Abstract
Autophagy is responsible for the degradation of protein aggregates and damaged organelles. Several studies have reported increased autophagic activity in tubular cells after kidney injury. Here, we examine the role of tubular cell autophagy in vivo under both physiological conditions and stress using two different tubular-specific Atg5-knockout mouse models. While Atg5 deletion in distal tubule cells does not cause a significant alteration in kidney function, deleting Atg5 in both distal and proximal tubule cells results in impaired kidney function. Already under physiological conditions, Atg5-null tubule cells display a significant accumulation of p62 and oxidative stress markers. Strikingly, tubular cell Atg5-deficiency dramatically sensitizes the kidneys to ischemic injury, resulting in impaired kidney function, accumulation of damaged mitochondria as well as increased tubular cell apoptosis and proliferation, highlighting the critical role that autophagy plays in maintaining tubular cell integrity during stress conditions.
Introduction
Autophagy is a bulk degradation pathway responsible for degrading protein aggregates and damaged organelles. Portions of the cytoplasm are sequestered within double-membraned cytosolic vesicles called autophagosomes that fuse with lysosomes to form single-membraned autolysosomes.Citation1 Basal autophagic activity and the upregulation of autophagy are relevant to many pathological and physiological conditions.Citation1 Increased autophagic activity has been observed in tubule cells obtained from human transplanted kidney biopsy specimensCitation2 and in renal proximal tubular epithelial cells obtained from patients with nephropathic cystinosis.Citation3 Several studies have reported the upregulation of autophagy in tubular cells in response to acute kidney injury caused by experimental nephrotoxic-, ischemia/reperfusion- or ureteral obstruction models.Citation2,Citation4-Citation8 In cultured proximal tubular cells, autophagy is induced prior to apoptosis.Citation4-Citation6 However, it is unclear whether autophagy supports survival or apoptosis of tubular epithelial cells. Several groups have shown that blocking autophagy with pharmacological inhibitors or genetic inhibition sensitizes both RPTC (a rat proximal tubular cell line) and LLC-PK1 (a pig proximal tubular cell line) cells to cisplatin-induced apoptosis, suggesting that autophagy is a prosurvival mechanism.Citation5,Citation6 In contrast, Inuoe et al. showed that inhibiting autophagy in NRK-52E cells, another rat proximal tubular cell line, suppresses cisplatin-induced apoptosis, suggesting that autophagy is involved in cell death.Citation4 The discrepancies might be due to differences in the cell lines used in these experiments. Interestingly, autophagy and apoptosis pathways can be regulated by common factors.Citation1 In renal proximal tubular cells, p53 inhibition partially suppresses cisplatin-induced autophagy, and the stable expression of Bcl-2 in renal cells attenuates autophagy after cisplatin administration.Citation5
Ischemia is a common cause of acute kidney injury,Citation9 and ischemia/reperfusion (I/R) injury plays a major role in delayed graft function and long-term changes after kidney transplantation.Citation10 Autophagy is induced in cultured proximal tubular cells and in ischemia/reperfusion models in mouse kidneys in response to hypoxic conditions.Citation2,Citation7 Furthermore, using pharmacological inhibitors or small-interfering RNA to block autophagy in vitro sensitizes the tubular cells to hypoxia-induced apoptosis,Citation7 whereas blocking autophagy protects the tubular cells from ROS-induced cell death after H2O2 treatment.Citation2 However, Suzuki et al. suggest that autophagy is part of the physiological response to I/R injury and that the rapid rate of autophagy during I/R injury may lead to autophagic cell death.Citation2 In agreement with these findings, in cold preservation ischemia of mouse kidneys, both autophagy and apoptosis are induced.Citation11
Previous studies have shown that low levels of autophagy generally serve a prosurvival function, while excessive autophagy is able to induce cell death.Citation1 The functional role of autophagy in kidney tubule cells remains controversial, particularly in models of acute kidney injury. Difficulties in controlling the level of autophagy, especially in cell culture, result in the possibility of generating misleading results. Blocking autophagy with pharmacological inhibitors might also impact other degradative pathways such as the lysosome and the ubiquitin-proteasome system (UPS).Citation12 Therefore, investigating tissue-specific autophagy-deficient mice is required to accurately identify the function of autophagy in the kidney.
Here, we examine the roles played by basal autophagy levels in tubular cell aging and upregulated autophagy in tubular cells under stress conditions using distal tubular-specific and doxycycline-inducible, time-specific Atg5-knockout mouse models.
Results
Autophagic activity in the tubule cell system
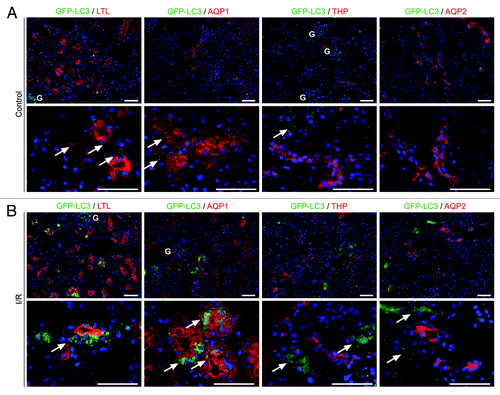
Previously, we demonstrated that glomerular podocytes exhibit high levels of basal autophagy and that podocyte-specific Atg5-deficiency results in age-related glomerulosclerosis and increased sensitivity to glomerular stress.Citation13 To investigate the role of autophagy in tubular cells, we analyzed the GFP-LC3 reporter mouse. Cytosolic LC3-I is lipidated into LC3-II and recruited into autophagosomes, which can be identified as GFP-positive dots in the GFP-LC3 mouse.Citation14 Staining kidney cryosections with the proximal tubule marker reagent Lotus Tetragonolobus lectin (LTL) and antibodies against the tubular marker proteins aquaporin 1 (AQP1), a marker for the proximal tubule, Tamm Horsfall protein (THP), a marker for the thick ascending limb of the loop of Henle, and aquaporin 2 (AQP2), a marker for the collecting duct, revealed that autophagosomes can rarely be detected under basal conditions in the tubular system (). However, 24 h after ischemia/reperfusion of the kidney, autophagosomes were extremely prevalent in the proximal tubule cells (), suggesting that autophagy might be essential for the tubular cell stress response.
Figure 1. Autophagic activity in the tubule system. (A) Under basal conditions, only a few autophagosomes can be detected in the tubule system of GFP-LC3 reporter mice. Kidney cryosections were stained with the proximal tubule marker reagent Lotus Tetragonolobus lectin (LTL) and antibodies against the proximal tubule marker AQP1, the thick ascending limb of loop of Henle marker THP and the collecting duct marker AQP2. (B) Twenty-four hours after ischemia/reperfusion injury, the kidney autophagosomes are upregulated predominantly in the proximal tubule colocalizing with LTL and AQP1. Arrows indicate autophagosomes. Glomeruli (marked with “G”) show high basal autophagosomal activity. Scale bars: 50 µm.
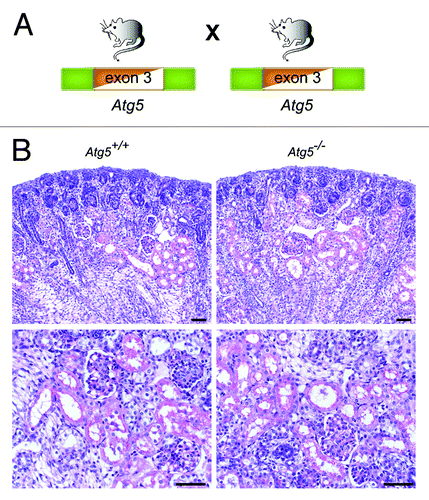
There is no detectable abnormality in Atg5−/− kidneys at E19.5
Atg5 deficiency results in the inability of cells to form autophagosomes. Constitutive Atg5-knockout mice die within 24 h after birth.Citation15 To analyze the role of autophagy for kidney tubular development, we bred heterozygous Atg5+/− mice to generate Atg5−/− mice (). We have previously confirmed that Atg5−/− kidneys lack Atg5 and LC3-I to LC3-II conversion by western blot.Citation13 No obvious histological phenotype could be detected in Atg5−/− kidneys at E19.5, indicating that autophagy is dispensable for tubular development ().
Figure 2. There is no detectable abnormality in Atg5−/− kidneys at E19.5. (A) Atg5+/− mice, missing exon 3 constitutively in one Atg5 allele, were bred to generate Atg5−/− mice. (B) No obvious histological alterations can be detected in Atg5−/− mice at E19.5. Scale bars: 50 µm.
The time-specific deletion of Atg5 in the kidney tubule system results in ultrastructural alterations and increased serum creatinine
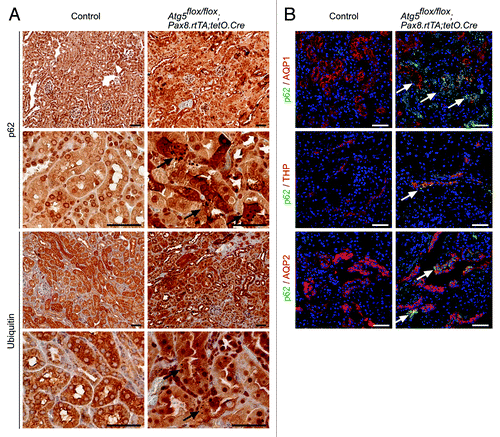
To analyze the impact of autophagy on the entire tubule system in adult mice we utilized the inducible Pax8.rtTA;tetO.Cre system. This system offers several important advantages. First, it has been demonstrated to be active in all tubular segments. Second, the inducible system prevents chronic adaptive pathways (e.g., upregulation of other protein degradative pathways) as it has been demonstrated in constitutive deletion of autophagy. Third, confounding factors due to developmental defects can be excluded. To generate a time-specific tubular Atg5 knockout, we crossed Atg5flox/flox mice to doxycycline-inducible Pax8.rtTA;tetO.Cre transgenic mice ().Citation16 In addition, we generated mT/mG;Pax8.rtTA;tetO.Cre reporter mice. mT/mG transgenic mice carry a loxP-flanked Tomato cassette.Citation17 Upon Cre-mediated excision of the membrane-targeted tandem dimer Tomato (mT), an alternate reporter protein, membrane-targeted GFP (mG), is expressed. The immunofluorescence of kidney sections two weeks after doxycycline induction indicated that all tubular segments were positive for the excision event (). Western blot analysis 1 mo after doxycycline administration confirmed a significant reduction in Atg5 and almost complete loss of LC3-I to LC3-II conversion in total kidney lysates as well as lysates of the cortex and the medulla (; Fig. S1). Further, western blot analysis revealed accumulation of p62 (). No obvious histological phenotype could be detected 5 mo after induction (). However, ultrastructural analysis detected concentric membrane bodies in Atg5 deficient tubular cells (). Interestingly, similar aberrant membrane structures were previously described in Atg7-null hepatic cells and Atg5-null Purkinje cells.Citation18,Citation19 Analysis of clinical parameters 5 mo after induction revealed a mild but significant increase in serum creatinine, with no significant difference in urinary levels of the tubular injury marker neutrophil gelatinase-associated lipocalin (NGAL) ().Citation20,Citation21 We next examined protein aggregation immunohistochemically using antibodies against p62 and ubiquitin. illustrates p62 and ubiquitin positive inclusion bodies in Atg5-knockout tubular cells 5 mo after doxycycline induction. Costaining for p62 and tubular marker proteins revealed the accumulation of p62 in the proximal tubules, the thick ascending limb of the loop of Henle, and the collecting duct ().
Figure 3. Time-specific deletion of Atg5 in the kidney tubule system results in ultrastructural alterations and increased serum creatinine. (A) Schematic illustration of the generation of doxycycline-inducible kidney tubule-specific Atg5-deficient mice (Atg5flox/flox;Pax8.rtTA;tetO.Cre). (B) Expression of GFP in the tubule system of mT/mG;Pax8.rtTA;tetO.Cre reporter mice 2 weeks after doxycycline induction. (C) Western blot analysis of total kidney lysate confirmed significant reduction of Atg5, almost complete loss of LC3-I to LC3-II conversion and accumulation of p62 4 weeks after doxycycline induction. (D) Densitometric analysis (n = 3 each; *p < 0.05). (E) No obvious histological lesions in Atg5flox/flox;Pax8.rtTA;tetO.Cre 5 mo after doxycycline induction. (F) Ultrastructural analysis revealed accumulation of concentric membrane bodies (arrowheads) in Atg5 deficient proximal tubule cells. (G–I) Significant increased serum creatinine, but no differences in serum urea and urinary NGAL 1 and 5 mo after doxycyclin induction (n = 7–11 Atg5flox/flox;Pax8.rtTA;tetO.Cre mice and n = 6–11 control mice; ***p < 0.001). (J) No significant difference in mouse body weight (male mice, n = 6 Atg5flox/flox;Pax8.rtTA;tetO.Cre mice and n = 6 control mice). Scale bars: 100 µm in (B), 50 µm in (E), 1 µm in (F).
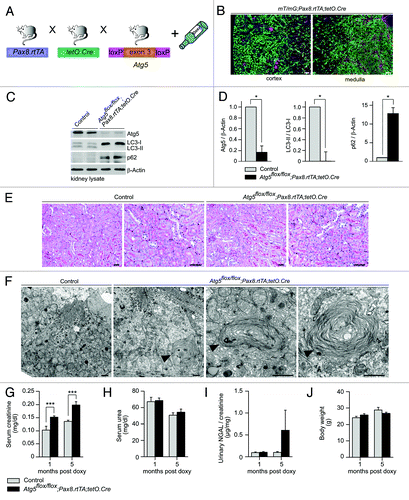
Figure 4. Accumulation of p62 and ubiquitin-positive inclusion bodies in Atg5-deficient tubule cells. (A) Immunohistochemistry staining of kidney sections of Atg5flox/flox;Pax8.rtTA;tetO.Cre mice and control littermates 5 mo after doxycycline induction for p62 and ubiquitin. Arrows indicate p62 or ubiquitin positive inclusion bodies, respectively. (B) Immunofluorescence staining of kidney cryosections as indicated. Arrows mark colocalization of p62 with AQP1, THP and AQP2. Scale bars: 50 µm.
Ischemia/reperfusion results in severe tubular injury in tubule cell-specific Atg5-deficient kidneys
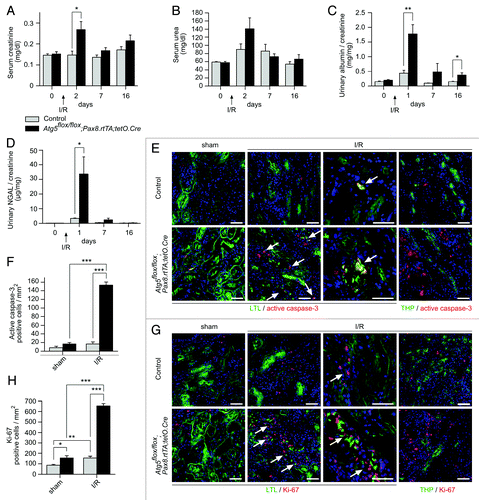
To investigate the role of autophagy in the tubular cell stress response, we performed kidney ischemia/reperfusion two weeks after doxycycline administration (). This resulted in severe tubular injury in the Atg5flox/flox;Pax8.rtTA;tetO.Cre mice, with sloughing of tubular cells and detached cells filling the tubular lumina (). Autophagosomes and autolysosomes partially filled with mitochondria could be detected in the proximal tubular cells of control mice, while Atg5-null tubular cells exhibited concentric membranes surrounding damaged mitochondria (). The costaining of kidney cryosections after ischemia/reperfusion revealed a significant increase of p62 accumulation in the proximal tubule and thick ascending limb of the loops of Henle in Atg5flox/flox;Pax8.rtTA;tetO.Cre mice compared with control littermates and sham-operated mice (Fig. S2). Atg5flox/flox;Pax8.rtTA;tetO.Cre mice exhibited transient elevations in serum creatinine and urinary albumin and a massive increase in urinary NGAL, a tubular cell injury marker (). In addition, these kidneys displayed an infiltration with T-cells and macrophages (Fig. S3). Immunofluorescent staining for active caspase-3 and the proliferation marker Ki-67 revealed significantly higher tubular cell apoptosis and proliferation predominantly in the proximal tubules in Atg5flox/flox;Pax8.rtTA;tetO.Cre kidneys compared with control kidneys (; Fig. S4 and S5) indicating that loss of autophagy dramatically sensitizes the tubular system toward ischemic injury.
Figure 5. Ischemia/reperfusion results in severe tubular injury in tubule-specific Atg5-deficient kidneys. (A) Two weeks after doxycycline administration Atg5flox/flox;Pax8.rtTA;tetO.Cre mice and control littermates were exposed to ischemia/reperfusion injury. (B) Severe tubular injury in Atg5flox/flox;Pax8.rtTA;tetO.Cre 2 and 7 d after ischemia/reperfusion. Arrows indicate sloughing of tubular cells and tubular lumina filled with detached cells, respectively. (C) Three days after ischemia/reperfusion, ultrastructural analysis revealed increase of autophagosomes and autolysosomes containing mitochondria in control proximal tubular cells. Knockout proximal tubular cells displayed accumulation of damaged mitochondria and concentric membranes surrounding mitochondria, protein aggregates and lipid inclusions. Arrows indicate autophagosomes with their surrounding double membrane and an autolysosome with a single membrane (right), arrowheads indicate concentric membranes, mitochondria are marked with “M” exemplarily, lipid inclusion is marked with “L.” Scale bars: 50 µm in (B), 1 µm in (C).
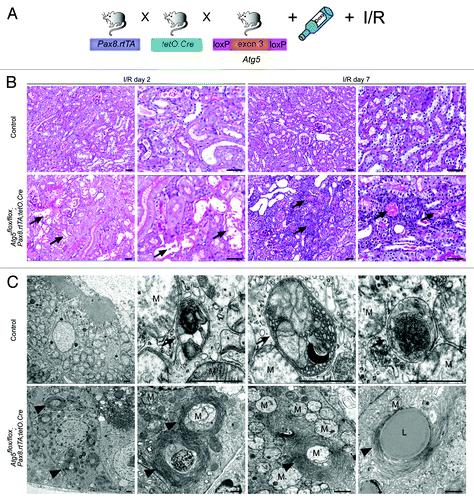
Figure 6. Characterization of tubular injury after ischemia/reperfusion. (A) Ischemia/reperfusion resulted in transiently elevated serum creatinine, (B) not significantly increased serum urea, (C) albuminuria and (D) massive increased urinary NGAL levels in Atg5flox/flox;Pax8.rtTA;tetO.Cre mice (n = 10 Atg5flox/flox;Pax8.rtTA;tetO.Cre mice and n = 9 control mice; **p < 0.01, *p < 0.05). (E and F) Increased number of apoptotic cells, stained by active caspase-3 antibody and (G and H) accelerated proliferation, marked by Ki-67 antibody, costained with the proximal tubule marker Lotus Tetragonolobus lectin (LTL) in Atg5flox/flox;Pax8.rtTA;tetO.Cre mice 7 d after ischemia/reperfusion (n = 3 each, *p < 0.05, **p < 0.01, ***p < 0.001). No costaining of active caspase-3 or Ki-67 respectively with the thick ascending limb of loop of Henle marker THP could be detected. Scale bars: 50 µm.
p62 and oxidative injury markers accumulate in Atg5 knockout distal tubule cells
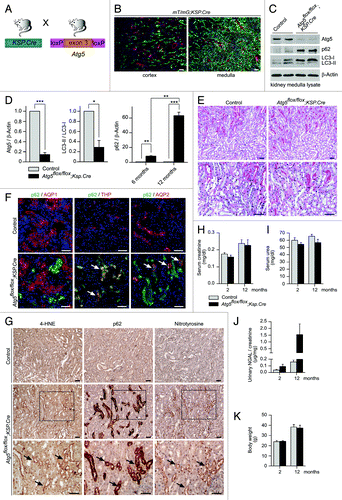
Autophagy seems to be of major importance for the proximal tubule system. Therefore, a complete tubular ATG5 knockout will always be dominated by the phenotype of proximal tubular dysfunction. To exclude that we do not miss a more subtle phenotype in the distale tubule, we generated a distal tubule specific Atg5 knockout by crossing Atg5flox/flox mice to Ksp.Cre transgenic mice ().Citation22 In addition, we crossed Ksp.Cre mice to the mT/mG transgenic reporter strain.Citation17 In agreement with previously published results, kidney sections were GFP-positive in distal tubular segments, confirming Cre expression in distal tubular segments ().Citation22 Western blot analysis confirmed almost complete loss of Atg5 and a significant reduction in LC3-I to LC3-II conversion in kidney medulla lysates, resulting in accumulation of LC3-I (; Fig. S1). Furthermore, the time-dependent accumulation of p62, an ubiquitin-binding scaffold protein that is degraded by autophagy,Citation23 could be detected (). Histological analysis of kidneys from 14-mo-old Atg5flox/flox;Ksp.Cre mice and control littermates did not reveal any obvious tubule structural differences between the two groups (). Costaining of kidney cryosections for p62 and tubular marker proteins showed colocalization of p62 with THP and AQP2, confirming the accumulation of p62 in the distal tubular segments of Atg5flox/flox;Ksp.Cre mice (). In addition, we performed immunohistochemical staining of consecutive kidney sections for p62 and the oxidative stress markers 4-hydroxynonenal (4-HNE) and nitrotyrosine and identified an increased signal for both markers in the tubular segments that accumulate p62 (). Our analysis of the clinical parameters for kidney function displayed no differences in serum creatinine and serum urea levels between Atg5flox/flox;Ksp.Cre mice and control littermates until 12 mo of age (). Furthermore, no significant differences in the expression of NGAL were detected in the urinary analysis () and no difference in body weight (). In summary, distal tubular deletion of autophagy leads to increased p62 accumulation and increased oxidative stress markers, but these changes do not reach the threshold for causing structural or clinical consequences under physiological conditions.
Figure 7. Distal tubule-specific Atg5 knockout results in accumulation of p62 and oxidative injury markers. (A) Schematic illustration of the generation of distal tubule-specific Atg5-deficient mice (Atg5flox/flox;Ksp-Cre). (B) Expression of GFP in the distal tubule system of mT/mG;Ksp.Cre reporter mice. (C) Western blot of kidney medulla lysate. (D) Densitometric analysis confirmed significant reduction of Atg5, reduced LC3-I to LC3-II conversion and time-dependant accumulation of p62 in kidney medulla of Atg5flox/flox;Ksp-Cre mice (n = 3 each; *p < 0.05, **p < 0.01, ***p < 0.001). (E) No obvious histological lesions in 14-mo-old Atg5flox/flox;Ksp-Cre mice. (F) Costaining of p62 with the proximal tubule marker AQP1, the thick ascending limb of loop of Henle marker THP and the collecting duct marker AQP2. Arrows indicate colocalization of p62 with THP and AQP2, but not with AQP1 in kidney cryosections of 6 mo old Atg5flox/flox;Ksp-Cre mice and control littermates. (G) Immunohistochemistry staining of consecutive kidney sections show accumulation of the oxidative stress markers 4-HNE and nitrotyrosine in the p62 positive tubule segments of 14 mo old Atg5flox/flox;Ksp-Cre mice. Arrows mark serial sections of the same tubule segment. (H–J) No significant differences in serum creatinine, serum urea and urinary NGAL levels of 12 mo old Atg5flox/flox;Ksp-Cre mice and control littermates (n = 8–9 Atg5flox/flox;Ksp-Cre mice and n = 9–11 control mice). (K) No significant difference in mouse body weight (male mice, n = 5 Atg5flox/flox;Ksp-Cre mice and n = 7 control mice). Scale bars: 100 µm in (B), 50 µm in (E–G).
Discussion
Autophagy is a cellular pathway involved in protein and organelle degradation that has an amazing number of connections to cellular homeostasis and human disease.Citation1 Our current results highlight the critical role autophagy plays in maintaining tubule homeostasis and integrity under conditions of stress.
Under normal conditions, tubular cells from GFP-LC3 transgenic mice exhibit very low levels of GFP-LC3-positive autophagosomes (), which has also been described recently.Citation14 However, in tubule cell-specific Atg5-deficient mice, the selective autophagy substrate p62Citation24 rapidly accumulates, resulting in oxidative stress and the formation of inclusion bodies (, and ). Such findings have been reported in many types of cells that have high basal levels of autophagy.Citation13,Citation25-Citation30,Citation31 The fast accumulation of p62 suggests that tubular cells might have higher levels of autophagic activity as estimated by the GFP-LC3 transgenic mouse system. The formation of p62-positive inclusion bodies or aggregates under autophagy-deficient conditions is thought to compromise the activity of the UPS.Citation32,Citation33 This causes cell dysfunction and death.Citation32,Citation33 For instance, autophagy-deficient podocytes in the kidney accumulate p62 and ubiquitin-positive inclusion bodies that impact the UPS, leading to the aggregation of oxidated proteins, ER stress, and consequently the loss of podocytes.Citation13
Although the lack of autophagy in the distal tubule segments of our mouse model leads to the accumulation of p62 and the deposition of oxidative stress markers, the kidney function of these mice remains stable until one year of age (). On the other hand, the time-specific deletion of Atg5 in the entire tubule system results in increased serum creatinine levels that are accompanied by the formation of p62 and ubiquitin-containing inclusion bodies ( and ) 4 to 5 mo after doxycycline administration. In mice creatinine is not only filtrated, but also significantly secreted by tubule cells. Therefore, it cannot be ruled out that differences in serum creatinine might be due to defective tubular secretion. Recently, Kimura et al. analyzed the role of autophagy in the proximal tubule system with a mouse line that expresses Cre under the control of the kidney androgen-regulated protein (KAP) gene promoter.Citation34 These mice specifically express Cre in the proximal tubule system. Similar to our observations, KAP.Cre Atg5flox/flox-knockout mice exhibit p62- and ubiquitin-positive inclusion bodies and the accumulation of an amorphous substrate in the cytoplasm of proximal tubule cells at 6 to 9 mo of age. In electron microscopy, these cells displayed crescent-like structures and deformed mitochondria, while we observed concentric membrane bodies in doxycycline-induced, time-specific Atg5-knockout tubular cells (). The accumulation of deformed mitochondria under autophagy-deficient conditions has been reported in hepatocytes,Citation18 neural cells,Citation26,Citation27 cardiomyocytesCitation28 and skeletal muscle cells.Citation29 The proximale tubule contains a large quantity of mitochondria, requiring an efficient intracellular quality control system of damaged mitochondria. Corresponding with the observation that deformed mitochondria primarily accumulate in aged or stressed proximal tubular cells (),Citation34 mice that are deficient in Atg5 throughout the entire tubule showed steadily increased serum creatinine, whereas mice deficient in Atg5 only in the distal tubule show oxidative stress in distal tubules but retain stable kidney function until one year of age (). These data suggest that proximal tubule cells depend more than any other tubule segment on basal autophagic activity.
To elucidate the role of autophagy in kidney tubular cells experiencing pathological conditions, we performed kidney ischemia/reperfusion experiments. Unlike Kimura et al.,Citation34 we used a time-specific, doxycycline-induced Atg5-knockout model, which appears to be a very useful system as we can thereby exclude the possibility of any confounding developmental or compensatory phenotypes. Ischemia/reperfusion experiments in Atg5-null tubular and control mice revealed the importance of autophagy for tubules under hypoxic conditions: First, the presence of p62 inclusion bodies in Atg5-null tubular cells increased greatly after ischemia/reperfusion, underlining the importance of autophagy induction in this stress model (Fig. S2). Second, in histological and immunofluorescence analysis, we detected severe tubular injury, high rates of apoptosis and proliferation, and transiently increased serum creatinine and urinary NGAL levels in tubule-specific Atg5-null animals ( and ). The reversibility of these observations underlines the fact that tubule cells can be renewed by cell division, unlike postmitotic cells like neurons, cardiomyocytes and glomerular podocytes. Third, in ultrastructural analysis studies, control proximal tubular cells exhibited high levels of autophagosomes containing mitochondria and altered cellular components () in response to ischemia/reperfusion, again suggesting that autophagy plays a protective role against mitochondrial damage and reactive oxygen species in tubular cells. Interestingly, in Atg5-null proximal tubular cells, mitochondria, protein aggregates and lipid inclusions were surrounded by concentric membranes, which might reflect cellular effort to isolate these damaged structures from the cytoplasm in the absence of autophagy. Interestingly, similar structures have also been observed in Atg7-null hepatic cells and Atg5-null Purkinje cells.Citation18,Citation19 Recent studies have revealed that various types of cargo-specific autophagy, including mitophagy, aggregophagy, and lipophagy, occur under nutrient-rich conditions.Citation35,Citation36 Our observations suggest that autophagy is upregulated following I/R injury in order to selectively degrade damaged mitochondria (mitophagy) and protein aggregates (aggregophagy), indicating that autophagy might be a protection mechanism utilized by tubular cells during stress conditions.
Interestingly, lysosomal storage diseases like Fabry disease and cystinosis cause impaired autophagic flux with defective autophagic maturation and abnormal mitochondria.Citation3,Citation37 However, it is conceivable to speculate that impairment of autophagy contributes not only to rare lysosomal storage diseases, but also to common kidney diseases. It will be interesting to see if future genome-wide association studies (GWAS) will link increased kidney disease susceptibility to autophagy related genes.
In summary, our data demonstrate that autophagy is an important homeostatic mechanism for kidney tubular cells. Autophagy is essential for the maintenance of tubular cell integrity, particularly under pathological conditions such as acute kidney injury caused by ischemia/reperfusion.
Materials and Methods
Mice
Mice bearing an Atg5flox allele,Citation27 in which exon 3 of the Atg5 gene is flanked by two loxP sequences, and Ksp.Cre transgenic miceCitation22 have been previously reported. Atg5-floxed mice (Atg5flox/flox) were crossed with Ksp.Cre mice to generate distal tubule-specific Atg5 knockout mice Atg5flox/flox;Ksp.Cre. Atg5flox/WT;Ksp-Cre and Atg5flox/flox littermates served as control. Constitutive Atg5 knockout mice (Atg5−/−),Citation15 GFP-LC3,Citation14 and Pax8.rtTA;tetO.Cre transgenic miceCitation16 have been previously reported. To generate doxycycline-inducible tubule-specific Atg5 knockout mice (Atg5flox/flox;Pax8.rtTA;tetO.Cre) Atg5-floxed mice (Atg5flox/flox) were crossed with Pax8.rtTA;tetO.Cre mice; Pax8.rtTA- or tetO.Cre- littermates served as control. All mice were crossed on a pure C57/Bl6 background. For the induction of Atg5 deletion 8 weeks old Atg5flox/flox;Pax8.rtTA;tetO.Cre mice received doxycycline hydrochloride (Fagron, 137087-0008) via the drinking water (2 mg/ml with 5% sucrose, protected from light) for a total of 14 d. Kidney ischemia/reperfusion injury was induced in male mice 2 weeks after doxycycline administration. Mice were anesthetized. Back incision was made and kidney arteries were clamped bilateral for 25 min.Citation20 To generate mT/mG;Pax8.rtTA;tetO.Cre and mT/mG;Ksp.Cre reporter mice, mT/mG miceCitation17 were crossed with Pax8.rtTA;tetO.Cre or Ksp.Cre mice, respectively. All animal studies were approved by the Committee on Research Animal Care Freiburg.
Urine and serum analyses
Urinary and serum albumin, creatinine and urea were measured using a fluorimetric albumin test kit (Progen, PR2005) and enzymatic colorimetric creatinine and urea kits (Labor+Technik, LT-CR0053, LT-UR0010) following the manufacturer’s instructions. Urinary NGAL was measured using a mouse NGAL specific ELISA kit (Bioporto, KIT042).
Morphological analysis
Kidneys were fixed in 4% paraformaldehyde in PBS, embedded in paraffin and further processed for PAS staining. For ultrastructural analysis kidneys were fixed in 4% paraformaldehyde and 1% glutaraldehyde in 0.1 M PB. Tissue blocks (1 mm3 blocks) were treated with OsO4, stained with uranyl acetate, dehydrated, and embedded in epoxy resin (Durcupan ACM Fluka, Sigma-Aldrich, 44610). Ultrathin sections were cut and analyzed with a Philips CM100 electron microscope.
Immunofluorescence staining of kidney sections
Kidneys were frozen in OCT compound and sectioned at 6 µm (Leica Kryostat). The sections were fixed with 4% paraformaldehyde, blocked in PBS containing 5% BSA and incubated for 1 h with primary antibodies as indicated. After PBS rinse for several times, fluorophore-conjugated secondary antibodies (Invitrogen) were applied for 30 min. Images were taken using a Zeiss laser scan microscope or a Zeiss fluorescence microscope equipped with a 20 × and a 63 × water immersion objective.
Immunohistochemistry staining of kidney sections
Kidneys were fixed in 4% paraformaldehyde in PBS, embedded in paraffin and sectioned at 4 µm (Leica Microtome). After deparaffinization, hydration and antigene retrieval in 10mM sodium citrate sections were blocked with PBS containing 2% BSA and incubated overnight with primary antibodies as indicated, followed by peroxidase blocking in 3% H2O2 for 10 min and incubation with HRP conjugated secondary antibodies (Dako, K4002, P0141) for 1 h. DAB (Dako, K3467) was applied for 6 min. Sections were counterstained with hematoxylin. Staining with mouse monoclonal primary antibodies was performed using a histomouse kit (Zymed, 85-9541) following the manufacturer’s instructions.
Western blot
Kidneys were glass-glass-homogenized in lysis buffer (containing 20 mM CHAPS and 1% Triton X-100). After centrifugation (15,000 × g, 15 min, 4°C) protein concentration was determined by Dc Protein-Assay (Bio-Rad, 500-0113, 500-0114, 500-0115). Equal amounts of protein were separated on SDS page. Western blots were densitometrically analyzed using LabImage software. Ratios of protein band intensity to loading control protein band intensity are shown.
Antibodies
Antibodies were obtained from Alomone labs (anti-aquaporin 1 rabbit pAb, AQP-001; anti-aquaporin 2 rabbit pAb, AQP-002), AbD serotec (anti-Tamm Horsfall glycoprotein sheep pAb, 8595-0054), MBL (anti-LC3 mouse mAb, M152-3), Progen (anti-p62 guinea pig pAb, GP62-C), Thermo Scientific (anti-Ki-67 rabbit pAb, RM-9106-S0), Millipore (anti-Nitrotyrosine rabbit pAb, AB5411; anti-Nidogen rat mAb, MAB1946), Oxis International (anti-4-HNE mouse mAb, 24325), Cosmo Bio (anti-Atg5 rabbit pAb, CAC-TMD-PH-AT5), Sigma-Aldrich (anti-Desmin mouse mAb, D1033), Santa Cruz Biotechnology (anti-CD3 rat mAb, sc-101442; anti-Macrophage marker rat mAb, sc-101447; anti-Neutrophil marker rat mAb, sc-71674) and R&D Systems (anti-active caspase-3 rabbit pAb, AF835). Proximal tubule staining reagent (fluorescein Lotus Tetragonolobus lectin, FL-1321) was obtained from Vector Laboratories. Nuclear staining reagent (To-Pro-3, T3605) and fluorophore conjugated secondary antibodies were obtained from Invitrogen.
Statistical Analyses
Data were expressed as the mean ± SEM. Statistical comparisons were performed using two-tailed Student’s t-test if not stated otherwise. Differences with p < 0.05 were considered significant.
| Abbreviations: | ||
| I/R | = | ischemia/reperfusion |
| AQP1 | = | aquaporin 1 |
| THP | = | Tamm Horsfall protein |
| AQP2 | = | aquaporin 2 |
| 4-HNE | = | 4-hydroxynonenal |
| NGAL | = | neutrophil gelatinase-associated lipocalin |
| UPS | = | ubiquitin-proteasome system |
| KAP | = | kidney androgen-regulated protein |
| LTL | = | Lotus Tetragonolobus lectin |
Additional material
Download Zip (4.5 MB)Acknowledgments
We thank Charlotte Meyer, Ann-Kathrin Fritz (Renal Division, University Hospital Freiburg), Sigrun Nestel (Department of Anatomy, Albert-Ludwigs-University Freiburg) and Evelyn Wätzig (Department of Pathology, University Hospital Freiburg) for excellent technical assistance. This study was supported by DFG (Deutsche Forschungsgemeinschaft) grant KFO 201 (to GW and T.B.H.), by the Excellence Initiative of the German Federal and State Governments EXC 294 (to T.B.H.) and GSC-4, Spemann Graduate School (to S.L. and T.B.H.) and BMBF GerontosysII?NephAge (031589GA) (to T.B.H.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Suzuki C, Isaka Y, Takabatake Y, Tanaka H, Koike M, Shibata M, et al. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun 2008; 368:100 - 6; http://dx.doi.org/10.1016/j.bbrc.2008.01.059; PMID: 18222169
- Sansanwal P, Yen B, Gahl WA, Ma Y, Ying L, Wong LJ, et al. Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J Am Soc Nephrol 2010; 21:272 - 83; http://dx.doi.org/10.1681/ASN.2009040383; PMID: 19959713
- Inoue K, Kuwana H, Shimamura Y, Ogata K, Taniguchi Y, Kagawa T, et al. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin Exp Nephrol 2010; 14:112 - 22; http://dx.doi.org/10.1007/s10157-009-0254-7; PMID: 20013139
- Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 2008; 74:631 - 40; http://dx.doi.org/10.1038/ki.2008.214; PMID: 18509315
- Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol 2008; 294:F777 - 87; http://dx.doi.org/10.1152/ajprenal.00590.2007; PMID: 18256309
- Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol 2010; 176:1181 - 92; http://dx.doi.org/10.2353/ajpath.2010.090594; PMID: 20075199
- Li L, Zepeda-Orozco D, Black R, Lin F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am J Pathol 2010; 176:1767 - 78; http://dx.doi.org/10.2353/ajpath.2010.090345; PMID: 20150430
- Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol 2011; 7:189 - 200; http://dx.doi.org/10.1038/nrneph.2011.16; PMID: 21364518
- Gueler F, Gwinner W, Schwarz A, Haller H. Long-term effects of acute ischemia and reperfusion injury. Kidney Int 2004; 66:523 - 7; http://dx.doi.org/10.1111/j.1523-1755.2004.761_11.x; PMID: 15253702
- Turkmen K, Martin J, Akcay A, Nguyen Q, Ravichandran K, Faubel S, et al. Apoptosis and autophagy in cold preservation ischemia. Transplantation 2011; 91:1192 - 7; http://dx.doi.org/10.1097/TP.0b013e31821ab9c8; PMID: 21577181
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest 2010; 120:1084 - 96; http://dx.doi.org/10.1172/JCI39492; PMID: 20200449
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101 - 11; http://dx.doi.org/10.1091/mbc.E03-09-0704; PMID: 14699058
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032 - 6; http://dx.doi.org/10.1038/nature03029; PMID: 15525940
- Traykova-Brauch M, Schönig K, Greiner O, Miloud T, Jauch A, Bode M, et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med 2008; 14:979 - 84; http://dx.doi.org/10.1038/nm.1865; PMID: 18724376
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis 2007; 45:593 - 605; http://dx.doi.org/10.1002/dvg.20335; PMID: 17868096
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425 - 34; http://dx.doi.org/10.1083/jcb.200412022; PMID: 15866887
- Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 2007; 3:591 - 6; PMID: 17912025
- Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol 2003; 14:2534 - 43; http://dx.doi.org/10.1097/01.ASN.0000088027.54400.C6; PMID: 14514731
- Mishra J, Mori K, Ma Q, Kelly C, Barasch J, Devarajan P. Neutrophil gelatinase-associated lipocalin: a novel early urinary biomarker for cisplatin nephrotoxicity. Am J Nephrol 2004; 24:307 - 15; http://dx.doi.org/10.1159/000078452; PMID: 15148457
- Shao X, Somlo S, Igarashi P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol 2002; 13:1837 - 46; http://dx.doi.org/10.1097/01.ASN.0000016444.90348.50; PMID: 12089379
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Kirkin V, Lamark T, Johansen T, Dikic I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 2009; 5:732 - 3; http://dx.doi.org/10.4161/auto.5.5.8566; PMID: 19398892
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149 - 63; http://dx.doi.org/10.1016/j.cell.2007.10.035; PMID: 18083104
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/10.1038/nature04723; PMID: 16625205
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007; 13:619 - 24; http://dx.doi.org/10.1038/nm1574; PMID: 17450150
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab 2009; 10:507 - 15; http://dx.doi.org/10.1016/j.cmet.2009.10.008; PMID: 19945408
- Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 2008; 8:325 - 32; http://dx.doi.org/10.1016/j.cmet.2008.08.009; PMID: 18840363
- Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 2008; 8:318 - 24; http://dx.doi.org/10.1016/j.cmet.2008.08.013; PMID: 18840362
- Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett 2010; 584:1374 - 8; http://dx.doi.org/10.1016/j.febslet.2010.02.017; PMID: 20153326
- Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 2010; 584:1393 - 8; http://dx.doi.org/10.1016/j.febslet.2009.12.047; PMID: 20040365
- Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 2011; 22:902 - 13; http://dx.doi.org/10.1681/ASN.2010070705; PMID: 21493778
- Wong E, Cuervo AM. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol 2010; 2:a006734; http://dx.doi.org/10.1101/cshperspect.a006734; PMID: 21068151
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nature reviews 2011; 12:9-14.
- Chévrier M, Brakch N, Céline L, Genty D, Ramdani Y, Moll S, et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010; 6:6; http://dx.doi.org/10.4161/auto.6.5.11943; PMID: 20431343