb.83.3.576; PMID: 574870
- Leach DH, Oliphant LW. Degradation of annular gap junctions of the equine hoof wall. Acta Anat (Basel) 1984; 120:214 - 9; http://dx.doi.org/10.1159/000145923; PMID: 6516782
- Mazet F, Wittenberg BA, Spray DC. Fate of intercellular junctions in isolated adult rat cardiac cells. Circ Res 1985; 56:195 - 204; PMID: 3971501
- Hesketh GG, Van Eyk JE, Tomaselli GF. Mechanisms of gap junction traffic in health and disease. J Cardiovasc Pharmacol 2009; 54:263 - 72; http://dx.doi.org/10.1097/FJC.0b013e3181ba0811; PMID: 19701097
- Laing JG, Tadros PN, Westphale EM, Beyer EC. Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp Cell Res 1997; 236:482 - 92; http://dx.doi.org/10.1006/excr.1997.3747; PMID: 9367633
- Leithe E, Rivedal E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J Cell Sci 2004; 117:1211 - 20; http://dx.doi.org/10.1242/jcs.00951; PMID: 14970263
- Musil LS, Le AC, VanSlyke JK, Roberts LM. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem 2000; 275:25207 - 15; http://dx.doi.org/10.1074/jbc.275.33.25207; PMID: 10940315
- Qin H, Shao Q, Igdoura SA, Alaoui-Jamali MA, Laird DW. Lysosomal and proteasomal degradation play distinct roles in the life cycle of Cx43 in gap junctional intercellular communication-deficient and -competent breast tumor cells. J Biol Chem 2003; 278:30005 - 14; http://dx.doi.org/10.1074/jbc.M300614200; PMID: 12767974
- Pfeifer U. Autophagic sequestration of internalized gap junctions in rat liver. Eur J Cell Biol 1980; 21:244 - 6; PMID: 7449766
- Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol 1998; 8:931 - 4; http://dx.doi.org/10.1016/S0960-9822(07)00375-2; PMID: 9707407
- Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell 2005; 16:5686 - 98; http://dx.doi.org/10.1091/mbc.E05-08-0737; PMID: 16195341
- Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem 1998; 273:12725 - 31; http://dx.doi.org/10.1074/jbc.273.21.12725; PMID: 9582296
- Severs NJ, Shovel KS, Slade AM, Powell T, Twist VW, Green CR. Fate of gap junctions in isolated adult mammalian cardiomyocytes. Circ Res 1989; 65:22 - 42; PMID: 2736737
- Semerdjieva S, Shortt B, Maxwell E, Singh S, Fonarev P, Hansen J, et al. Coordinated regulation of AP2 uncoating from clathrin-coated vesicles by rab5 and hRME-6. J Cell Biol 2008; 183:499 - 511; http://dx.doi.org/10.1083/jcb.200806016; PMID: 18981233
- Ravikumar B, Imarisio S, Sarkar S, O’Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J Cell Sci 2008; 121:1649 - 60; http://dx.doi.org/10.1242/jcs.025726; PMID: 18430781
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol 2004; 36:2491 - 502; http://dx.doi.org/10.1016/j.biocel.2004.02.005; PMID: 15325587
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 77; http://dx.doi.org/10.1016/S1534-5807(04)00099-1; PMID: 15068787
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861 - 73; http://dx.doi.org/10.1101/gad.1599207; PMID: 18006683
- Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelárová H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 1997; 243:240 - 6; http://dx.doi.org/10.1111/j.1432-1033.1997.0240a.x; PMID: 9030745
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A 1982; 79:1889 - 92; http://dx.doi.org/10.1073/pnas.79.6.1889; PMID: 6952238
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 1998; 23:33 - 42; http://dx.doi.org/10.1247/csf.23.33; PMID: 9639028
- Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2001; 2:330 - 5; http://dx.doi.org/10.1093/embo-reports/kve061; PMID: 11306555
- Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J 2007; 26:313 - 24; http://dx.doi.org/10.1038/sj.emboj.7601511; PMID: 17245426
- Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 2008; 4:141 - 50; PMID: 17986870
- Pohl C, Jentsch S. Midbody ring disposal by autophagy is a post-abscission event of cytokinesis. Nat Cell Biol 2009; 11:65 - 70; http://dx.doi.org/10.1038/ncb1813; PMID: 19079246
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Girão H, Catarino S, Pereira P. Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp Cell Res 2009; 315:3587 - 97; http://dx.doi.org/10.1016/j.yexcr.2009.10.003; PMID: 19835873
- Kjenseth A, Fykerud T, Rivedal E, Leithe E. Regulation of gap junction intercellular communication by the ubiquitin system. Cell Signal 2010; 22:1267 - 73; http://dx.doi.org/10.1016/j.cellsig.2010.03.005; PMID: 20206687
- Leithe E, Kjenseth A, Sirnes S, Stenmark H, Brech A, Rivedal E. Ubiquitylation of the gap junction protein connexin-43 signals its trafficking from early endosomes to lysosomes in a process mediated by Hrs and Tsg101. J Cell Sci 2009; 122:3883 - 93; http://dx.doi.org/10.1242/jcs.053801; PMID: 19808888
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119:753 - 66; http://dx.doi.org/10.1016/j.cell.2004.11.038; PMID: 15607973
- Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, et al. Autophagy defends cells against invading group A Streptococcus. Science 2004; 306:1037 - 40; http://dx.doi.org/10.1126/science.1103966; PMID: 15528445
- Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science 2005; 307:727 - 31; http://dx.doi.org/10.1126/science.1106036; PMID: 15576571
- Hung SY, Huang WP, Liou HC, Fu WM. Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy 2009; 5:502 - 10; http://dx.doi.org/10.4161/auto.5.4.8096; PMID: 19270530
- Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2001; 2:195 - 201; http://dx.doi.org/10.1038/35056583; PMID: 11265249
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol 2003; 19:141 - 72; http://dx.doi.org/10.1146/annurev.cellbio.19.110701.154617; PMID: 14570567
- Schnell DJ, Hebert DN. Protein translocons: multifunctional mediators of protein translocation across membranes. Cell 2003; 112:491 - 505; http://dx.doi.org/10.1016/S0092-8674(03)00110-7; PMID: 12600313
- Shih SC, Katzmann DJ, Schnell JD, Sutanto M, Emr SD, Hicke L. Epsins and Vps27p/Hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nat Cell Biol 2002; 4:389 - 93; http://dx.doi.org/10.1038/ncb790; PMID: 11988742
- Stahl PD, Barbieri MA. Multivesicular bodies and multivesicular endosomes: the “ins and outs” of endosomal traffic. Sci STKE 2002; 2002:pe32; http://dx.doi.org/10.1126/stke.2002.141.pe32; PMID: 12122203
- Belouzard S, Rouillé Y. Ubiquitylation of leptin receptor OB-Ra regulates its clathrin-mediated endocytosis. EMBO J 2006; 25:932 - 42; http://dx.doi.org/10.1038/sj.emboj.7600989; PMID: 16482222
- Geetha T, Jiang J, Wooten MW. Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol Cell 2005; 20:301 - 12; http://dx.doi.org/10.1016/j.molcel.2005.09.014; PMID: 16246731
- Barriere H, Nemes C, Lechardeur D, Khan-Mohammad M, Fruh K, Lukacs GL. Molecular basis of oligoubiquitin-dependent internalization of membrane proteins in Mammalian cells. Traffic 2006; 7:282 - 97; http://dx.doi.org/10.1111/j.1600-0854.2006.00384.x; PMID: 16497223
- Hawryluk MJ, Keyel PA, Mishra SK, Watkins SC, Heuser JE, Traub LM. Epsin 1 is a polyubiquitin-selective clathrin-associated sorting protein. Traffic 2006; 7:262 - 81; http://dx.doi.org/10.1111/j.1600-0854.2006.00383.x; PMID: 16497222
- Madshus IH. Ubiquitin binding in endocytosis--how tight should it be and where does it happen?. Traffic 2006; 7:258 - 61; http://dx.doi.org/10.1111/j.1600-0854.2006.00393.x; PMID: 16497221
- Ciani B, Layfield R, Cavey JR, Sheppard PW, Searle MS. Structure of the ubiquitin-associated domain of p62 (SQSTM1) and implications for mutations that cause Paget’s disease of bone. J Biol Chem 2003; 278:37409 - 12; http://dx.doi.org/10.1074/jbc.M307416200; PMID: 12857745
- Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 2004; 24:8055 - 68; http://dx.doi.org/10.1128/MCB.24.18.8055-8068.2004; PMID: 15340068
- Wilkinson CR, Seeger M, Hartmann-Petersen R, Stone M, Wallace M, Semple C, et al. Proteins containing the UBA domain are able to bind to multi-ubiquitin chains. Nat Cell Biol 2001; 3:939 - 43; http://dx.doi.org/10.1038/ncb1001-939; PMID: 11584278
- Leithe E, Rivedal E. Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J Biol Chem 2004; 279:50089 - 96; http://dx.doi.org/10.1074/jbc.M402006200; PMID: 15371442
- Leithe E, Sirnes S, Fykerud T, Kjenseth A, Rivedal E. Endocytosis and post-endocytic sorting of connexins. [Epub ahead of print] Biochim Biophys Acta 2011; PMID: 21996040
- Su V, Lau AF. Ubiquitination, intracellular trafficking, and degradation of connexins. Arch Biochem Biophys 2012; In press; PMID: 22239989
- Lichtenstein A, Minogue PJ, Beyer EC, Berthoud VM. Autophagy: a pathway that contributes to connexin degradation. J Cell Sci 2011; 124:910 - 20; http://dx.doi.org/10.1242/jcs.073072; PMID: 21378309
- Falk MM. Connexin-specific distribution within gap junctions revealed in living cells. J Cell Sci 2000; 113:4109 - 20; PMID: 11058097
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, et al. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods 2008; 5:545 - 51; http://dx.doi.org/10.1038/nmeth.1209; PMID: 18454154
- Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol 2004; 22:445 - 9; http://dx.doi.org/10.1038/nbt945; PMID: 14990965