Abstract
The mammalian target of rapamycin (MTOR) protein kinase complex is a key component of a pathway that regulates cell growth and proliferation in response to energy levels, hypoxia, nutrients and insulin. Inhibition of MTORC1 strongly induces autophagy by regulating the activity of the ULK protein kinase complex that is required for the formation of autophagosomes. However, the participation of MTORC1 in the expression of autophagy genes has not been characterized. Here we show that MTORC1 regulates nuclear localization and activity of the transcription factor EB (TFEB), a member of the bHLH leucine-zipper family of transcription factors that drives expression of autophagy and lysosomal genes. Under normal nutrient conditions, TFEB is phosphorylated in Ser211 in an MTORC1-dependent manner. This phosphorylation promotes association of TFEB with members of the YWHA (14-3-3) family of proteins and retention of the transcription factor in the cytosol. Pharmacological or genetic inhibition of MTORC1 causes dissociation of the TFEB/YWHA complex and rapid transport of TFEB to the nucleus where it increases transcription of multiple genes implicated in autophagy and lysosomal function. Active TFEB also associates with late endosomal/lysosomal membranes through interaction with the LAMTOR/RRAG/MTORC1 complex. Our results unveil a novel role for MTORC1 in the maintenance of cellular homeostasis by regulating autophagy at the transcriptional level.
Introduction
The MTOR kinase signaling pathway coordinates energy, growth signals, and nutrient abundance with cell growth and division.Citation1 MTOR responds to numerous stresses and its dysregulation leads to cancer, metabolic disease and diabetes. In cells, MTOR exists as two structurally and functionally distinct complexes termed MTOR complex 1 (MTORC1) and MTOR complex 2 (MTORC2).Citation2-Citation4 MTORC1 couples energy and nutrient abundance to cell growth and proliferation by balancing anabolic (protein synthesis and nutrient storage) and catabolic processes (autophagy and the utilization of energy stores). MTORC1 localizes to late endosomes/lysosomes and this distribution is thought to be critical for the ability of MTORC1 to sense and respond to variations in the levels of amino acids.Citation5
MTORC1 is considered a transcription-independent regulator of autophagy. Under rich-nutrient conditions, MTORC1 is active and directly phosphorylates and inhibits ATG proteins involved in autophagy induction such as ATG13 and ATG1 (ULK1/2).Citation6,Citation7 Under starvation conditions when MTORC1 is inactivated, MTORC1 dissociates from the ULK complex, thus leading to autophagy induction.Citation8 In addition, many cellular pathways known to regulate autophagy merge on MTORC1.Citation9 Despite these insights, the contribution of MTORC1 to the regulated expression of autophagy genes has not been characterized.
Recently, a new transcription-dependent mechanism regulating autophagy has been identified. The transcription factor EB (TFEB) is a member of the basic helix-loop-helix leucine-zipper family of TFs that controls lysosomal biogenesis and autophagy by positively regulating genes belonging to the Coordinated Lysosomal Expression and Regulation (CLEAR) network.Citation10-Citation12 TFEB is normally localized to the cytosol but mobilizes into the nucleus under starvation conditions or when lysosomal function is compromised.Citation10,Citation13 TFEB overexpression induces increased number of autophagosomes and autophagic flux, generation of new lysosomes, and leads to clearance of storage material in several lysosomal storage disorders (LSDs) by promoting lysosomal secretion.Citation12,Citation14 In this study we show that MTORC1 drives expression of autophagy and lysosomal genes by regulating phosphorylation-dependent association of TFEB with YWHA (14-3-3) proteins. These findings have broad implications for our understanding of MTORC1 function and provide new insights into the complex regulation of autophagy.
Results
MTOR regulates the subcellular distribution of TFEB
We hypothesized that MTORC1 might regulate the distribution and activation of TFEB. To test this possibility we infected ARPE-19 cells with adenovirus expressing TFEB-Flag and treated the cells with a number of MTORC1 inhibitors. As predicted, inhibition of MTORC1 rapidly induced nuclear translocation of TFEB (). In control conditions (cells infected with Ad-TFEB and incubated with DMSO), less than 20% of the cells (18.34% SD ± 1.08, n = 322) showed staining of TFEB in the nucleus. In contrast, treatment of cells with the MTOR catalytic inhibitors LY294002, wortmannin or PP242 induced a significant accumulation of TFEB in the nucleus in almost 100% of the cells (97.08% SD ± 0.87, n = 308 for LY294002; 95.46% SD ± 0.5, n = 309 for wortmannin; 92.2% SD ± 1.31, n = 312 for PP242). Similar results were obtained with Torin-1 (data not shown). The allosteric MTORC1 inhibitor rapamycin also induced a significant accumulation of TFEB in the nucleus although the effect was slightly less robust (70.4% SD ± 1.97, n = 321). This is probably due to the fact that only partial inhibition of MTORC1 is achieved by treatment with this drugCitation15-Citation17 (). The redistribution of TFEB to the nucleus upon MTOR inactivation was observed in various cells types, including human fibroblasts and a clone of HeLa cells (CF7) stably expressing TFEBCitation10 (Fig. S1). As a control experiment, we tested inhibition of other cellular kinases, including PtdIns 3-kinase type I, AKT1, AMPK, MAPK1/3, MAPK14 and MAPK8. These did not alter the distribution of TFEB (). Delivery of TFEB to the nucleus was independent of autophagosome formation, as both LY294002 and wortmannin (but not PP242) blocked formation of autophagosomes due to their inhibitory effect on PtdIns 3-kinase type III but still induced TFEB activation (Fig. S2).
Figure 1. MTORC1 regulates the subcellular distribution of TFEB. (A) Immunofluorescence confocal microscopy showing nuclear localization of recombinant TFEB-Flag in ARPE-19 cells incubated with MTORC1 inhibitors. (B) Quantification of the nuclear localization of TFEB in ARPE-19 cells incubated with different kinase inhibitors. Values are means ± SD of three independent experiments. (C) Immunofluorescence confocal microscopy showing colocalization between TFEB-Flag and MTORC1 in ARPE-19 cells incubated with PP242 for 2 h. (D) Quantification of the lysosomal localization of TFEB in ARPE-19 cells incubated with PP242. (E) Still images of a time-lapse movie showing the sequential association of TFEB-GFP with lysosomes and subsequent accumulation in the nucleus. Scale bars: 10 μm. ***p < 0.001; n.s.: not significant.
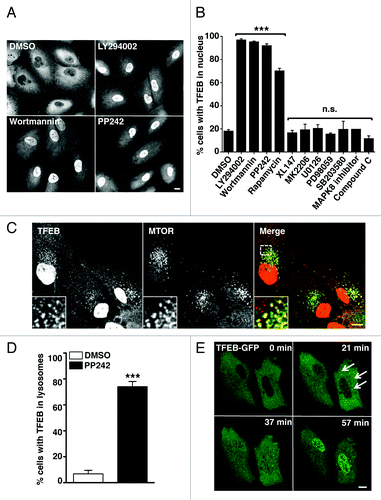
Figure 2. Pharmacological or genetic inhibition of MTORC1 induces transport of TFEB to the nucleus. (A) HeLa cells stably expressing TFEB (CF7) were incubated with the indicated kinase inhibitors and the electrophoretic mobility of TFEB was monitored by immunoblotting. (B) Subcellular fractionation of HeLa (CF7) cells incubated with different kinase inhibitors. (C) Immunofluorescence confocal microscopy showing TFEB localization in ARPE-19 cells depleted of either RPTOR or RICTOR. Scale bar: 10 μm. (D) Quantification of the nuclear localization of TFEB in ARPE-19 cells depleted of either RPTOR or RICTOR. Values are means ± SD of three independent experiments. ***p < 0.001, **p < 0.01. (E) Immunoblotting analysis of TFEB electrophoretic mobility in lysates of ARPE-19 cells depleted of either RPTOR or RICTOR.
Following MTORC1 inactivation, we also observed increased colocalization between TFEB and MTORC1 into vesicular structures (). These vesicles were positive for LAMP1, suggesting that they correspond to late endosomes/lysosomes (Fig. S3). In control conditions only 6% of cells showed noticeable association of TFEB with late endosomes/lysosomes (6.79% SD ± 2.75, n = 704), while this number increased to almost 75% in cells incubated with PP242 (74.05% SD ± 4.02, n = 1183) (). Similar results were obtained when the cells were incubated with LY294002, wortmannin or Torin-1 (data not shown). The association of TFEB with lysosomes was further confirmed by isolation of lysosomal membranes in Optiprep gradients (Fig. S4). Activation of TFEB was also followed by time-lapse microscopy. ARPE-19 cells were transiently transfected with a plasmid encoding TFEB-GFP. In untreated cells or cells incubated with DMSO, TFEB remained in the cytosol. In contrast, just 30 min after adding PP242, TFEB was recruited to vesicular structures and then progressively accumulated in the nucleus (; Fig. S5A and S5B; Movies S1 and S2. Therefore, our data suggest that the transport of TFEB to the nucleus upon MTORC1 inactivation is quite rapid and preceded by the association of TFEB with lysosomal membranes.
Pharmacological or genetic inactivation of MTORC1 changes the electrophoretic motility of TFEB
To further analyze how MTORC1 regulates the distribution of TFEB, we analyzed lysates of TFEB-expressing cells incubated with different kinase inhibitors by SDS-PAGE. As shown in , inhibition of MTORC1 by treatment with LY294002, wortmannin, PP242, rapamycin, or starvation changed the electrophoretic mobility of TFEB, which now appeared as a fast-migrating form. In contrast, no molecular weight shift was observed in cells treated with DMSO or with inhibitors of PtdIns 3-kinase type I, AKT1, AMPK, MAPK1/3, MAPK14 and MAPK8 (). We observed increased levels of phospho-MAPK1/3 in cells treated with MTOR inhibitors. These results are in agreement with previous reports indicating that inhibition of MTOR leads to activation of the Ras-Raf1-MEK1/2-MAPK pathway.Citation18 The change on TFEB motility was reversible, and TFEB recovered its normal molecular weight and cytosolic distribution 4 h after the MTORC1 inhibitor was removed (Fig. S6A and S6B). The recovery was not affected by the addition of inhibitors of MAPK1/3 or AMPK to the normal medium but was inhibited by rapamycin (Fig. S6A and S6B). To address whether the electrophoretic mobility of TFEB was linked to its nuclear localization we performed subcellular fractionation experiments and found that, upon MTORC1 inactivation, only the fast-migrating TFEB was present in the nucleus ().
MTOR exists as two distinct complexes within cells, MTORC1 and MTORC2, depending on its association with RPTOR (RAPTOR) or RICTOR, respectively. To corroborate that the activity of TFEB is regulate by MTORC1, we genetically inhibited either MTORC1 or MTORC2 by depleting RPTOR or RICTOR with specific siRNAs. In cells depleted of RPTOR, TFEB primarily accumulated in the nucleus and exclusively appeared as the fast-migrating form both in the absence or the presence of PP242 (). Downregulation of MTORC1 in the absence of RPTOR was assessed by immunoblotting (Fig. S7). In contrast, inactivation of MTORC2 by depletion of RICTOR did not change the distribution or electrophoretic motility of TFEB (). All together, our results reveal a clear correlation between the activity of MTORC1 and the motility and subcellular distribution of TFEB.
Identification of YWHA proteins as novel binding partners of TFEB
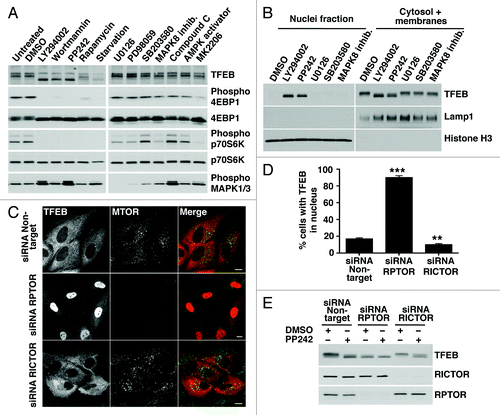
To further understand the mechanism that regulates retention of TFEB in the cytoplasm we searched for proteins that interact with TFEB. Recombinant TFEB was immunoprecipitated with antibodies against the Flag epitope and the samples were separated by SDS-PAGE, and visualized by Coomassie staining. Importantly, a band of approximately 27 kDa was observed to co-immunoprecipitate with TFEB in cells treated with DMSO but it nearly disappeared in cells treated with PP242. The band was excised from the gel, trypsinized, subjected to mass spectrometry analysis, and identified as YWHA (). The identification of YWHA as a novel binding partner of TFEB was highly encouraging considering that the YWHA family of proteins plays a key regulatory role in nutrient-sensing pathways and in nuclear transport of several transcription factors.Citation19,Citation20 The interaction of TFEB with endogenous YWHA was confirmed by immunoblotting with anti-YWHA antibodies (). This experiment also corroborated that treatment of cells with PP242 significantly reduced the amount of YWHA co-immunoprecipitated by TFEB (). Moreover, depletion of RPTOR (but not RICTOR) nearly abolished the interaction, thus corroborating that inactivation of MTORC1 dissociated the TFEB/YWHA complex (Fig. S8). In mammals, at least seven different isoforms of YWHA have been identified. Our mass spectrometry data suggested a preferential binding of TFEB to the YWHAG (gamma) and YWHAZ (zeta/delta) isoforms (data not shown). However, most of the YWHA isoforms were found to co-immunoprecipitate with TFEB (Fig. S9). This is not surprising considering that little specificity has been demonstrated for YWHA isoforms and that many of the isoforms can form heterodimers with each other.
Figure 3. S211 is a novel MTORC1-dependent phosphorylation site in TFEB required for the binding to YWHA. (A) Coomassie blue stained SDS-PAGE gel of co-immunopreciptated TFEB and YWHA from HeLa (CF7) cells treated with either DMSO or PP242. The asterisk (*) indicates IgG heavy and light chains. The arrowhead denotes actin. (B) Immunoblotting analysis of immunoprecipitates obtained as in (A). (C) Immunoblotting analysis of immunoprecipitates obtained as in (A) and incubated with or without calf intestinal alkaline phosphatase (CIAP). (D) Multi-sequence alignment of human TFEB S211 phosphorylation site with MITF and TFEB orthologs. (E) Immunoblotting analysis of immunoprecipitates from ARPE-19 cells overexpressing amino acid specific mutants and truncated forms of TFEB. (F) Collision-induced dissociation (CID) spectrum obtained for the TFEB S211 containing peptide (VGVTSSpSCPADL) showing phosphorylation at Serine 211. (G) Extracted ion chromatograms (XIC) of the peptide (m/z = 636.766) from cells treated with either DMSO or PP242.
The binding of TFEB to YWHA is phosphorylation-dependent and requires Ser211
The phosphorylation-dependent binding of YWHA to target proteins is a common mechanism to achieve phosphorylation-based inactivation. However, YWHA can also bind proteins in a phosphorylation-independent manner.Citation21 To distinguish between these two possibilities, TFEB was dephosphorylated by treatment with alkaline phosphatase. As shown in , dephosphorylation of TFEB abolished the interaction with YWHA, indicating that the binding is dependent on phosphorylation. The microphthalmia-associated transcription factor (MITF) is a transcription factor closely related to the TFE family that also interacts with YWHA in a phosphorylation-dependent manner.Citation22 Sequence alignment revealed a high degree of homology in the YWHA binding motif between MITF and TFEB, and identified S211 as a putative YWHA binding site in TFEB (). This motif was also well conserved among different TFEB orthologs (). Moreover, analysis of the TFEB sequence by using a peptide motif based scanning algorithm (scansite.mit.edu)Citation23 also identified S211 as a weakly related YWHA binding motif (Fig. S10).
Based on this data, we analyzed whether mutations that mimicked the unphosphorylated state for this site (S211A) affected the interaction with YWHA. As predicted, punctual mutation of S211 to Ala completely abolished the interaction of TFEB with YWHA (). In contrast, mutation of adjacent Ser residues S209 and S210 did not have any effect. Several additional serine/threonine residues in TFEB have been suggested to be phosphorylated in response to specific signaling pathways, including S142 by MAPK1Citation13 and T330, T331, S332 and S334 in response to rapamycin.Citation24 However, mutation of these residues to Ala (mutants S142 > A and TS > A) had no effect in the formation of the TFEB/YWHA complex (). Finally, deletion or punctual mutation of a serine-rich region in the C-terminus of TFEB (462SSRRSSFS469) (mutants 1–358, 1–459 and 5S > A) neither changed the ability of TFEB to bind YWHA (). Therefore, our data corroborate that the interaction between TFEB and YWHA is mediated by S211.
MTORC1 regulates phosphorylation-dependent binding of TFEB to YWHA
S211 has never been identified as a site of phosphorylation in TFEB. This may be due to the high reliance on trypsin for polypeptide cleavage in previous mass spectrometry studies.Citation13 To obtain a more suitable digestion pattern, we digested our samples with chymotrypsin and analyzed them by nano scale high performance liquid chromatography-mass spectrometry (nanoLCMS). Our results confirmed that S211 is indeed phosphorylated under control conditions () (an additional example showing the CID spectrum of a different peptide is shown in Fig. S11). Notably, label free quantification revealed that inactivation of MTORC1 by treatment with PP242 caused a 3.6 fold decrease in the phosphorylation levels of S211 (; Fig. S11). These results suggest that MTORC1 induces phosphorylation of TFEB at S211, thus promoting binding of TFEB to YWHA.
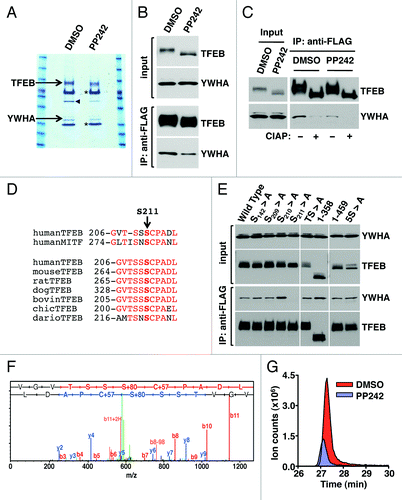
Next, we tested whether binding to YWHA is enough to retain TFEB in the cytosol. In agreement with this idea, we found that mutation of S211 to alanine (TFEB-S211A) caused a very significant accumulation of TFEB into the nucleus of most cells in the absence of MTORC1 inhibitors (). TFEB-S211A also was efficiently recruited to late endosomes/lysosomes (). Inhibition of MTORC1 by starvation further increased the number of cells with TFEB-S211A in the nucleus (from 70% in untreated cells to 80% in starved cells). Therefore, we cannot rule out that additional Ser/Thr residues, other than S211, might also participate in the regulation of TFEB.
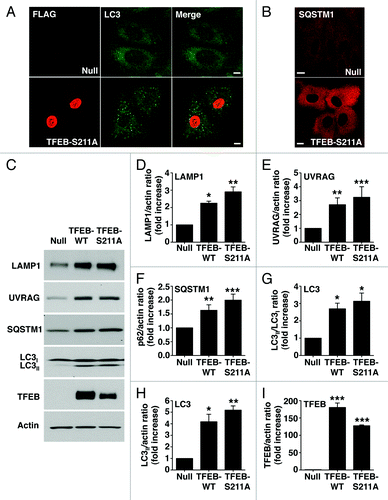
Figure 4. Dissociation of the TFEB/YWHA complex results in transport of TFEB to the nucleus and increased expression of autophagic and lysosomal genes. (A) Immunofluorescence confocal microscopy showing nuclear and late endosomal/lysosomal localization of TFEB-S211A in ARPE-19 cells. (B) Quantification of the nuclear localization of TFEB as shown in (A). Values are means ± SD of five independent experiments. (C–F) Relative quantitative RT-PCR analysis of the mRNA expression of autophagy (ATG9B and UVRAG) and lysosomal (LAMP1 and MCOLN1) genes in ARPE-19 cells infected with control adenovirus (Null) or adenovirus expressing TFEB-wt or TFEB-S211A. Values are means ± SD of three independent experiments. Scale bar: 10 μm. ***p < 0.001; **p < 0.01; *p < 0.05; n.s., not significant.
It was previously described that overexpression of TFEB increases transcription of several autophagy and lysosomal genes.Citation10,Citation13 To address whether the TFEB-S211A mutant is capable to induce transcription, or if additional modifications induced by MTORC1 inactivation are required, we measured expression of several known TFEB targets, including autophagy (ATG9B and UVRAG) and lysosomal (LAMP1 and MCOLN1) genes. As shown in , overexpression of TFEB-S211A significantly increased transcription of autophagy and lysosomal genes despite MTORC1 remaining active (Fig. S12). TFEB-S211A was more efficient than TFEB wild-type inducing transcription of specific genes, thus suggesting that transport of TFEB to the nucleus is critical in the regulation of TFEB activity. Moreover, overexpression of TFEB-S211A caused a significant accumulation of autophagosomes and cytosolic SQSTM1 (). Increased proteins levels of LC3-II, SQSTM1, UVRAG and LAMP1, as well as increased LC3-II/LC3-I and LC3-II/actin ratios were also observed upon TFEB-wt and TFEB-S211A overexpression (). All together, our data show that dissociation of the TFEB/YWHA complex results in transport of TFEB to the nucleus and increased autophagy.
Figure 5. TFEB-S211A induces accumulation of autophagosomes. Immunofluorescence confocal microscopy showing autophagosome accumulation (A) and increased cytosolic SQSTM1 (B) in ARPE-19 cells overexpressing TFEB-S211A. (C) Immunoblotting of ARPE-19 cell lysates infected with either adenovirus Null or adenovirus expressing TFEB-wt or TFEB-S211A. Protein bands were detected using the indicated antibodies. (D–I) Quantification of protein bands as shown in (C). Bars represent the ratio of LAMP1/actin (D), UVRAG/actin (E), SQSTM1/actin (F), LC3II/LC3I (G), LC3II/actin (H), and TFEB-Flag/actin (I) expressed as fold increase of the ratio from cells infected with adenovirus Null. Values are means ± SD of three independent experiments. Scale bar: 10 μm. ***p < 0.001, **p < 0.01, *p < 0.05.
TFEB interacts with the MTORC1 complex
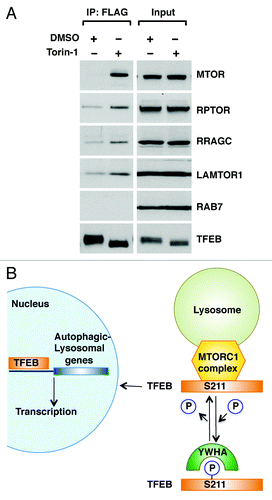
Finally, we addressed the mechanism that regulates association of TFEB with late endosomal/lysosomal membranes. Recent evidence shows that MTORC1 is recruited to late endosomes/lysosomes in response to aminoacids, where it is assembled into an activated MTORC1 complex. This amino acid-dependent recruitment requires Rag GTPases and LAMTOR, a scaffolding complex that comprises the LAMTOR1/2/3 proteins and anchors the Rag GTPases to the lysosome.Citation25,Citation26 Importantly, TFEB was capable to co-immunoprecipitate several components of the endogenous LAMTOR/RRAG/MTORC1 complex, including MTORC1, RPTOR, RRAGC and LAMTOR1. Moreover, the interaction was significantly stronger in cells treated with Torin-1, a MTORC1 inhibitor that promotes efficient shuttling of TFEB from cytosol to late endosomes/lysosomes (; Fig. S13). Therefore, our data suggest that TFEB is recruited to lysosomal membranes through a direct interaction with the MTORC1 complex, further corroborating the important role played by MTORC1 on TFEB activation.
Figure 6. TFEB interacts with the MTORC1 complex. (A) HeLa (CF7) cells treated with either DMSO or Torin-1 (250 nM) for 1 h at 37°C were lysed and TFEB-Flag was immunoprecipitated using anti-FLAG antibody. Immunoprecipitates were then subjected to immunoblotting analysis using the indicated antibodies against components of the MTORC1 complex and RAB7. Note that inputs represent 3% of the total lysates and immunoprecipitated TFEB represents 10% of the total immunoprecipitated material. (B) Model depicting the proposed mechanistic regulation of TFEB by MTORC1.
Discussion
The present study shows for the first time that TFEB is phosphorylated at S211 in an MTORC1-dependent manner, and provides mechanistic information on the retention of this transcription factor in the cytosol and the significance of these events in the regulation of the autophagic pathway. We find that MTORC1 promotes phosphorylation-dependent association of TFEB with YWHA proteins that function as cytoplasmic repressors of TFEB. Our data further reveal that upon MTORC1 inactivation, the TFEB/YWHA complex dissociates, thus allowing transport of TFEB to the nucleus and transcription of multiple genes implicated in the formation of autophagosomes and lysosomes (). Given the therapeutic potential of TFEB in the treatment of storage disorders,Citation14 the elucidation of the mechanisms that regulate TFEB activation is of critical importance.
Recently, two studies have addressed the mechanisms of TFEB activation and produced somewhat contradictory results. The Ballabio’s group found that starvation causes transport of TFEB to the nucleus and increases transcription of autophagy genes both in vitro and in vivo. They proposed that phosphorylation of S142 by MAPK1 causes retention of TFEB in the cytosol, while mutation of S142 to Ala leads to accumulation of TFEB in the nucleus.Citation13 In contrast, the Brugarolas’ group suggested that in cells with high MTORC1 activity (TSC2-null murine embryonic fibroblasts), phosphorylation of a serine-rich region between amino acids 462–469 triggers transport of TFEB to the nucleus. In addition, they reported that mutation of S142 to Ala does not affect the subcellular distribution of TFEB.Citation27 Therefore, the apparent contradictory data showing that TFEB localizes to the nucleus either upon starvation or when MTORC1 activity is high requires further clarification.
Our study provides critical confirmation for some of the results previously published and adds crucial and novel information to clarify and extend our understanding of TFEB function. In agreement with Settembre et al., we found that starvation induces accumulation of TFEB in the nucleus, thus confirming that nutrient levels play a major role in the activation of TFEB. We also confirmed that overexpression of TFEB increases transcription of autophagy and lysosomal genes and leads to the accumulation of autophagosomes. However, we suggest that MTORC1 is the primary regulator of TFEB activity. In this regard, it is important to note that the activity of MTORC1 is also regulated by nutrients and MTORC1 is inactivated under starvation conditions. In agreement with Peña-Llopis et al., we show MTORC1 is a key regulator in the activation of TFEB. However, there is a critical difference between the Peña-Llopis study and ours. While Peña-Llopis et al., suggest that active MTORC1 induces transport of TFEB to the nucleus, we find that active MTORC1 promotes sequestration of TFEB in the cytosol and that inactivation of MTORC1 must occur to trigger transport of TFEB to the nucleus and transcription of autophagy genes. In fact, it is difficult to reconcile why active MTORC1 (an inhibitor of autophagy) would promote TFEB-dependent transcription of autophagy genes. Peña-Llopis et al. suggest that this process may be implicated in reformation of lysosomes from autophagosomes following MTORC1 reactivation.Citation28 However, we observe redistribution of TFEB to the nucleus just 30 min after MTORC1 inactivation, a time at which autolysosomes formation and MTORC1 reactivation has not yet occurred. In addition, accumulation of TFEB in the nucleus is independent of autophagosome formation as evidenced by our results showing that both wortmannin and LY294002 prevent formation of autophagomes through their inhibitory effect on PtdIns 3-kinase type III, but still induce TFEB activation. The study from Peña-Llopis et al. does not address what genes are targeted by TFEB (or whether autophagy is augmented) in TSC2-deficient cells. Therefore, one possibility is that reactivation of MTORC1 after autophagy modifies TFEB in a different way, thus promoting transcription of a specific set of TFEB-responsive genes.
Importantly, our study is the first one to elucidate the mechanism that regulates retention of TFEB in the cytosol and to identify S211 as a novel phosphorylation site in TFEB. Previous mass spectrometry studies have relied on trypsin for polypeptide cleavage.Citation13 However, we found that digestion of TFEB with trypsin results in the generation of large fragments that are not optimal for mass spectrometry analysis. In contrast, digestion with chymotrypsin allowed us to unambiguously uncover MTORC1-dependent phosphorylation of TFEB at S211. Our study also identifies YWHA as a new binding partner of TFEB for the first time, and shows that the interaction between YWHA and TFEB is phosphorylation dependent and mediated by S211.
Finally, here we report the initial description of recruitment of TFEB to late endosomes/lysosomes upon activation through the use of fixed cells and live imaging. The physiological significance of this association is currently unknown but there is growing evidence showing that endocytic compartments function as membrane platforms for specific signaling complexes.Citation29 Recruitment of TFEB to late endosomes/lysosomes, the compartment where active MTORC1 resides, may greatly improve its phosphorylation potential. It is possible that TFEB shuttles continuously between the cytosol and late endosomes/lysosomes as a means to constantly monitor MTORC1 activity. Alternatively, additional modifications or heteroligomerization of TFEB may occur at lysosomes, thus providing further checkpoints for the activation of this transcription factor. Importantly, we observed co-immunoprecipitation of TFEB with several components of the LAMTOR/RRAG/MTORC1 complex. Future studies will help to discern whether TFEB may be part of the lysosome-associate machinery for amino acid sensing.
The present study offers important new insights into the mechanism by which TFEB is activated under conditions that induce lysosomal stress.Citation10 Defective protein degradation in lysosomes results in decreased levels of amino acids in the cell and accompanying inactivation of MTORC1.Citation30 This would also explain the initial observation that TFEB localizes to the nucleus in LSDs cells,Citation10 as the lack of specific lysosomal hydrolases in these disorders leads to the disruption of lysosomal degradation. In addition, it is tempting to speculate that additional stressors that inhibit MTORC1 function, such as mitochondrial dysfunction or ER stress, might also activate TFEB. Finally, it is important to keep in mind that TFEB does not only activate autophagy genes but also genes involved in lysosomal biogenesis, regulation of endo- and exocytosis, and expression of nonlysosomal enzymes implicated in the degradation of essential proteins.Citation12 Therefore, in agreement with the critical role played by MTORC1 in the control of catabolic processes, our data suggest that MTORC1 is a major regulator of the main degradative pathways of the cell.
Overall, this work improves our understanding of MTOR biology and its mechanistic roles in the complex cellular coordination of multiple metabolic and trafficking pathways. It is well established that MTOR regulates the activity of several transcription factors implicated in mitochondrial metabolism and lipid synthesis. Here we unveil a new and novel role for this kinase in the maintenance of cellular homeostasis by regulating expression of genes essential for the execution of the autophagic program.
Materials and Methods
Cell lines and drug treatments
ARPE-19 cells (American Type Culture Collection, CRL-2302) were grown at 37°C in a 1:1 mixture of DMEM and Ham’s F12 media supplemented with 10% fetal bovine serum (Invitrogen, 21041-025), 2 mM Glutamax™ (Gibco, 35050) 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco, 2114) in a humidified 5% CO2 atmosphere. Cells were transiently transfected using FuGENE-6 reagent (Roche Applied Science, 11988387001) following manufacturer’s recommendations. Transfected cells were analyzed 18–24 h post-transfection. For infection experiments, cells were infected with adenoviruses according to the manufacturer’s recommendations. Analyses were performed 16–24 h post-infection. HeLa (CF7) cells stably expressing TFEB-FLAGCitation10 were a kind gift of Dr. Andrea Ballabio (Baylor College of Medicine, Houston, TX and Telethon Institute of Genetics and Medicine, Napoli, Italy). CF7 cells were grown at 37°C in DMEM media (Gibco, 10569) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 0.5 mg/ml geneticin (Invitrogen, 10131-035) in a humidified 5% CO2 atmosphere. Cells were incubated for 2 h at 37°C in medium containing one of the following reagents: DMSO (Fisher Scientific, BP231-1), LY294002 (50 μM), Wortmannin (2 μM), Rapamycin (0.2 μM), U0126 (20 μM) and PD98059 (50 μM) were from Cell Signaling Technology (9901, 9951, 9904, 9903 and 9900, respectively); PP242 (1 μM), MK2206 (1 μM) and XL147 (10 μM) were from Selleck (S2218, S1078 and S1118, respectively); SB203580 (20 μM), MAPK8 inhibitor II (20 μM), Compound C (40 μM) and AMPK activator (20 μM) were from Calbiochem (559398, 420119, 171261 and 171256, respectively); Torin-1 (250 nM) was from TOCRIS (4247). For starvation experiments cells were washed three times in Hank’s balanced salt solution (Invitrogen, 14175) and incubated for 4 h at 37°C in Earle’s balanced salt solution (Sigma-Aldrich, E2888).
Antibodies
The following mouse monoclonal antibodies were used: clone 14 to EEA1, clone Ab5 to actin and clone 3/P62 LCK ligand to SQSTM1 (BD Transduction Laboratories, 610457, 612656 and 610832), clone 4C5 to Flag (Origen, TA50011), clone H4A3 to LAMP1 (Developmental Studies Hybridoma Bank, CD107a) and clones M2 and M5 to Flag (Sigma-Aldrich, F3165, F4042). The following polyclonal antibodies were also used: anti-MTOR, anti-RPTOR, anti-RICTOR, anti-Phospho-p70 S6 kinase, anti-p70 S6 kinase, anti-phospho-4E-BP1, anti-4E-BP1, phospho-MAPK1/3, anti-YWHA, anti-Histone H3, anti-RAB7, anti-UVRAG and anti-RRAGC were from Cell Signaling Technology (2983, 2280, 2114, 9205, 2708, 9451, 9644, 4695, 8312, 4499, 2094, 5320 and 5466 respectively) anti-LC3 and anti-LAMTOR1 (Sigma-Aldrich, L7543 and HPA002997), anti-Porin and anti-LAMP1 (Abcam, ab15895 and ab24170) and anti-Flag (Covance, PRB-132C). Alexa Fluor 568-conjugated goat anti-mouse IgG, Alexa Fluor 488-conjugated goat anti-rabbit IgG, were purchased from Invitrogen (A21090 and A-11008). HRP-conjugated anti-mouse or anti-rabbit IgG were acquired from Cell Signaling Technology (7076 and 7074).
Adenovirus
Adenovirus expressing either TFEB or TFEB-S211A were prepared, amplified and purified by Welgen, Inc.
Plasmids
The plasmid encoding the full-length human TFEB has been previously describedCitation10 and was obtained from Dr. Andrea Ballabio (Baylor College of Medicine, Houston, TX and Telethon Institute of Genetics and Medicine, Napoli, Italy). Amino acid substitutions in TFEB were made using the QuickChange Lightning site-directed mutagenesis kit (Stratagene, 200522/200521) according to the manufacturer's instructions.
Immunofluorescence confocal microscopy
For immunofluorescence, cells grown on coverslips were washed with PBS and fixed with 4% formaldehyde for 15 min at room temperature or with methanol/acetone (1:1, v/v) for 10 min at -20°C. For monitoring nuclear localization of TFEB, cells were permeabilized in PBS containing 0.2% Triton X100 for 10 min at room temperature. Cells were then incubated with the indicated primary antibodies in PBS containing 10% fetal bovine serum and 0.1% (wt/v) saponin for 1h at room temperature, followed by incubation with the corresponding secondary antibodies conjugated to Alexa Fluor-568 or Alexa Fluor-488. After staining, the coverslips were mounted onto glass slides with Fluoromount-G (Southern Biotech, 0100-01). Images were acquired on a Zeiss LSM 510 confocal system with a 63X NA 1.4 oil immersion objective using 488 nm and 543 nm laser excitation (Carl Zeiss).
Subcellular fractionation
Cells were lysed in NP-40 lysis buffer (l0 mM Tris, pH 7.9, 140 mM KCI, 5 mM MgCl2 and 0.5% NP-40) supplemented with protease and phosphatase inhibitors and kept on ice for 15 min. The lysates were then centrifugated at 1000 x g for 3 min. The supernatant represents the cytosolic plus the membrane fraction. The pellets (nuclear fraction) were washed twice in NP-40 lysis buffer and sonicated in Laemmli sample buffer. For lysosomal membrane isolation, cells were grown in 150 mm dished, lysed and processed using a commercial lysosomal enrichment kit (Thermo Scientific, 89839).
RNA interference (RNAi)
For siRNA knockdown, cells were transfected using DharmaFECT transfection reagent with ON-TARGETplus nontargeting pool siRNA duplexes or ON-TARGETplus smart pool siRNA duplexes targeted against RPTOR or RICTOR genes (Dharmacon-Thermo Scientific, D-001810-10-20, L-004107-00-005 and L-016984-00-005, respectively). Treated cells were analyzed 72 h after transfection.
Mass spectrometry
Immunoprecipitated proteins were sequentially reduced with dithiothreitol and alkylated with iodoacetamide. Proteins were then digested with trypsin or chymotrypsin. The resulting peptide mixtures were analyzed with an LTQ Orbitrap Velos (Thermo Fisher Scientific) equipped with a nanoLC system (Eksigent). For phosphorylation site identification, TiO2 columns were used to enrich phosphopeptides prior to mass spectrometric analysis. Peptide IDs and phosphorylation sites were assigned with Mascot 2.3 (Matrix Science) and manually validated using Scaffold 3 software (Proteome Software). For label-free quantitation, peptide peak areas were calculated with Proteome Discoverer 1.3 (Thermo Fisher Scientific).
Co-immunoprecipitation, electrophoresis and immunoblotting
Cells were washed with ice-cold PBS, resuspended in lysis buffer (25 mM Hepes-KOH, pH 7.4, 250 mM NaCl, 1% Triton X-100 (wt/v) supplemented with protease and phosphatase inhibitors cocktail, and lysed by passing the samples 10 times through a 25 gauge needle. Cell lysates were centrifuged at 16,000 x g for 15 min at 4°C, and the soluble fractions were collected. For immunoprecipitation, soluble fractions were incubated with 2 μl of anti-FLAG antibody, and protein G-Sepharose beads (Amersham, 17-0618-01) for 2 h at 4°C. Immunoprecipitates bound to beads were collected, washed four times with lysis buffer, and proteins were eluted with Laemmli sample buffer. Samples were analyzed by SDS-PAGE (4–20% gradient gels, Invitrogen, EC61385BOX) under reducing conditions and transferred to nitrocellulose. Membranes were immunoblotted using the indicated antibodies. Horseradish peroxidase-chemiluminiscence was developed by using Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Sciences, NEL 104001EA).
RNA isolation and relative quantitative real-time polymerase chain reaction
RNA was isolated from cells by using PureLink RNA Mini Kit (Invitrogen, 12183018A) following manufacturer recommendations. RNA yield was quantified using a Nanodrop ND-1000 spectrophotometer (Thermo Scientific). Reverse transcription of RNA (2–4 μg) was made in a 20 μl reaction using oligo(dT)20 and SuperScript III First-Strand Synthesis System (Invitrogen, 18080400). Relative Quantitative Real Time PCR was performed using 5 μl SYBR GreenER qPCR SuperMix (Invitrogen, 11760100), 2 μl cDNA, 1 μl gene specific primer mix (QuantiTect primer Assays) and 2 μl water for a total reaction volume of 10 μl. Quantification of gene expression was performed using 7900HT Fast Real-Time PCR System (Applied Biosystems). The thermal profile of the reaction was: 50°C for 2 min, 95°C for 10 min and 35 cycles of 95°C for 15 sec followed by 60°C for 1 min. All samples were run in triplicate. Amplification of the sequence of interest was normalized with a reference endogenous gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The value was expressed as a ratio to RNA from cells infected with control adenovirus (Ad-Null). The 7900HT Fast Real-Time PCR System Software was used for data analyses (Applied Biosystems).
Statistical analysis
Data were processed in Excel (Microsoft Corporation) then Prism (GraphPad Software) to generate curve and bar charts and perform statistical analyses. One-way ANOVA and pairwise post-tests were performed for each dependent variable. *p < 0.05 was considered statistically significant; **p < 0.01, very significant; and ***p < 0.001, extremely significant. p > 0.05 was considered not significant (n.s.).
Additional material
Download Zip (34 MB)Acknowledgments
We thank B. Lelouvier and C. Mullins for assistance. This project was supported by the Intramural Research Program of the NIH, National Heart, Lung, and Blood Institute (NHLBI).
Author Contribution: J.M. was involved in experimental strategy, performed the experiments and analyzed the data. Y.C. and M.G. designed, help to perform, and interpret the mass spectrometry analysis. R.P. designed the research, performed experiments, analyzed the data, supervised the project, and wrote the manuscript. All the authors reviewed the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12:21 - 35; http://dx.doi.org/10.1038/nrm3025; PMID: 21157483
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002; 110:177 - 89; http://dx.doi.org/10.1016/S0092-8674(02)00833-4; PMID: 12150926
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002; 110:163 - 75; http://dx.doi.org/10.1016/S0092-8674(02)00808-5; PMID: 12150925
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 2004; 14:1296 - 302; http://dx.doi.org/10.1016/j.cub.2004.06.054; PMID: 15268862
- Flinn RJ, Yan Y, Goswami S, Parker PJ, Backer JM. The late endosome is essential for mTORC1 signaling. Mol Biol Cell 2010; 21:833 - 41; http://dx.doi.org/10.1091/mbc.E09-09-0756; PMID: 20053679
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981 - 91; http://dx.doi.org/10.1091/mbc.E08-12-1248; PMID: 19211835
- Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009; 5:973 - 9; http://dx.doi.org/10.4161/auto.5.7.9296; PMID: 19597335
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814 - 22; http://dx.doi.org/10.1038/ncb0910-814; PMID: 20811353
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science 2009; 325:473 - 7; PMID: 19556463
- Sardiello M, Ballabio A. Lysosomal enhancement: a CLEAR answer to cellular degradative needs. Cell Cycle 2009; 8:4021 - 2; http://dx.doi.org/10.4161/cc.8.24.10263; PMID: 19949301
- Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 2011; 20:3852 - 66; http://dx.doi.org/10.1093/hmg/ddr306; PMID: 21752829
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429 - 33; http://dx.doi.org/10.1126/science.1204592; PMID: 21617040
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell 2011; 21:421 - 30; http://dx.doi.org/10.1016/j.devcel.2011.07.016; PMID: 21889421
- Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A 2008; 105:17414 - 9; http://dx.doi.org/10.1073/pnas.0809136105; PMID: 18955708
- Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol 2009; 7:e38; http://dx.doi.org/10.1371/journal.pbio.1000038; PMID: 19209957
- Nyfeler B, Bergman P, Triantafellow E, Wilson CJ, Zhu Y, Radetich B, et al. Relieving autophagy and 4EBP1 from rapamycin resistance. Mol Cell Biol 2011; 31:2867 - 76; http://dx.doi.org/10.1128/MCB.05430-11; PMID: 21576371
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008; 118:3065 - 74; PMID: 18725988
- Muslin AJ, Xing H. 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell Signal 2000; 12:703 - 9; http://dx.doi.org/10.1016/S0898-6568(00)00131-5; PMID: 11152955
- Kleppe R, Martinez A, Døskeland SO, Haavik J. The 14-3-3 proteins in regulation of cellular metabolism. Semin Cell Dev Biol 2011; 22:713 - 9; http://dx.doi.org/10.1016/j.semcdb.2011.08.008; PMID: 21888985
- Bridges D, Moorhead GB. 14-3-3 Proteins: A Number of Functions for a Numbered Protein. Sci STKE 2004; 242:re10; http://dx.doi.org/10.1126/stke.2422004re10; PMID: 15266103
- Bronisz A, Sharma SM, Hu R, Godlewski J, Tzivion G, Mansky KC, et al. Microphthalmia-associated transcription factor interactions with 14-3-3 modulate differentiation of committed myeloid precursors. Mol Biol Cell 2006; 17:3897 - 906; http://dx.doi.org/10.1091/mbc.E06-05-0470; PMID: 16822840
- Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 2003; 31:3635 - 41; http://dx.doi.org/10.1093/nar/gkg584; PMID: 12824383
- Chen RQ, Yang QK, Lu BW, Yi W, Cantin G, Chen YL, et al. CDC25B mediates rapamycin-induced oncogenic responses in cancer cells. Cancer Res 2009; 69:2663 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-08-3222; PMID: 19276368
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010; 141:290 - 303; http://dx.doi.org/10.1016/j.cell.2010.02.024; PMID: 20381137
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008; 320:1496 - 501; http://dx.doi.org/10.1126/science.1157535; PMID: 18497260
- Peña-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, et al. Regulation of TFEB and V-ATPases by mTORC1. EMBO J 2011; 30:3242 - 58; http://dx.doi.org/10.1038/emboj.2011.257; PMID: 21804531
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465:942 - 6; http://dx.doi.org/10.1038/nature09076; PMID: 20526321
- Hupalowska A, Miaczynska M. The New Faces of Endocytosis in Signaling. Traffic 2012; 13 - 9 - 18; PMID: 21752167
- Christie GR, Hajduch E, Hundal HS, Proud CG, Taylor PM. Intracellular sensing of amino acids in Xenopus laevis oocytes stimulates p70 S6 kinase in a target of rapamycin-dependent manner. J Biol Chem 2002; 277:9952 - 7; http://dx.doi.org/10.1074/jbc.M107694200; PMID: 11788584