Abstract
Autophagy is a highly conserved process that degrades cellular long-lived proteins and organelles. Accumulating evidence indicates that autophagy plays a critical role in kidney maintenance, diseases and aging. Ischemic, toxic, immunological, and oxidative insults can cause an induction of autophagy in renal epithelial cells modifying the course of various kidney diseases. This review summarizes recent insights on the role of autophagy in kidney physiology and diseases alluding to possible novel intervention strategies for treating specific kidney disorders by modifying autophagy.
Introduction
Autophagy is a highly conserved, physiological, catabolic process, involving the bulk lysosomal degradation of cytosolic components, including macromolecules (such as proteins and lipids) and cytosolic organelles.Citation1 These are broken down to their basic components, which are then reutilized. Autophagy is believed to be essential for the maintenance of cellular homeostasis, for a number of fundamental biological activities, and as an important component of the complex response of cells to multiple forms of stress.Citation1 Unlike the ubiquitin-proteasome system (UPS), which selectively degrades short-lived proteins,Citation2 autophagy is thought to be a “nonselective” degradation system for long-lived cytoplasmic proteins and dysfunctional organelles.Citation1,Citation3 So far, at least, five steps have been characterized as part of the autophagic mechanism: (1) the initiation of a double-membrane structure, which is called the phagophore, (2) the expansion of the phagophore, (3) the maturation of this structure into the autophagosome, (4) the fusion of autophagosomes with lysosomes, resulting in autolysosomes, and, finally, (5) the degradation of the ingested biological materials.Citation3-Citation5
Autophagy is involved in the pathogenesis of a number of clinically important disorders; however, until recently, little was known about the connection between autophagy and kidney diseases. However, there is now growing evidence that autophagy is specifically linked to the pathogenesis of important renal diseases such as acute kidney injury (AKI), diabetic nephropathy (DN) and polycystic kidney disease (PKD). However, an understanding of the precise role of autophagy in the course of kidney diseases is still in its infancy. The kidney offers an opportunity to study autophagy in the context of highly specialized epithelia, with unique functions in solute transport and barrier function. Dysregulation of autophagy can result in the accumulation of autophagosomes, an event that has been noted in several kidney diseases. Depending upon the disorder involved, the accumulation of autophagosomes may be due to their increased formation or the result of their reduced degradation.
This review, after briefly discussing the important role played by the mechanistic target of rapamycin (MTOR) in the regulation of autophagy, discusses the current state of knowledge regarding the role of autophagy in renal physiology, renal aging and important kidney diseases, including AKI, glomerular diseases (such as DN) and PKD. Available information indicates that the role played by autophagy in the kidney is complex, since both the up- and downregulation of autophagy have been shown to be protective against different kidney diseases. Therefore, further research is required to determine the specific contribution of autophagic dysregulation to individual renal diseases. Also, increased understanding of many issues, with regard to autophagy and the kidney, including the factors that regulate autophagy, the timing of onset of dysregulated autophagy in disease states, and whether these changes in autophagy are primary or compensatory mechanisms, is required in each form of renal disease. This review points out areas of ongoing controversy and areas of particular interest for future research, and also discusses the importance of such information on whether the pharmacological agents that modulate autophagy are potentially feasible as novel forms of treatment for various kidney diseases.
Regulation of Autophagy in the Kidney
Autophagy regulation by MTOR
MTOR pathway
MTOR exists in at least two different functional complexes termed MTOR complex 1, MTORC1, and MTOR complex 2, MTORC2 ().Citation6-Citation8 MTORC1 consists of RPTOR/RAPTOR, a scaffolding protein that recruits MTORC1 substrates, and several additional components such as MLST8/GβL, AKT1S1/PRAS40 and DEPTOR.Citation9-Citation14 MTORC1 is a rapamycin-sensitive complex as the rapamycin-FKBP1A/FKBP12 complex directly associates with and acutely inhibits MTORC1. MTORC1 is activated by multiple extra- and intracellular cues such as amino acids, growth factors, glucose, oxidative stress and cellular redox.Citation15-Citation17 Upon stimulation, MTORC1 directly phosphorylates several substrates including RPS6KB1/S6K, EIF4EBP1, LPIN1/lipin1, GRB10 and Unc-51-like kinase 1 (ULK1),Citation18-Citation24 and stimulates protein translation and lipid biogenesis while inhibiting autophagy induction.Citation25 It is now evident that constitutive activation of MTORC1 suppresses phosphatidylinositol 3-kinase (PtdIns3K) activity, forming a “negative feed-back loop” through GRB10 phosphorylation and the downregulation of insulin receptor substrate (IRS) function.Citation21,Citation24,Citation26-Citation28
Figure 1. MTOR signaling pathway. (A) Two MTOR complexes (MTORC1 and MTORC2) are stimulated by multiple extra- and intracellular factors. MTORC1 activates translation and cell growth, whereas it inhibits autophagy. (B) Amino acids activate RRAG small GTPases thereby recruiting MTORC1 to the lysosomes where MTORC1 is activated by the RHEB small GTPase.
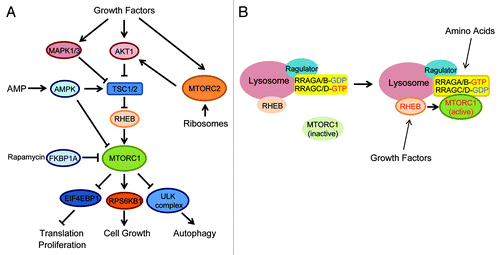
MTORC2 includes RICTOR, MAPKAP1/mSin1, MLST8, DEPTOR and PRR5, and is relatively rapamycin insensitive.Citation6,Citation8,Citation12,Citation29-Citation31 Although MTORC2 is primarily activated by growth factors but not nutrients, a recent study showed an unexpected regulation mechanism, whereby association of MTORC2 with ribosomes triggers its activation to phosphorylate downstream targets such as AKT1, thereby modulating cell survival.Citation32,Citation33
It has been well known that MTORC1 requires both growth factors and amino acids to be fully activated, and lack of one of these inputs largely downregulates its activity. In addition, two small Ras-related GTPases, RRAG/Rag and RHEB, play critical roles in proximal regulation of MTORC1 activation ().Citation34-Citation39 The RRAG small GTPases exist as a heterodimer consisting of RRAGA/B and RRAGC/D and bind to the LAMTOR1/p18-LAMTOR2/p14-LAMTOR3/MP1 complex (Ragulator), which is predominantly expressed at the lysosomal membrane.Citation40,Citation41 Upon amino acid stimulation, the RRAG heterodimer is activated and recruits MTORC1 to the lysosomal membrane where MTORC1 is able to gain access to RHEB.Citation37 A direct interaction with RHEB activates MTORC1 ().Citation13,Citation42,Citation43 Thus, RRAG small GTPases function as essential spatial regulators for mTORC1 localization. On the other hand, RHEB is regulated by the specific GTPase activating protein (GAP) activity of TSC2, which is part of the TSC1-TSC2 complex ().Citation34,Citation35,Citation38,Citation39,Citation44,Citation45 It has been postulated that upon growth factors stimulation, multiple kinases such as AKT1 and MAPK1/3 (ERK) phosphorylate TSC2 and may inhibit its GAP activity, thereby activating MTORC1, although the precise roles of TSC2 phosphorylation in the regulation of TSC2 GAP activity remain elusive ().Citation46-Citation48
MTORC1 inhibits autophagy
Numerous studies in S. cerevisiae have shown that TORC1, the yeast counterpart of MTORC1, negatively regulates autophagy at the initial step called autophagy induction.Citation3,Citation49 Genetic and biochemical studies in yeast indicate that Atg1 is an autophagy-initiating kinase and its activity is downregulated by TORC1. In yeast, the atg1 mutant is defective in autophagy induction even under rapamycin treatment or stress conditions, providing key evidence that Atg1 acts downstream of TORC1 for autophagy induction.Citation50,Citation51 Atg1 forms a large complex with other autophagy-related proteins such as Atg13 and Atg17, and the formation of this complex appears to be important for Atg1 kinase activity. It has been suggested that TORC1 phosphorylates Atg13 and disrupts the Atg1 complex thereby inhibiting autophagy induction.Citation50,Citation52,Citation53 Importantly, the function of TORC1 in the regulation of autophagy is conserved in mammals. MTORC1 also inhibits autophagy induction in many types of mammalian cells.Citation49 Recent studies reveal that MTORC1 interacts with the ULK1-ATG13-RB1CC1/FIP200 complex (a mammalian functional homolog of the Atg1 complex) and phosphorylates ULK1 and ATG13.Citation19,Citation20,Citation22 Although the precise roles of ULK1 and ATG13 phosphorylation by MTORC1 are unclear, the inhibition of the ULK complex is likely to be the mechanism underlying MTORC1-dependent autophagy inhibition.Citation49
MTORC1 is a scoundrel in the glomerular podocyte for disease
Beyond unraveling the biochemical mechanisms by which MTORC1 inhibits autophagy induction, the emerging studies have started revealing the function of MTOR as well as autophagy in human diseases, such as cancer, metabolic disorders and renal diseases.Citation54,Citation55 Diabetic nephropathy is a complication associated with both type 1 and type 2 diabetes and is the most common cause of end-stage renal disease (ESRD) in the USA.Citation56 Recent studies have shown that injuries to podocytes play a critical role in the development of many glomerular diseases including DN.Citation57-Citation59 Interestingly, previous work on mouse models of both type 1 and type 2 diabetes have shown that systemic administration of rapamycin, a specific inhibitor of MTORC1, prevents the development of DN.Citation60-Citation64 However, the molecular mechanism and site of action by which rapamycin prevents the development of DN have not been well understood.
Genetic studies have begun to unveil the roles of the MTORC1-autophagy system in maintaining glomerular function.Citation65-Citation67 MTORC1 activity is enhanced in podocytes of diabetic animals as well as human DN. Specific activation of MTORC1 in podocytes recapitulates many DN phenotypes including glomerular membrane thickening, mesangial expansion, podocyte loss and proteinuria in nondiabetic podocyte-specific Tsc1 knockout mice (Tsc1 PcKO mice).Citation67 In contrast, a reduction of MTORC1 expression in podocytes (podocyte-specific Rptor heterozygous mice) prevents the development of DN in both type 1 and type 2 diabetic animal models, providing the evidence that hyperactivation of MTORC1 is a critical event for the onset or development of DN.Citation65,Citation67 As also seen in the podocytes of diabetic animals and human DN, Tsc1 PcKO podocytes display overgrowth, foot processes effacement, EMT-like phenotypic change and mislocalization of membrane proteins such as NPHS1/nephrin and NPHS2/podocin that are essential slit diaphragm (SD) proteins forming the filtration barrier. Importantly, lack of autophagy in podocytes (podocyte-specific Atg5 knockout mice: Atg5 PcKO mice) also causes podocyte loss and glomerulosclerosis in older mice, suggesting that defective autophagy may be the mechanism underlying the injury of podocytes induced by MTORC1 activation.Citation66 However, the phenotypes associated with podocyte injury seen in Atg5 PcKO mice are much weaker than those in Tsc1 PcKO mice. For instance, lack of autophagy does not cause mislocalization of SD proteins or severe proteinuria. These observations suggest that besides a blockade of autophagy, MTORC1 activation induces additional deteriorative processes in podocyte injury.
TASCC formation: A plant for quality control of protein synthesis
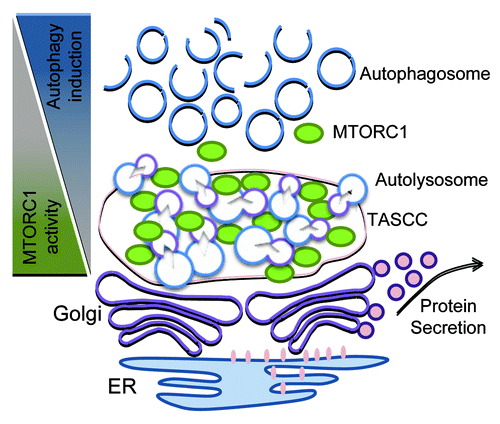
Higher autophagic activity compared with that in other glomerular cells is observed not only in maturing podocytes but also in developed and differentiated podocytes.Citation68-Citation70 Furthermore, podocytes have higher MTORC1 activity compared with other glomerular cells, suggestive of the existence of a special mechanism involving the collaboration of MTORC1 and autophagy in podocytes.Citation65,Citation67 Recently, the formation of a distinct cytoplasmic compartment termed TOR-autophagy spatial coupling compartment (TASCC)Citation71,Citation72 has been identified. TASCC was originally found in human diploid fibroblasts undergoing cellular senescence induced by oncogenic protein HRASV12A.Citation71 During the transition to senescence, cells display dramatic changes of transcriptional profile, replication program and cell shape. Furthermore, cells start producing a large amount of secretory proteins (SASP), which are essential for establishing the senescence phenotype. The TASCC is detectable as a distinct cellular compartment at the trans side of the Golgi apparatus, and is enriched for MTOR and autolysosomes but largely excludes autophagosomes. Indeed, secretory proteins are actively synthesized, especially in the rER-Golgi system close to the TASCC. Since a large amount of cellular MTORC1 is sequestered by enriched autolysosomes within the TASCC in a manner dependent on RRAG GTPase activity, the environment out of TASCC is preferable for autophagy induction. Thus, TASCC formation creates a spatio-temporal arrangement of MTORC1 and autophagy that enables activation of both pathways within the cell. It was anticipated that TASCC formation could be observed in other systems when cells need to produce specific secretory proteins and change their phenotypes. Interestingly, it was found that a similar compartment (TASCC-like structure) characterized by enriched expression of MTOR, lysosome-associated membrane protein type 2A (LAMP2A), and LC3 is present close to the Golgi apparatus in glomerular podocytes ().Citation71 Besides constituting the essential component in filtration barriers, podocytes play a crucial role in the constant turnover of glomerular basement membranes and endothelium by supplying matrix proteins and secreting growth factors, such as vascular endothelial growth factor (VEGF).Citation73 It has also been shown that the Golgi apparatus is enlarged in podocytes during their differentiation.Citation74 Moreover, podocytes display high autophagy and MTORC1 activity.Citation66,Citation67 These observations are consistent with the original findings that the formation of TASCC provides an environment where MTORC1 and autophagy are mutually regulated, thereby working as an important compartmentalized plant for secretory protein synthesis. Accordingly, a strong VEGF expression is detected at the TASCC-like structure in matured podocytes.Citation71 Although it is still unclear as to when the TASCC-like structure is formed during podocyte maturation, and whether this structure plays a critical role in the differentiation of podocytes, the formation may facilitate synthesis of secretory proteins for stimulating or maintaining their specific phenotype as well as glomerular homeostasis.
Figure 2. Spatial regulation of MTORC1 and autophagy in podocytes. The proposed functions of the TASCC-like structure in podocytes is depicted. The formation of the TASCC-like structure in podocytes may provide a unique environment producing specific secretory proteins. In the TASCC-like structure, the enriched autolysosomes and lysosomes generate cellular amino acids that may further recruit MTORC1 into the TASCC-like structure via RRAG GTPases. This process creates mutually reciprocal gradients for MTORC1 and autophagic activity within a podocyte, and may allow the cells to convert unnecessary proteins into the special ones that are crucial for keeping or transforming their phenotypes.
Conclusions and perspectives
A series of biochemical, biological, genetic and clinical studies have demonstrated the double-edged role of MTORC1 in glomerular function.Citation60,Citation65,Citation67 From a clinical point of view, this suggests that rapamycin can be both an excellent medicine and poison. It is conceivable that the benefit from rapamycin would be both context- and timing-dependent. In podocytes, both genetic activation and inhibition of MTORC1 deteriorate those functions leading to glomerulosclerosis with proteinuria. However, the pathomechanism of podocyte dysfunction caused by MTORC1 activation and inhibition is clearly different. For instance, podocytes with hyperactive MTORC1 display hypertrophy, and their foot processes are entirely effaced with the redistribution of SD proteins.Citation67 Podocytes without MTORC1 activity show little change in their cell volume and partially effaced secondary foot processes without redistribution of SD proteins.Citation65 It is likely that hyperactivation of MTORC1 causes postmitotic dedifferentiation whereas loss of MTORC1 hampers normal differentiation of podocytes. Although physiological as well as pathological roles of MTORC1 in other glomerular and renal cells are areas that need to be explored, further understanding of MTORC1 regulation during normal podocyte maturation/differentiation and the course of disease development would provide a better approach for MTORC1 inhibitors in kidney diseases.
Chaperone-mediated autophagy in the kidney
Chaperone-mediated autophagy (CMA) is one of the three lysosomal degradation pathways described in mammals to date, others being macroautophagy (often referred to as just autophagy) and microautophagy.Citation75 In contrast to the “in-bulk” degradation of cytosolic components by macroautophagy, CMA is a more selective pathway that has a certain subset of cytosolic protein substrates.Citation76,Citation77 There is a functional crosstalk between macroautophagy and CMA.Citation78 Upon inhibition of one of the pathways the other one will be constitutively upregulated, so that a cell can partially maintain long-lived protein degradation at least in basal conditions.Citation78,Citation79 Similar to macroautophagy, CMA is stimulated by starvation or serum removal in culture but it maintains its activity for much longer than macroautophagy (up to 72 h), providing some source of energy and amino acids, as well as quality control against misfolded proteins.Citation80 In addition CMA is also increased in response to oxidative stress.Citation81 Together with other proteolytic systems, CMA activity decreases with aging,Citation82 contributing to the impaired functioning and homeostasis of aging cells and aging-related pathologies.
The selectivity of CMA is achieved via the first known step of the process—the recognition of a specific KFERQ-like motif on a substrate by a molecular chaperone, HSPA8/HSC70.Citation83 Together with other cochaperones, which include HSP90, HSPA8 delivers the substrate to the LAMP2A complex, which, upon stimulation by other lysosomal membrane resident proteins and signaling molecules, forms a translocation channel for CMA substrates.Citation84,Citation85 Therefore, similar to the ubiquitin-proteasome system, CMA substrates need to be unfolded to be transported into the lysosomal lumen for degradation.Citation86 Up to 30% of cytosolic proteins can be putative CMA substrates and the list grows longer if we include the proteins that acquire the motif after a post-translational modification,Citation87 such as phosphorylation and acetylation. Some of the known CMA substrates are GAPDH, RNASE A,Citation88 NFKBIA/IκB, MEF2D,Citation89 SNCA/α-synuclein,Citation90 and PKM2.Citation91
Whereas the regulation of the CMA pathway has been the subject of recent investigations, not much is known about the upstream regulatory signaling pathways. It has been established that the rate-limiting step in the process is the binding of substrate to the LAMP2A receptor. Therefore, CMA activity is dependent on the amount of functional LAMP2A receptor on the lysosomal membrane.Citation92 LAMP2A levels are regulated in several ways depending on the cellular stressor that stimulates CMA: de novo synthesis of the protein, reinsertion of LAMP2A from the lumenal “reserve” into the membrane, or decreased degradation by lysosomal cathepsins.Citation92
CMA deregulation has been implemented in different pathologies, such as lysosomal storage disorders and especially neurodegenerative disorders, since nondividing neurons are particularly dependent on homeostatic mechanisms to guarantee their survival during stress.Citation76 In particular, the role of CMA has been revealed when known pathologically-related proteins such as SNCA, APP, MAPT/TAU and PARK2/PARKIN turned out to have the KFERQ motif and undergo degradation through this pathway, which often is aberrant in Alzheimer or Parkinson diseases.
There has not been a systematic analysis examining the role of CMA in the kidney. Using cultured rat renal epithelial cells, Sooparb et al.Citation93 showed that EGF in cultured renal cells regulates the breakdown of proteins targeted for destruction by CMA. If CMA is suppressed they find an increase in PAX2 expression. PAX2 is an important factor in kidney development, indicating the possibility that CMA and PAX2 could play roles in epithelial cell growth.
Sooparb et al. found that proteolysis is decreased early in diabetic rats followed by an increase and subsequent return to baseline. The abundance of proteins in diabetic kidneys containing the CMA KFERQ signal motif increases by 38% and individual KFERQ-containing proteins (e.g., PKM2, GAPDH and PAX2) are more abundant in diabetic rats. LAMP2A and HSPA8 decrease in cortical lysosomes from diabetic vs. control rats, indicating the possibility that CMA might be increased in diabetic kidneys. There are several potential avenues of crosstalk between the insulin and macroautophagy signaling pathways. It is not known if insulin signaling or hyperglycemia have a direct effect on CMA, but it could definitely have a secondary effect via interfering with downstream signaling. For example inhibition of macroautophagy has been reported in rat diabetic tubule samples,Citation94 and insulin injections reverse macroautophagy inhibition in distal tubular cells.
Conclusions and perspectives
In the future it is possible that the role of CMA in degrading other proteins and regulating kidney function will be discovered. For example, it has been already shown that CMA is responsible for the direct uptake of A2M/α-2-microglobulin—a protein that accumulates in lysosomes of kidneys during chemical-induced nephropathy.Citation95 Manipulation of post-translational modifications of certain proteins that are therapeutic targets can make them resistant or susceptible to CMA degradation, depending on the desired level of the protein in the context of disease. Since CMA in general is crucial for homeostasis maintenance, it will be important to look at its activity levels and deregulation in different kinds of kidney pathologies, such as nephrotic syndrome, since NPHS2 carries the CMA degradation sequence.
Role of Autophagy in the Tubulo-Interstitial Compartment
Autophagy in acute kidney injury
Acute kidney injury
Acute kidney injury is characterized by a rapid loss of kidney function that leads to the decline of glomerular filtration, accumulation of nitrogenous wastes such as urea nitrogen, and perturbation of extracellular fluid volume as well as electrolyte and acid-base balance. AKI is one of the most common and serious clinical syndromes in hospitalized patients, in particular for the patients in the intensive care unit. Despite advances in the understanding of the pathophysiology of AKI and in the technology of clinical care for AKI patients, the outcomes of this disease, as well as preventive and therapeutic strategies for it, have not improved over the past several decades. In-hospital mortality rates of patients with AKI remain unacceptably high, at 40% to 80% in the intensive care setting.Citation96-Citation99 Approximately 13% of patients with AKI develop chronic kidney disease over a 3-y period.Citation100 In addition, the incidence of AKI has increased in recent years,Citation101,Citation102 which further strains an already burdened health care system with patients in need of renal replacement therapies.
Multiple causative factors contribute to the etiology of AKI. From the clinical viewpoint, the largest number of cases of AKI are caused by renal ischemia-reperfusion, sepsis and nephrotoxins.Citation96-Citation99,Citation103 The pathogenesis of AKI is very complex and multifactorial, with emerging tubular, microvascular and inflammatory mechanisms that interplay with and amplify each other.Citation96,Citation97,Citation99,Citation103,Citation104 Renal tubular cell injury and death are the key pathological features in AKI, which results in the generation of inflammatory and vasoactive mediators. These chemical mediators induce vasoconstriction and tissue inflammation, further exacerbating tubular damage and renal dysfunction. In experimental models of AKI, the proximal tubules, especially the S3 segment located within the outer medulla of the kidney, suffer the most severe injury.Citation105,Citation106
Autophagy induction in proximal tubular cells during AKI
Autophagy was originally defined as a “self-eating” process of the cell under starvation. However, recently numerous studies have established autophagy as a cellular response to stress. In the last few years, a growing body of evidence has demonstrated the induction of autophagy in proximal tubular cells and kidneys during AKI induced by renal ischemia-reperfusion and nephrotoxins such as cisplatin, cyclosporine and environmental toxins.
Lai and colleagues showed an increased expression of autophagy-related (ATG) proteins (BECN1 and LC3) in renal tubules during ischemia-reperfusion in rats, suggesting autophagy activation in this renal pathology.Citation107,Citation108 Suzuki et al. further showed increased numbers of LC3- and LAMP2-positive vacuoles in a human kidney proximal tubular cell line (HK-2) following hypoxia incubation, and in mouse kidneys during ischemia-reperfusion. Under these conditions, LC3-positive vacuoles colocalize with LAMP2-positive vacuoles, suggesting the fusion of autophagosomes with lysosomes for degradation.Citation109 Notably, the formation of autophagic vacuoles was also shown in tubular cells in transplanted human kidneys by electron microscopy (EM).Citation109 A recent study systematically analyzed autophagy using in vitro and in vivo models of renal ischemia-reperfusion.Citation110 It was shown that autophagy is induced by hypoxia in a rat proximal tubular cell line (RPTC), as indicated by the formation of fluorescent puncta in GFP-LC3-transfected cells and the accumulation of LC3-II, a lipidated form of LC3 that localizes on phagophores and autophagosomes. Importantly, induction of autophagy was shown to be an early response to hypoxic stress, prior to tubular cell apoptosis. In anoxia-reoxygenation, an in vitro model of ischemia-reperfusion, anoxia alone induces LC3-II accumulation but not formation of GFP-LC3 puncta. Nonetheless, autophagy is induced undoubtedly during reoxygenation as indicated by both GFP-LC3 punctate cells and LC3-II accumulation.Citation110 These observations are consistent with an autophagic response during in vitro ischemia-reperfusion of a cardiac cell line.Citation111 In C57BL/6 mice, autophagy is induced in kidneys by renal ischemia-reperfusion. Although not obvious during the ischemic period, LC3-II accumulates in a time-dependent fashion in renal tissues during reperfusion (). The appearance of autophagic vacuoles in proximal tubular cells is further revealed by EM ().Citation110 Consistently, later studies using GFP-LC3 transgenic mice showed an increased formation of GFP-LC3 dots in proximal tubules during renal ischemia-reperfusion.Citation113,Citation114 It is noteworthy that autophagic flux was also determined by comparisons of LC3-II level and autophagosome number in the presence and absence of lysosomal inhibitors. Lysosomal inhibitors increase LC3-II accumulation in RPTC cells during hypoxic or anoxic incubation, and in kidney tissues during renal ischemia-reperfusion.Citation110 Similarly, Suzuki et al. showed that the number of LC3-positive vacuoles is significantly increased by lysosomal inhibitors during hypoxic treatment of HK-2 cells.Citation109 Together, these studies indicate that autophagy is indeed induced in tubular cells during renal ischemia-reperfusion.
Figure 3. Autophagy in acute kidney injury. (A) Immunoblot analysis of LC3-II accumulation in kidney tissues after 30 min of bilateral renal ischemia, followed by 0 to 48 h of reperfusion. The figure is adopted from Jiang et al.,Citation110 with permission from Elsevier. (B) Formation of autophagosomes and autolysosomes in tubular cells of kidney tissues during ischemia-reperfusion. Kidney tissues were fixed for EM examination of autophagosomes (left panel, arrows: double or multiple membrane structures containing cytoplasm; middle panel, arrows: undigested organelles such as mitochondria) and autolysosomes (right panel, arrowheads: single membrane structures with remnants of cytoplasmic components). The figure is adopted from Jiang et al.,Citation110 with permission from Elsevier. (C) Cisplatin-induced autophagy in proximal tubular cells in kidney tissues. GFP-LC3 transgenic mice were treated with 20 mg/kg cisplatin for 24 h to harvest kidney tissues for immunostaining of aquaporin-1 (AQP1; marker of proximal tubules). Co-localization of GFP-LC3 dots with AQP1 suggests autophagy induction in proximal tubular cells. The figure is adopted from Inoue et al.,Citation112 with permission from Springer.
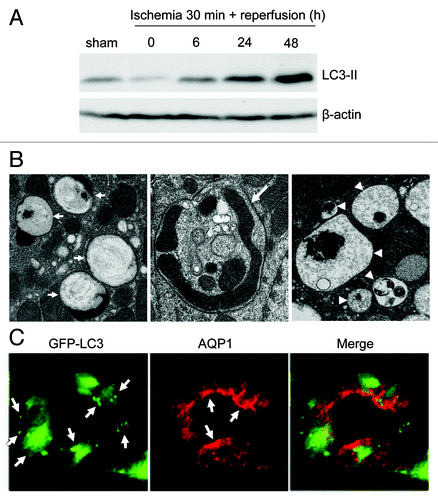
In nephrotoxic models of AKI, cisplatin was shown to induce autophagy in different renal proximal tubular cell lines including RPTC (rat),Citation115 LLC-PK1 (porcine),Citation116 and NRK-52E (rat).Citation112 In all three cell lines, cisplatin induces autophagy in a treatment time-dependent manner, as shown by autophagosome formation and LC3-II accumulation. In addition, EM of cisplatin-treated RPTC cells identifies various characteristic structures that may represent the maturation of autophagic vesicle from phagophore through the autolysosome.Citation115 Notably, autophagy is activated by cisplatin within hours, earlier than apoptosis.Citation112,Citation115,Citation116 Using GFP-LC3 transgenic mice, Inoue et al. further monitored autophagy in kidney tissues during cisplatin nephrotoxicity, unveiling autophagy mainly in proximal tubules ().Citation112 These observations are consistent with a recent examination by EM, which demonstrated a time-dependent increase of autophagic vacuoles in proximal tubular cells following cisplatin treatment of C57BL/6 mice.Citation115 Autophagy is also induced in proximal tubular cells during cyclosporine nephrotoxicity.Citation117 In addition, some environmental toxins, such as cadmium and arsenic, were also shown to induce autophagy in proximal tubular cells and kidneys.Citation118,Citation119
Signaling regulation of tubular cell autophagy during AKI
The regulation of autophagy has been extensively studied in the past few years, and tremendous progress has recently been made in understanding the molecular mechanism and signaling pathways of autophagy from yeast to mammals.Citation120-Citation123 The ATG proteins constitute the core molecular machinery of autophagy and function at several successive steps of the autophagy cascade to orchestrate the process. Upstream of the core machinery, autophagy is regulated by a complex signaling network of multiple stimulatory and inhibitory inputs. Diverse signaling pathways regulate autophagy in response to various stresses. Moreover, these signaling pathways may interact with each other to determine the specificity and magnitude of autophagy. Despite these significant advances, signaling pathways that lead to tubular cell autophagy in AKI remain unclear. Several recent studies have just started the journey of exploration.
Oxidative stress
Bolisetty et al. examined the regulation of autophagy by HMOX1/heme oxygenase 1 during cisplatin-induced AKI.Citation124 They found that cisplatin induces HMOX1, oxidative stress and autophagy in cultured renal tubular cells and mouse kidneys. In HMOX1-null models, oxidative stress is elevated, resulting in an increased sensitivity to autophagy and tubular cell death. Restoring HMOX1 in tubular cells reverses the autophagic response. Furthermore, overexpression of HMOX1 reduces oxidative stress, delays autophagy induction and protects against cisplatin-induced cell death. These results suggest that oxidative stress may lead to autophagy in renal tubular cells.
ER stress
In cyclosporine-treated human renal tubular cells, endoplasmic reticulum (ER) stress is induced along with autophagy activation. Salubrinal, an ER stress inhibitor, suppresses cyclosporine-induced autophagy, suggesting the involvement of ER stress in tubular cell autophagy during cyclosporine nephrotoxicity.Citation117 In mammals, ER stress through the unfolded protein response (UPR) and intracellular calcium has been implicated in autophagy regulation.Citation125 The signaling pathways by which ER stress induces tubular cell autophagy were then studied in the models of tunicamycin-induced kidney injury. Gozuacik et al. showed that death-associated protein kinase (DAPK1) is activated by ER stress through protein phosphatase 2A dephosphorylation. Importantly, ER stress-induced autophagy is inhibited in Dapk1-null cells, suggesting that DAPK1 may be a positive mediator of autophagy.Citation126 Using inhibitors of the mitogen-activated protein kinases (MAPKs), Kawakami et al. demonstrated that activation of MAPK1/3, but not MAPK8 (c-Jun N-terminal kinase, JNK) or MAPK14/p38, is necessary for the induction of tubular cell autophagy by ER stress.Citation127 Further investigations should test how DAPK1 and MAPK1/3 regulate autophagy in tubular cells. Given that ER stress is activated by a variety of insults that cause AKI,Citation128 it would also be important to examine if ER stress is a common modulator of tubular cell autophagy.
TP53
In cisplatin-treated renal tubular cells, autophagy was partially suppressed by chemical inhibition of TP53/p53, suggesting that TP53 may positively regulate autophagy in this experimental condition.Citation115 The pro-autophagy role of TP53 has been demonstrated in several studies. It has been suggested that, upon cellular stress, TP53 is activated and accumulates in the nucleus, where it transactivates 5′-AMP activated protein kinase (AMPK) to inhibit MTOR, resulting in autophagy.Citation129 Alternatively, TP53 can induce autophagy by transcriptional activation of damage-regulated autophagy modulator (DRAM1), a lysosomal membrane protein that stimulates autophagosome-lysosome fusion.Citation130 While promoting autophagy in these studies, TP53 may induce TP53-induced glycolysis and apoptosis regulator (C12orf5/TIGAR) to suppress oxidative stress and inhibit autophagy via an MTOR-independent pathway.Citation131 Tasdemir et al. suggested that the regulation of autophagy by TP53 may depend on its subcellular localization.Citation132 Whether these mechanisms are responsible for autophagy regulation by TP53 in tubular cells during AKI remains to be determined.
BCL2 family proteins
Inhibitory effects of BCL2 or BCL2L1/Bcl-xL on tubular cell autophagy during AKI have been suggested by several studies. In rats, intrarenal arterial delivery of the adenoviral Bcl2l1 gene reduces autophagy in tubular cells following ischemia-reperfusion.Citation107 Suppression of autophagy by BCL2 was also shown in BCL2 GFP-LC3 double transgenic mice under this condition.Citation133 Moreover, cisplatin-induced autophagy is almost completely blocked in a BCL2 stable expression tubular cell line.Citation115 A well-recognized mechanism by which BCL2-BCL2L1 downregulates autophagy is that BCL2-BCL2L1 binds BECN1 through a BCL2 homology 3 (BH3) domain to prevent BECN1 from assembling the class III PtdIns3K complex that is indispensable for autophagosome formation.Citation134,Citation135 Notably, only ER-targeted BCL2 inhibits autophagy.Citation134 However, in BCL2-overexpressing renal tubular cells BCL2-BECN1 interaction is not detected by co-immunoprecipitation assay.Citation115 Recently, Chang et al. showed that nutrient-deprivation autophagy factor 1 (NAF1) binds BCL2 at the ER and is required for the interaction of BCL2 with BECN1, and BCL2 inhibition of autophagy.Citation136 In addition to the association with BECN1, ER-localized BCL2 may also suppress autophagy by diminishing stress-induced Ca2+ release and consequent activation of the Ca2+/calmodulin-dependent protein kinase-AMPK signaling pathway.Citation137 These interesting possibilities need to be investigated in experimental models of AKI.
ULK1 and hypoxic response
ULK1 is a mammalian homolog of yeast Atg1, which functions as an initiating kinase in the autophagy core machinery.Citation138 Preliminary work showed that ULK1 is partially dephosphorylated during severe hypoxia or anoxia in renal tubular cells through dissociation from the MTOR inhibitory complex (Jiang M, Dong Z, unpublished data). Under nutrient starvation, the physical interactions and/or mechanistic interplays of ULK1 with AMPK and MTOR have been suggested by several groups.Citation139-Citation144 A later study has further shown that ULK1 may be transcriptionally upregulated by TP53 for sustained autophagy and subsequent cell death induced by DNA damage.Citation145 Furthermore, ULK1 phosphorylates AMBRA1, a BECN1 interacting protein that is necessary for activity of the class III PtdIns3K complex, releasing the complex from dynein and thereby inducing autophagy.Citation146 These findings will certainly inspire research into the regulation mechanisms of ULK1 and its role in tubular cell autophagy during AKI.
Pathological role of tubular cell autophagy in AKI: Pro-survival or pro-death?
Clearly, the observations that autophagy occurs prior to apoptosis in renal tubular cells during AKI suggest that autophagy is an early response of the cells to stress and not a result of apoptosis. However, what role autophagy plays under this condition is still controversial.
In cisplatin-treated RPTC cells, inhibition of autophagy by pharmacological inhibitors (3-methyladenine or bafilomycin A1) or genetic knockdown of Becn1 or Atg5 with short hairpin RNAs increases apoptosis, suggesting a protective role for autophagy in cisplatin-induced tubular cell injury.Citation115 Similarly, Yang et al. demonstrated that cisplatin-induced autophagy in LLC-PK1 cells acts as a prosurvival mechanism against cell apoptosis.Citation116 Later in vivo work further confirmed and extended the in vitro findings. In a mouse model of cisplatin nephrotoxicity, pharmacological blockade of autophagic flux by chloroquine significantly enhanced cisplatin-induced kidney injury (Jiang M, Dong Z, unpublished data). A cytoprotective role of autophagy was also shown in primary culture of human proximal tubular cells during cyclosporine nephrotoxicity.Citation117 Using both in vitro and in vivo experimental models, a recent study examined the role of autophagy in renal ischemia-reperfusion. In vitro, pharmacological or genetic suppression of autophagy sensitizes tubular cells to apoptosis induced by hypoxia incubation or anoxia-reoxygenation. Inhibition of autophagy in vivo by chloroquine or 3-methyladenine worsens ischemia-reperfusion renal injury, as indicated by renal function, histology, and tubular apoptosis. Together, these results suggested that autophagy is a renoprotective mechanism for cell survival in acute ischemic kidney injury.Citation110
In contrast, several studies have also demonstrated that tubular cell autophagy may contribute to cell death during AKI. In a rat model of renal ischemia-reperfusion, Lai and colleagues showed increased BECN1 and LC3 expression as well as apoptosis in injured renal tubules. Both autophagy and apoptosis are suppressed by BCL2L1 overexpression or ischemic preconditioning, accompanied by the amelioration of kidney injury.Citation107,Citation108 Similarly, Suzuki et al. found that autophagy occurs in renal tubules with disrupted morphology in GFP-LC3 transgenic mice after renal ischemia-reperfusion. While autophagy is reduced in BCL2-GFP-LC3 double transgenic mice, tubular damage is also attenuated. Along with the in vitro observation that autophagy inhibitors protect HK2 cells from H2O2-induced cell death, they concluded that autophagy might be detrimental during renal ischemia-reperfusion.Citation109 In tunicamycin-treated mice, Gozuacik et al. showed that ER stress induces apoptosis and autophagy concomitantly in the same damaged tubular cells. Interestingly, inhibition of autophagy by itself does not change cell death or survival; however, in combination with caspase blockade, autophagy inhibition increases cell viability. Furthermore, Dapk1 knockout mice are resistant to ER stress-induced kidney injury as tubular cell autophagy is suppressed in those mice. Based on these results, it was suggested that autophagy may serve as a second cell killing mechanism that acts in concert with apoptosis to trigger kidney injury during ER stress.Citation126 A cell death-promoting role of autophagy was also suggested by Inoue et al. showing that pharmacological or genetic inhibition of autophagy suppresses cisplatin-induced caspase activation and apoptosis in NRK-52E cells.Citation112
The cause of the obvious discrepancy among the aforementioned studies is unclear, although it is generally believed that, depending on experimental conditions, autophagy can be either protective or detrimental. Considering that pharmacological inhibitors may have nonspecific effects on cellular process other than autophagy, and genetic knockdown of ATG proteins is sometimes unable to block autophagy completely, in vivo tests using tissue-specific Atg gene knockout animals should be a preferable strategy to determine the role of autophagy in renal pathology. To this end, Kimura et al. and Liu et al. established tubule-specific Atg5 knockout mouse models, which show the accumulation of deformed mitochondria, aberrant concentric membranous structures, and cytoplasmic inclusions including SQSTM1- and ubiquitin-positive protein aggregates in renal tubules, leading to cellular degeneration. During renal ischemia-reperfusion, tubular cell autophagy is inhibited in the Atg5 conditional knockout mice and, importantly, more severe kidney injury is induced by renal ischemia-reperfusion in these mice compared with wild-type animals.Citation113,Citation114 Lately, a mouse model of Atg7 knockout in proximal tubules (Atg7 PTKO) was established (Jiang M, Dong Z, unpublished data). Knockout of Atg7 leads to impairment of the autophagy-conjugation systems, resulting in inhibition of autophagy and accumulation of autophagy-selective substrates such as SQSTM1 during cisplatin treatment. Compared with their wild-type littermates, Atg7 PTKO mice show accelerated loss of renal function, aggravated kidney tissue damage and tubular apoptosis. Primary proximal tubular cells isolated from Atg7 PTKO mice are more sensitive to cisplatin-induced caspase activation and apoptosis than the cells from wild-type mice. Atg7 PTKO mice are also more sensitive to renal ischemia-reperfusion injury than their wild-type littermates (Jiang M, Dong Z, unpublished data). Further, Takahashi et al. most recently demonstrated that Atg5 knockout in proximal tubule cells results in a more severe cisplatin-induced injury in vivo, accompanied by accelerated DNA damage and TP53 activation.Citation147 Together, these studies have demonstrated definitive evidence for a renoprotective role of tubular cell autophagy during AKI.
It is not yet understood how autophagy protects tubular cells from injury or apoptosis. Upon metabolic stress in which the availability of oxygen and nutrient is poor, this catabolic pathway can generate amino acids and lipids that can be reused for protein synthesis and ATP production, which are essential for the adaptation to bioenergetic catastrophe.Citation148 As a cellular housekeeping process, autophagy can clear misfolded proteins and damaged organelles to maintain cellular homeostasis and thereby set a higher threshold against apoptosis induction.Citation148,Citation149 Indeed, when autophagy is impaired in proximal tubular cells by Atg5 knockout, damaged mitochondria and abnormal protein aggregates accumulate in the cells. As a result, these autophagy-deficient mice are more sensitive to kidney injury than wild-type mice that have intact autophagy.Citation113,Citation114,Citation147 Similar findings are also shown in a study when autophagy is inhibited by chloroquine or ATG7 deficiency (Jiang M, Dong Z, unpublished data). In addition, it is plausible that signaling activated during autophagy can interfere with or compromise cell death pathways. This possibility has been implicated in the studies showing that, on the one hand, liberation of BCL2 and CFLAR/FLIP from activated autophagy protein complexes may block the intrinsic and extrinsic pathways of apoptosis;Citation134,Citation150 on the other hand, autophagic degradation of active CASP8/caspase 8 is responsible for the inhibition of apoptotic cell death.Citation151 Moreover, certain stress stimuli can activate other cytoprotective responses to cooperate with autophagy to achieve optimal cellular repair and adaptation.Citation123 Finally, data obtained from studying programmed cell death in embryo development indicate that autophagy is required for the maintenance of high ATP levels that may in turn facilitate the elimination of apoptotic cells. This function of autophagy could prevent a detrimental inflammatory response both during normal development and after exposure to pathological stimuli.Citation149 Along these lines, recent studies have demonstrated that autophagy negatively regulates inflammatory response, protecting cells from ischemic brain injury and endotoxin-induced intestinal epithelium injury.Citation152,Citation153 Further research is needed to gain insights into underlying mechanisms for the renoprotection of tubular cell autophagy in AKI.
It is generally acknowledged that autophagy is induced to serve primarily as an adaptive and defense mechanism for cell survival, because Atg gene knockdown or knockout accelerates rather than delays cell death; however, in certain settings,Citation154 uncontrolled massive autophagy may lead to cell death. How this prosurvival attempt fails and then switches to an alternative cell death pathway is unclear. It possibly is a result of an irreversible collapse of cell viability due to nonspecific destruction of large proportions of cytoplasmic contents or a result of selective degradation of cytoprotective elements.Citation149 Some molecules, for example, Draper, MAPK8 and DAPK1, have been shown to direct autophagy from a survival to a death pathway, although the exact mechanisms are unknown.Citation155 Notably, many examples of Atg gene-dependent cell death occur in cells whose apoptotic machinery is compromised; however, a caveat should be considered since knockout or overexpression of a single Atg gene could have unknown indirect effects beyond autophagy.Citation156 In addition to triggering cell death on its own, autophagy may also join apoptosis to coordinately determine a cell’s fate. The functional relationship between autophagy and apoptosis is complex and generally presents as three scenarios.Citation149,Citation154,Citation157 First, the two pathways share common regulatory signals and each can regulate and modify the activity of the other. Second, autophagy acts either upstream of apoptosis to enable apoptotic signaling, or during the final stage of apoptosis to participate in certain morphological changes by providing ATP. Third, autophagy and apoptosis develop in a mutually exclusive manner under certain conditions, probably as a result of distinct thresholds for each process or mutual inhibition between the two processes. The intricate interplay and the cross-regulation between autophagy and apoptosis pathways further complicate the conundrum of how autophagy contributes to the life and death decisions of a stressed cell.
Conclusions and perspectives
Despite some controversies, pharmacological and genetic knockdown or knockout studies have suggested a renoprotective role of autophagy in renal tubular cells in AKI. The mechanism by which autophagy protects tubular cells is currently unclear. In addition, whether and how autophagy changes its role from a pro-survival mechanism to a pro-death factor are currently unknown. The key signaling pathways that induce and regulate autophagy in AKI are also poorly understood. Further research should focus on these areas to elucidate the mechanism of autophagy induction in tubular cells in AKI, delineate the underlying signaling pathways, and define the precise roles played by autophagy in tubular regulation in this disease. A comprehensive understanding of the regulatory network of tubular cell autophagy will facilitate the discovery of genetic and pharmacological modulators for the prevention and treatment of kidney diseases including AKI.
Autophagy in polycystic kidney disease
There are reasons to believe that autophagy is present in polycystic kidneys and that it may even play a role in cyst formation and growth. First, cyst formation in polycystic kidney disease results in localized areas of hypoxia as evidenced by increased EPO levels and HIF1A/HIF-1α in PKD kidneys.Citation158 Autophagy can be induced by physiological stress stimuli such as hypoxia.Citation159 Second, human and experimental data provide strong evidence that abnormal proliferation and apoptosis in tubular epithelial cells plays a crucial role in cyst development and/or growth in PKD. Apoptosis and autophagy are intricately related. Third, there is evidence for activation of the PtdIns3K-AKT1-TSC-MTOR pathway in PKD and a therapeutic effect of the MTOR inhibitor rapamycin in animal models of PKD. In general, activation of MTOR inhibits autophagy and the MTORC1 inhibitor rapamycin induces autophagy. As hypoxia, apoptosis and MTOR signaling are modulators of autophagy and increased hypoxia, apoptosis and MTOR signaling are also features of PKD kidneys, a connection between autophagy and PKD has been proposed.Citation158
Apoptosis and autophagy in PKD
There is much evidence that apoptosis worsens cyst formation in PKD:Citation160 (1) Pharmacological inhibition of apoptosis (treatment of mice with caspase inhibitiors)Citation161 or genetic inhibition of apoptosis (Casp3 knockout)Citation162 results in less PKD. (2) BCL2-deficient mice have increased apoptosis and develop cysts in the kidney.Citation163 In this regard, BCL2 has been described as a “new guest at the autophagy table.”Citation164 BCL2 downregulation causes autophagy in a caspase-independent manner in human leukemia cells.Citation165 Also induction of autophagy may also be regulated by other BCL2 family members such as BCL211/BIM, BAD and BCL2L1.Citation166 The presence of autophagy in polycystic kidneys of Bcl2 knockout mice has not been reported and represents an interesting line of future research. (3) Apoptosis is essential for MDCK cell cyst cavitation in collagen type 1 matrix, and cystogenesis in this system is inhibited by overexpression of the anti-apoptotic gene, BCL2.Citation167 (4) The cell cycle inhibitor roscovitine results in long-lasting arrest of cystogenesis with decreased apoptosis.Citation168
Could there be a connection between apoptosis and autophagy in the cells lining the cysts? Many of the signals that regulate apoptosis in PKD, e.g., the BCL2 family of proteins, also regulate autophagy.Citation169 It is not well understood how cells respond to similar stimuli by undergoing autophagy or apoptosis. Caspase inhibitors prevent apoptosis but may induce autophagy.Citation170 The relationship between apoptosis and autophagy is complex depending on the type of cell, the nature of the injury or the timing of the injury.Citation171 For example, autophagy may result in a delay of apoptosis.Citation156,Citation172 Normal cells vs. cancer cells respond differently to apoptosis or autophagy-inducing stimuli. The genetic deletion of genes encoding key autophagy proteins increases rather than decreases apoptotic cell death.Citation156,Citation173 However, autophagy plays an important cell survival role. The relationship between apoptosis and autophagy and the effects of autophagy inhibition or autophagy activation remains to be determined in PKD.
MTOR signaling and autophagy in PKD
There is increased MTORC1 signaling in PKD in rodents and in humans.Citation174-Citation177 The MTORC1 inhibitor rapamycin or its analogs are protective against PKD and renal failure in the Han:SPRD rat and PKD1- or PKD2-deficient models of PKD.Citation178-Citation180 In general, activation of MTOR inhibits autophagic flux. Intracellular lysosomal positioning coordinates MTOR and autophagy signaling.Citation181 However, the mechanism of MTOR inhibition of autophagy is still not clear. The MTORC1 inhibitor rapamycin induces autophagy in a wide variety of cell types and species.Citation60 Thus, rapamycin may increase autophagy in polycystic kidneys. The increased autophagy induced by rapamycin may have deleterious effects, especially in view of the fact that rapamycin does not result in complete protection against PKD in animal models and does not work well in human studies.Citation182,Citation183 The effect of rapamycin on autophagy may rest on its effect on apoptosis in PKD. In this regard, Weimbs et al. have found that a high dose of rapamycin increases apoptosis of cysts lining the epithelium in PKD.Citation180 However, othersCitation179,Citation184 have shown that a low dose of rapamycin results in effective blockade of PKD without an effect on apoptosis.Citation184 Thus, rapamycin may turn out not to have an effect on autophagy in PKD.
In addition to increased MTORC1 signaling in PKD, there is also increased MTORC2 signaling as indicated by increases in pAKT1 Ser473.Citation180,Citation185 MTOR kinase inhibitors (TORKs) that directly inhibit both MTORC1 and MTORC2 have a potent effect to induce autophagy.Citation186 The effect of TORKs on apoptosis, autophagy and PKD remains to be determined.
Studies of autophagy in PKD
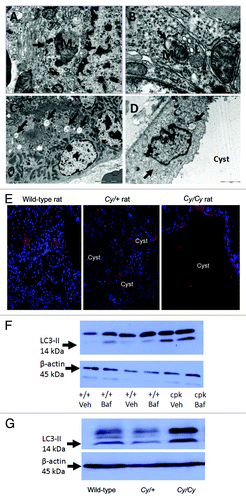
In a recently published study using EM, immunofluorescence and immunoblot for LC3-II and BECN1, autophagy was described in polycystic kidneys in rats and mice.Citation158 With EM, features of autophagy such as autophagosomes, mitophagy and autolysosomes are seen in wild-type rat and mouse kidneys as well as kidneys from Han:SPRD rats and cpk mice with PKD (). However, specific to PKD, autophagosomes are found in the tubular cells lining the cysts in PKD, and LC3 staining is seen on immunofluorescence in cells lining the cysts (). However, the presence of autophagosomes in tubular cells does not necessarily mean increased autophagy. Thus, BECN1 and LC3-II were examined. BECN1, a BH3-only protein, regulates formation and maturation of autophagosomes.Citation159 BECN1 plays a causative role in autophagy as evidenced by studies in which overexpression of BECN1 in cells promotes autophagy and BECN1-deficient mice have reduced autophagy.Citation188 BECN1 is increased at a late stage of PKD in homozygous Han:SPRD rats and cpk mice suggesting a possible causative role in the later stages of PKD. On immunoblot, the increase in LC3-II and BECN1 is seen in cpk mice and homozygous Han:SPRD rats at a late stage of the disease. Autophagic flux in PKD is measured by treating wild-type and cpk mice in vivo with bafilomycin A1 at a dose of 2 mg/kg/d ip for 4 d and measuring the amount of LC3-II with or without bafilomycin A1. In wild-type mice treated with bafilomycin A1 there was a definite and marked increase in LC3-II in the kidneys suggesting increased autophagic flux (). LC3-II was increased in PKD kidneys of cpk mice compared with wild-type kidneys (). The increase in LC3-II in PKD vs. wild-type kidneys suggests either increased autophagosome synthesis in PKD or decreased degradation in the lysosome in PKD. To investigate these possibilities, cpk mice were treated in vivo with bafilomycin A1 at the same dose of 2 mg/kg/d. Bafilomycin A1 had no effect on LC3-II in the polycystic kidneys of cpk mice (). The lack of effect of the lysosomal inhibitor bafilomycin A1 on LC3-II in PKD kidneys of cpk mice suggests a defect in autophagy in PKD resulting from a block of autophagosome-lysosome fusion and degradation. Apoptosis has been described at an early stage of PKD in heterozygous Han:SPRD rat kidneys.Citation189 Increased LC3-II is not found in heterozygous Han:SPRD rat kidneys (Cy/+) at an early stage of the disease, but is found in homozygous PKD kidneys (Cy/Cy) at a late stage of the disease (). These studies suggest that apoptosis precedes autophagy in PKD kidneys of the Han:SPRD rat.
Figure 4. Autophagy in PKD. Autophagosomes (arrows) are demonstrated in kidneys of +/+ rats (A), Cy/+ rats (B), +/+ mice (C) and in the tubular cells lining the cysts in cpk mice (D). Lysosomes (L) fusing to autophagosomes are shown in (B). Mitochondria (M) within a autophagosome (mitophagy) is shown in (A and B). Reproduced from Belibi et al.,Citation158 with permission from the American Physiological Society. Magnification is X20000 in (A), X40000 in (B), X6000 in (C) and X12000 in (D). (E) To demonstrate that LC3 is present in the cells lining the cysts, IF was performed. There is LC3 stainingCitation187 in cells lining the cysts in Cy/+ and Cy/Cy rats. Nuclei are represented by blue (DAPI) staining. LC3 staining is cytosolic. Reproduced from Belibi et al.,Citation158 with permission from the American Physiological Society. (F) The intensity of the LC3-II bands was increased in cpk mice compared with wild type (+/+) mice. In +/+ mice treated with bafilomycin A1 (+/+ Baf), there was an increase in LC3-II. In cpk mice treated with bafilomycin A1 (cpk Baf), LC3-II was not increased. (G) LC3-II was increased in Cy/Cy rats compared with +/+ and Cy/+ rats. ACTB/β-actin used as a loading control was not different between the groups. Reproduced from Belibi et al.,Citation158 with permission from the American Physiological Society.
While there is only one reported study that shows abnormalities of autophagy and autophagy-related proteins specifically in PKD kidneys,Citation158 there are proteins that are known to mediate autophagy, that are present in PKD kidneys or cells. Demonstration of these proteins in PKD kidneys or cells may suggest a relationship between autophagy and PKD. For example, sustained activation of the transcription factor, signal transducer and activator of transcription 1 (STAT1), that is known to mediate apoptosis, necrosis and autophagy, has been described in Pkd1 knockout polycystic kidneys and human ADPKD kidneys.Citation190 However, Stat1 knockout mice do not have a cystic phenotype,Citation190 suggesting that STAT1 is not a mediator of PKD.
Also, MAPK8 is a mediator of apoptosis and autophagy. Increased MAPK8 activity-mediated apoptosis has been shown in PC-1 knockdown MDCK cells.Citation191 This study suggests a possible connection between MAPK8 and autophagy in PKD. Also, AMPK has been described as a universal regulator of autophagy.Citation192 Metformin, an inducer of AMPK, has mostly but not exclusively been described to increase autophagy.Citation193,Citation194 Metformin slows cyst formation in in vitro and in vivo models of cyst formation.Citation195 This study suggests that autophagy induction by metformin may be therapeutic in PKD. Future studies of STAT1, MAPK8 or AMPK and their relationship to autophagy in PKD models will be interesting.
ADPKD is also a feature of tuberous sclerosisCitation183 and Von Hippel-Lindau disease (VHL). Tumorigenesis in TSC is autophagy-dependent, as ATG4 or BECN1 inhibition decreases tumorigenesis.Citation196 In VHL-deficient renal cancer cells, the small molecule STF-62247 induces autophagic cell death, and the reduction of ATG5 reduces the sensitivity of VHL-deficient cells to killing by STF-62247.Citation197 Whether the role of autophagy in tumorigenesis in TSC and VHL also applies to PKD in these models remains to be determined.
Conclusions and perspectives
A function of autophagy is to keep cells alive under “stressful” conditions. Autophagy in the apoptotic tubular epithelial cells lining the cysts may be a response to the cell death in these cells. Bafilomycin A1 resulted in an increase in LC3-II in wild-type but not in PKD kidneys ().Citation158 Thus, autophagy suppression may be a feature of disease progression in PKD. Further study of autophagy in PKD is merited especially as increased MTOR signaling and increased apoptosis are features of PKD, and there is important crosstalk between MTOR signaling, apoptosis and autophagy.Citation149 In summary, before using drugs in human PKD studies that have a profound effect on autophagy, it may be important to determine the effect of these drugs on autophagy in animal models of PKD.
Autophagy in renal transplantation
The transplanted kidney is permanently stressed
Transplanted kidney tissue permanently faces both acute and chronic stress signals that may contribute to cell death, inflammation, alloimmunity, cell senescence and fibrogenesis. In a pathological setting, such insults contribute to acute and chronic rejection, tissue remodeling, interstitial fibrosis, tubular atrophy, function deterioration and allograft loss.Citation198-Citation200
The number of stresses that a transplanted kidney encounters is considerable, and these stresses include ischemia (cold preservation and warm ischemia), reperfusion injury, toxicity (calcineurin inhibitors nephrotoxicity), immunological assaults (inflammation and cellular and humoral alloimmunity), hemodynamics (hypo- and hypertension), infections (BK virus and bacterial nephritis) and metabolic stresses (hyperglycemia and dyslipidemia). In response to these stresses, cells develop adaptive responses, most of which are evolutionarily conserved and include stabilization of HIF1A, the unfolded protein response, the integrated stress responses and MTOR signaling,Citation16,Citation123,Citation201 among many others. The decision between cell survival and death depends on integrating adaptive signals and cell death programs that are engaged in parallel and regulated by the duration and intensity of the stress. In addition to these cell life and death decisions, such adaptive responses often activate biological pathways that are involved in the regulation of innate and adaptive immunity,Citation202,Citation203 cellular phenotypic changes,Citation204 fibrogenesis,Citation205 and senescence,Citation205,Citation206 that will fuel tissue remodeling.
Autophagy emerges as an important adaptive process in transplantation
Mounting experimental evidence has identified autophagy as a master adaptive program that responds to a considerable number of insults whose consequences far exceed life and death decisions. The main biological activators of autophagy include starvation and inflammatory mediators.Citation123,Citation202 Strikingly, kidney allografts are almost always ischemic because of ischemia-reperfusion, arteriolar vasoconstriction, capillary destruction, rarefaction and interstitial fibrosis.Citation207 Moreover, the transplanted kidney is a pro-inflammatory milieu that promotes the release of danger-associated molecular patterns during ischemic insults, which are followed by the activation of humoral and cellular alloimmunity.Citation203,Citation207 Thus, there is little doubt that autophagy regulates kidney allograft tissue homeostasis more than is actually known. However, experimental and clinical evidence that implicates autophagy as an important stress response in the transplanted kidney has emerged.
Evidence for autophagy in kidney transplantation
Among insults that challenge a kidney allograft, immunosuppressive drugs and ischemic stresses (either cold ischemia or ischemia-reperfusion injury) promote autophagy, which can be either cytoprotective or cytotoxic depending on the nature of the stress.
Cold preservation
Cold ischemia is the period of time between kidney retrieval and transplantation and may vary from a few hours up to 36 h. During this period, the kidney is placed on ice in a preservation solution that contains nutrients and solutes that are designed to limit the cellular consequences of nutrient derivation associated with ischemia (but not hypoxia). Despite these procedures, cold ischemia is a deleterious process that promotes cell death and activates innate immunity.Citation208 In rat kidneys, cold preservation using University of Wisconsin (UW) solution (a solution that is widely used for kidney preservation) increases autophagic flux and apoptotic cell death.Citation209 Inhibiting autophagic flux by adding bafilomycin A1 to the UW solution reduces autophagic flux and apoptosis (and unexpectedly decreases LC3-II formation). Although the consequences of adding bafilomycin A1 on kidney pathology and function remain to be determined, these findings highlight the potential benefits of modulating autophagy during cold preservation.
Ischemia-reperfusion injury
After cold preservation, reperfusion injury occurs once kidney blood flow is established. Reperfusion stress promotes the release of reactive oxygen species that increase the ischemic stress and lead to so-called ischemia-reperfusion (IR) injury. IR injury potentiates the activation of autophagy induced by cold preservation. Indeed, oxidative stress upregulates the expression of autophagic regulators, including LC3 and BECN1,Citation134,Citation210 and promotes autophagy.Citation107,Citation109,Citation133 Therapeutically modulating autophagy during kidney IR injury gives promising results. The arterial delivery of a recombinant human adenovirus containing BCL2L1 cDNA before kidney IR can protect the kidney by inhibiting both apoptosis and autophagy during IR, as BCL2L1 inhibits mitochondrial outer membrane permeabilization and reactive oxygen species production.Citation133 Moreover, BCL2L1 augmentation reduces acute tubular necrosis and ameliorates renal dysfunction. Similar results were found in a BCL2 transgenic mouse model, which overexpresses BCL2.Citation133 These mice are protected against IR damage, as their tubular cells exhibit fewer apoptotic and autophagic features. A limitation of these two studies is that the respective roles of apoptosis and autophagy inhibition in mediating cytoprotection against IR were not delineated.
The occurrence and deleterious consequences of autophagy during IR in the kidney have also been found in the transplanted liver. Indeed, the transplanted liver is also subjected to IR injury,Citation211,Citation212 and autophagy seems to be deleterious in this setting. Cold preservation and reperfusion injury promote autophagic features in rat hepatocytes, and inhibiting autophagic flux (either by phosphatidylinositol 3-kinase inhibitors or aspartic and cysteine protease inhibitors) decreases liver injury and rat mortality.Citation211
Immunosuppressive drugs
Calcineurin inhibitors (cyclosporine A, CsA and tacrolimus) are powerful immunosuppressive drugs that constitute the cornerstone of immunosuppressive regimens in solid organ transplantation for 30 y, but their use is associated with many side effects, including acute and chronic nephrotoxicity, whose mechanisms are not fully understood. Recent data suggest that tubular cells play a central role in the pathogenesis of chronic nephropathies. CsA activates autophagy and protects against cell death via endoplasmic reticulum stress induction. Interestingly, an immunohistochemical analysis of rat kidneys has revealed positive LC3 staining in injured tubular cells, suggesting that CsA can activate autophagy in vivo.Citation117 Rapamycin (sirolimus, SRL) is another immunosuppressive drug that is used to prevent acute rejection. SRL strongly induces autophagy by inhibiting MTOR signaling. SRL does not inhibit calcineurin; thus, it was anticipated that SRL would lack the nephrotoxic profile of the calcineurin inhibitors. In clinical practice, SRL was initially introduced as an adjunct to calcineurin inhibitors, and it is now frequently used in regimens that minimize or avoid these nephrotoxic drugs. However, SRL is associated with adverse renal events, including proteinuria and glomerulonephritis.Citation213 High doses of SRL are also associated with systemic inflammatory syndrome, whose biological mechanisms are poorly understood.Citation214 There is actually no evidence that autophagy promotes these side effects, but given the importance of MTOR and autophagy in podocyte responses to stress on the one handCitation65,Citation66 and in regulating immune responses on the other hand,Citation215,Citation216 one can speculate that autophagy mediated by pharmacological MTOR inhibition is involved in such side effects.
Recent reports have shown that other immunosuppressive drugs, including mycophenolate mofetil and sphingosine kinase inhibitors, may be involved in regulating autophagy-related processes,Citation217,Citation218 but their mechanisms and functional consequences in kidney transplantation remain to be examined.
Conclusions and perspectives
Autophagy in the transplanted kidney has been implicated in the response to toxic and ischemic stresses, with opposing effects on cell fate. Autophagy is cytoprotective during cyclosporine nephrotoxicity, but its inhibition promotes cell death and aggravates tissue injury after IR. Autophagy is an exciting therapeutic target, but the complexity of its regulation and its unclear effects on cell viability indicate that its modulation will require fine-tuning that depends on factors such as the type of stress and its duration.
Our understanding of the implications of autophagy as a stress response during injured kidney transplantation is just beginning. The pathology of the transplanted kidney is promoted by inflammatory and ischemic stresses as well as infectious and toxic conditions that are also autophagic inducers. The effects of autophagy on regulating innate and adaptive immunity and cell viability might be of importance in transplantation biology.
Autophagy in Glomerular Health, Aging and Disease
Ultrafiltration of plasma is the major and life-sustaining function of kidneys. The filtration is performed by a unique sieving structure being composed of the fenestrated endothelium of the glomerular capillaries, the glomerular basement membrane, and the slit diaphragm, a specialized cell-cell contact bridging the processes of neighboring visceral epithelial cells—the podocytes.Citation219 To maintain the complex architecture of the filtration barrier each glomerulus consists of four resident cell types: glomerular endothelial cells, podocytes, mesangial cells, and parietal epithelial cells lining the Bowman's capsule. Recent experimental and clinical advances have identified the podocyte as the most vulnerable component of the glomerulus, being responsible for the progression of glomerular diseases characterized by proteinuria and loss of glomerular filtration rate. Accordingly, a decrease in the number of glomerular podocytes predicts the progression of renal diseases and renal aging.Citation58,Citation59,Citation220 More recently, an elegant podocyte-specific toxin model has proven that podocyte loss is sufficient to cause glomerulosclerosis.Citation221 Although the molecular programs of podocyte damage are incompletely understood, oxidative stress, ER stress, mitochondrial damage and cytoskeletal derangement are critical injury mechanisms in most forms of glomerulosclerosis.Citation4 Since podocytes are postmitotic cells and the capacity for replacement appears to be limited,Citation222 programs that help to counteract cellular stress events and thereby prevent podocyte detachment are the basis for the maintenance of the glomerular podocyte compartment. Interestingly, recent data suggest that glomerular autophagy might be a central theme in preventing podocyte degeneration.
Autophagosomes in podocytes
The first evidence for autophagy in podocytes came from Asanuma et al., who detected an upregulation of LC3-positive autophagosomes during the recovery from puromycin aminonucleoside-induced nephrosis in rats in vivo and in immortalized mouse podocytes in vitro.Citation68 Transgenic mouse models using GFP-LC3 confirmed that glomerular podocytes display significant levels of autophagosomes even under basal conditions.Citation66,Citation69 Although, it has been postulated for some tissues that removal of organelles through autophagy contributes to differentiation and development, autophagosomes in podocytes are predominantly detected at late developmental stages or in mature podocytes,Citation66 suggesting that autophagy might rather be important for differentiated podocytes. In agreement, constitutive embryonic deletion of Atg5 does not impair glomerular development.Citation66 Similar observations could be made in vitro, where immortalized mouse podocytes upregulate autophagy during cell differentiation.Citation68 Recently, autophagic flux experiments implied that the levels of basal autophagic activity in podocytes are higher compared with other renal epithelial cell lines.Citation66 Taken together these data suggested that basal autophagy is a prominent feature of differentiated postmitotic podocytes in vivo and in vitro.
Subsequently, human biopsy studies detected autophagosome formation in the course of glomerular diseases.Citation223 Ultrastructural and immunofluorescence analysis revealed increased numbers of autophagosomes in podocytes in IgA nephropathyCitation224 and membranous glomerulopathy.Citation66
Role of autophagy in podocyte maintenance and aging
While human biopsy studies suggested a role of autophagy in glomerular diseases, they could not define whether autophagy is protective or rather disease mediating for podocytes. However, in a recent study podocyte-specific deletion of the Atg5 gene resulted in proteinuria, loss of podocytes and aging-related glomerulosclerosis indicating the critical importance of autophagy for glomerular maintenance.Citation66 Atg5-deficient podocytes phenocopy typical age-related alterations including accumulation of lipofuscin, the occurrence of damaged mitochondria, an increase in the total load of oxidized proteins and the formation of ubiquitin and SQSTM1-positive protein aggregates.Citation66 Notably, ubiquitin and SQSTM1-positive inclusion bodies have also been identified in various neurodegenerative diseases (i.e., Alzheimer disease).Citation225 Autophagy deficiency in podocytes therefore appears to delay the global turnover of cytoplasmic components, resulting in accumulation of misfolded proteins followed by the formation of inclusion bodies and deformed organelles. Furthermore, an increase of ER stress markers could be detected; similar observations were made in other autophagy-deficient tissues such as cardiomyocytes.Citation226 Unlike other glomerular cells that are permanently renewed, podocytes seem to depend on autophagy to prevent the process of cellular degeneration. This might also relate podocyte autophagy to renal aging. In fact, it is well known that the age-related decline of kidney functionCitation220,Citation227,Citation228 is at least partially caused by the loss of podocytes.Citation220,Citation229 Like other tissues,Citation82,Citation230 podocytes display diminished autophagosome formation with age (Hartleben B, Huber TB, personal observation), which likely contributes to an age-related glomerulosclerosis.
Role of autophagy in podocyte disease
Basal autophagy seems to act as quality control machinery, being essential for the homeostasis of postmitotic podocytes. Accordingly, autophagy in podocytes is a predominantly cytoprotective process that likely mediates protective effects in both glomerular maintenance and glomerular injury. Consistently, under pathophysiological conditions, loss of autophagy in podocytes results in a dramatically increased susceptibility to various models of glomerular diseaseCitation66 highlighting the particular importance of autophagy as a key homeostatic mechanism not only under physiological but also under stress conditions.
Another recent observation underlines the importance of continuous protein degradation by the autophagosomal-lysosomal system in podocytes: Reduced lysosomal acidification by the knockout of the gene encoding the prorenin receptor (Atp6ap2) in podocytes results in a functional block of autolysosome formation.Citation231 This leads to a rapid accumulation of ubiquitinated proteins and SQSTM1, followed by severe podocyte damage and early lethality of mice.Citation231 Strikingly, several human lysosomal storage diseases are associated with an accumulation of storage products in podocytes, specifically leading to podocyte damage, proteinuria and kidney failure.Citation232-Citation236 One such example is Morbus Fabry, which is caused by deficiency of the lysosomal α-galactosidase resulting in renal dysfunction, systemic vasculopathy and cardiomyopathy.Citation237 Characteristic histological features of Morbus Fabry are cellular vacuolizations that are predominantly found in podocytes.Citation232,Citation238 These vacuolizations appear to be associated with reduced autophagic flux rates.Citation238 Defective proteins are not only degraded by the autophagy-lysosome pathway but also by the ubiquitin proteasome system.Citation2 In the podocyte the UPS and the autophagy-lysosome system might be functionally coupled, as autophagy deficiency causes increased proteasome activity, and an inhibition of the UPS results in an increased autophagic activity. In later stages, autophagy deficiency is accompanied by accumulation of SQSTM1 and ubiquitinated proteins and a decrease of UPS activity.Citation66 In general it has been shown that in autophagy-deficient tissue accumulating SQSTM1 binds to ubiquitinated proteins, forming protein aggregates and preventing them from degradation by the UPS, which leads to toxic levels of certain UPS substrates such as TP53.Citation239 Interestingly, it has recently been shown, that next to the protein degradation by autophagy and UPS, cytosolic proteases such as CTSL1/cathepsin L1 are also critical for podocyte homeostasis. Podocyte injury can induce cytosolic CTSL1 expression, which results in proteolytic degradation of podocyte cytoskeletal components.Citation240-Citation242 However, it is not clear yet if there is a functional coupling between autophagy and CTSL1. Molecular details and function of presumptive interactions of protein degradative pathways in podocytes will have to be addressed in future studies.
Since autophagy seems to be a general cellular stress surveillance factor for podocytes, it will be interesting, if genome-wide association studies identify disease susceptibility loci in autophagosomal regulatory genes. Recently, a seminal study identified polymorphisms in the APOL1 gene to be associated with significantly increased rates of focal segmental glomerulosclerosis and hypertension-attributed end stage renal disease in African Americans.Citation243 While the molecular mechanism leading to an increased glomerular disease predisposition remains unclear, there is evidence that APOL1 might directly regulate autophagy.Citation244
Regulation of autophagy in podocytes
Very little is known about the regulation of autophagy in podocytes. In general, the MTOR pathway is the best-described pathway regulating mammalian autophagy. However, podocytes seem to exhibit a quite unique feature, where high basal autophagy rates can be seen despite activation of MTOR (unpublished observation), suggesting that cytosolic autophagy might be regulated independently from MTORC1. A possible explanation for this could be the recently identified TOR-autophagy spatial coupling compartment in podocytes.Citation71 TASCC represents a distinct cellular compartment. By sequestering MTORC1 complexes with autolysosomes at the Golgi apparatus, the cytosolic concentrations of MTORC1 are lowered, supporting cytosolic autophagosome induction (for details see the chapter “Regulation of autophagy in the kidney”). This points to the question of whether MTOR-independent pathways contribute to the regulation of autophagy in podocytes. The first evidence for the existence of MTOR-independent regulation of mammalian autophagy came from studies showing that autophagy is negatively regulated by G protein-coupled receptor-mediated activation of phospholipase C.Citation225 In addition, increased autophagic activity in response to ROS generation has been described as an MTOR-independent pathway.Citation225 Both pathways would be very well conceivable as regulators of podocyte autophagy. However, future studies will have to elucidate the molecular details of the regulation of autophagy in the podocyte.
Conclusions and perspectives
Recent studies have shed light on the role of constitutive autophagy for the cellular homeostasis of podocytes in health and disease. Autophagy appears to be an important cytoprotective process that mediates protective effects in both glomerular maintenance and glomerular injury. Therefore, influencing autophagy could be a promising strategy for the treatment of glomerulopathies.
Future studies will have to elucidate the upstream regulation of autophagy, the interplay of autophagy with other degradative pathways in podocytes and the role of autophagy in other glomerular cell types.
Autophagy in Kidney Aging
Increasing age causes a progressive post-maturational deterioration of an organ’s functions. Accumulation of intracellular damaged proteins and organelles, such as mitochondria, is considered to be a pathogenesis underlying age-associated malfunctioning of tissues.Citation82 Consequently, inadequate removal of these damaged molecules leads to the development of age-associated diseases.Citation82
Over recent decades, the roles of autophagy in the aging processes of various species from yeasts to mammals have been extensively studied. The findings have revealed that both macroautophagy and chaperone-mediated autophagy decline with age,Citation245,Citation246 and that autophagy deficiency causes age-related waste accumulation in cells, leading to a progressive aging process.Citation82
Among the organs, the kidney is a typical target organ of aging. An analysis in the National Health and Nutrition Examination Survey (NHANES) showed that people older than 70 y of age have a higher prevalence of chronic kidney disease (CKD),Citation247 while an analysis in the US Renal Data System (USRDS) revealed that the incidence rates of end-stage kidney disease rise after 45 y of age and reach the maximum peak over 75 y of age.Citation248 Furthermore, the incidence of hospital-acquired acute kidney injury (AKI) is higher in elderly subjects.Citation249 Therefore, the increased incidences of CKD and stress susceptibility to drug- or ischemia-mediated AKI in elderly individuals are health problems worldwide.Citation247-Citation250 Consequently, better understanding of the mechanism underlying the aging process in the kidney is required.
Glomerulosclerosis and tubulointerstitial fibrosis together with podocyte damage and tubular damage, respectively, are typical histological changes in the aging kidney.Citation228,Citation251 Glomerular podocytes are terminally differentiated and long-lived postmitotic cells.Citation252 Therefore, the fate of podocytes is largely dependent on their capacity to cope with stress. In addition, proximal tubular cells contain large quantities of organelles, such as mitochondria and endoplasmic reticulum, and reabsorb large amounts of proteins filtered from the glomeruli.Citation253 These findings easily lead us to hypothesize that autophagy plays essential roles in maintaining the homeostasis and functions of both podocytes and proximal tubular cells, and that its insufficiency with aging might contribute to the progression of kidney aging. In fact, recent reports have shown renoprotective roles of autophagy against aging in both podocytes and proximal tubular cells.Citation4,Citation66,Citation113,Citation254 In this section, based on recently published studies, we review and discuss the role of autophagy in kidney aging.
The kidneys of aging rats and mice show altered mitochondrial morphology and accumulation of age-associated proteins, such as SQSTM1, together with a decrease in autophagy activity, suggesting that the aging kidney has an impaired ability to induce autophagy.Citation254,Citation255 ATG5 plays a role in the conversion of LC3-I to LC3-II, which is essential for autophagosome formation, and conditional Atg5 knockout mice represent an excellent tool for studying the role of autophagy in a tissue-specific manner.Citation5 Two recent studies using conditional Atg5 knockout mice have shown the relevance of autophagy to the aging process of both podocytes and proximal tubular cells.Citation66,Citation113 Podocyte-specific deletion of the Atg5 gene results in the accumulation of damaged mitochondria and ubiquitinated proteins in podocytes, thereby leading to exacerbation of age-dependent albuminuria and glomerulosclerosis.Citation66 Proximal tubule-specific deletion of the Atg5 gene also leads to significant increases in age-dependent accumulation of abnormal mitochondria, ubiquitinated proteins and SQSTM1, which significantly increases apoptosis in proximal tubular cells.Citation113 These findings clearly show that deficiency of basal autophagy in both podocytes and proximal tubular cells is associated with the development of kidney aging.
How can we activate autophagy in the aging kidney and protect the kidney against aging? Calorie restriction (CR) may provide an answer to this issue. CR, which can physiologically induce autophagy, exhibits anti-aging effects across eukaryotic species.Citation256,Citation257 Interestingly, CR can ameliorate age-associated kidney damage, including albuminuria, glomerulosclerosis and tubulo-interstitial lesions, in animal experimental models.Citation254,Citation258,Citation259 A more recent report showed that autophagy is essential for CR-mediated renoprotection in aged mice.Citation254 Renal hypoxia has recently been recognized as a pathogenesis and a therapeutic target for CKD, including kidney aging,Citation260 and is also an inducer of autophagy.Citation261 Interestingly, the aging kidney fails to induce autophagy in proximal tubular cells even under hypoxic stress conditions, which is associated with age-dependent kidney injury.Citation254 In contrast, CR restores autophagy activity against hypoxia, even in the aging kidney, and protects the kidney against aging.Citation254 As a molecular mechanism underlying the CR-mediated restoration of autophagy in the aging kidney, the antiaging molecule SIRT1 has a critical role. SIRT1, a mammalian homolog of the yeast silent information regulator 2 (Sir2), was originally identified as an NAD+-dependent deacetylase that is required for CR-mediated life-span elongation in the lower species.Citation254 SIRT1-mediated deacetylation of FOXO3/FOXO3A, a stress-responsible transcription factor, was shown to be essential for CR-mediated restoration of autophagy against hypoxia in the aging kidney.Citation254
In addition to age-associated CKD, susceptibility to drug- or ischemia-induced AKI in elderly individuals is also focused upon as an important health problem.Citation249 As described in another section, several AKI models, such as ischemic-reperfusion injury and cisplatin nephropathy, in mice induce autophagy to protect proximal tubular cells.Citation113,Citation115,Citation172 As mentioned in this section, autophagy activity in proximal tubular cells declines with age,Citation254,Citation255 which may be a reason why the kidney in elderly subjects shows stress susceptibility to AKI, such as that caused by ischemia and nephrotoxic agents.
Recent genetic evidence supports a critical role of autophagy as an anti-aging mechanism, since blockage of autophagy genes in long-lived Caenorhabditis elegans mutants or diet-restricted C. elegans prevents life-span extension.Citation262-Citation264 Furthermore, recent mouse studies have shown a role of autophagy in the organ aging of mammals. The roles of autophagy in age-associated neurodegenerative disorders have been extensively examined.Citation230 Since neural cells are terminally differentiated and long-lived postmitotic cells, similar to podocytes, an age-dependent decline in autophagy activity causes neuronal cell impairments, leading to the development of neurodegenerative disorders, such as Alzheimer and Huntington diseases. Heart failure is a progressive disease that is common in elderly patients. The basal autophagy in the heart antagonizes ventricular hypertrophy by increasing protein degradation, which decreases the tissue mass.Citation265 The rate of protective autophagy in the heart declines with age, which is associated with an age-dependent decline in cardiac function.Citation265 Muscle weakness is also a harmful symptom in elderly individuals. Mice lacking macroautophagy activity by deletion of the Atg7 gene in skeletal muscle show accumulation of abnormal mitochondria, which causes age-dependent muscle atrophy and a decrease in force.Citation266 Consequently, autophagy deficiency is likely to be associated with the pathogenesis of age-dependent dysfunctions in various organs. Interestingly, restoration of chaperone-mediated autophagy attenuates the age-dependent decline in hepatic function,Citation267 suggesting that restoration of degradation systems, including autophagy, should represent a therapeutic target against aging in various tissues.
Conclusions and perspectives
shows the relevance of autophagy in kidney aging. Two studies using Atg5 knockout mice have clearly shown that autophagy has a renoprotective role against aging.Citation66,Citation113 In addition, CR mimetics, including SIRT1 activation, may be new therapeutic strategies for protecting the kidney against aging.Citation254 Although further studies are needed, restoration of the autophagic activity during aging should contribute to renoprotection and subsequent successful aging in elderly people.
Figure 5. Autophagy in kidney aging. In both podocytes and proximal tubular cells, autophagy maintains cellular and organelles homeostasis under both basal and stress conditions. Normal aging process or a deletion of Atg5 gene alters autophagy in podocytes and proximal tubular cells, leading to kidney aging. Calorie restriction prevents the progression of kidney aging in a part through SIRT1-dependent autophagy.
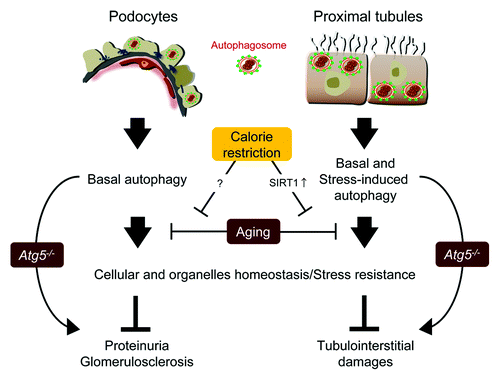
Future Directions
This review discusses the relatively recent developments regarding the role of autophagy in a number of renal diseases. There is now substantial evidence that autophagy is induced during AKI caused by ischemic or toxic forms of injury to the kidney (). Autophagy appears to be an early stress response in renal cells and other cell types, and appears to precede apoptosis in most settings. One important controversy in this field is whether autophagy promotes or prevents apoptosis after acute injury or stress. It seems likely that some of this controversy was incurred by the use, in many experiments, of nonspecific pharmacological inhibitors of autophagy. The “off-target” effects of such pharmacological agents may be responsible, at least in part, for the contradictory results regarding the role of autophagy in determining cell fate.
A major advance in the field of autophagy in general has been the availability of mice deficient in some of the genes essential for the autophagic response. For example, this review discusses the evidence in mice deficient in genes encoding either ATG5 or ATG7, showing that both types of knockouts are more susceptible to AKI than their wild-type controls. These studies suggest that, in most situations at least, autophagy protects tubular cells from apoptosis. However, this review discusses evidence suggesting that, in certain circumstances, and for reasons that remain unresolved, autophagy may occasionally either fail to prevent apoptosis, or may even cooperate with the apoptotic machinery to promote cell death. One of the important areas for future research in AKI and many other forms of renal disease is to determine when and how the autophagic response in kidney tubular cells acts as a protective as opposed to a pro-death response.
The influence of autophagy on cell survival in the kidney is likely to be an important component of any forms of stress to which the kidney is subjected. It is therefore not surprising that there is evidence suggesting that autophagy plays a role in the transplanted kidney, which is subjected to a host of different stresses. These include, in addition to AKI due to ischemia-reperfusion injury during the peri-transplant period, toxicity caused by pharmacological agents used for immunosuppression, immunological injury due to sometimes-recurrent episodes of transplant rejection, and renal injury due to some opportunistic infections. Available evidence suggests that the autophagic response is likely to play an important role in life and death decisions of individual renal cells exposed to these threats. As this review makes clear, information regarding the role of autophagy following renal transplantation has only recently begun to emerge. The role played by autophagy in each kind of stress to which the transplanted kidney is exposed, still remains to be elucidated. Clearly, though, this knowledge could prove extremely useful in the many therapeutic decisions that have to be made throughout the functional life of a renal transplant.
This review also discusses in some detail a major paradox in the field of autophagy. This regards the relationship between the activity of the MTOR pathway and autophagy ( and ). This topic is discussed in this review in greatest detail with regard to podocytes, but may be true of other glomerular cells. It is now well established that MTORC1 is activated in glomerular cells, including podocytes in DN, and that inhibition of MTORC1 with rapamycin ameliorates DN in a number of different animal models of this disease. Since it is now well established that, in all cells studied thus far, MTORC1 negatively regulates autophagy, the reported increased activity of MTORC1 in podocytes would predict that autophagy should be suppressed in these and other glomerular cells in DN. In contrast, as this review points out, available evidence suggests that autophagy is activated in response to stress, particularly in mature, differentiated podocytes. These cells appear to be able to exhibit high autophagic flux despite activation of MTOR. Some authors of this review speculate that MTORC1 could have different effects on autophagy in podocytes in different situations, while others suggest that autophagy could even be regulated independently from MTORC1 in these cells. However, there is not, as yet, published information to support either of these possibilities. Along these lines, this review discusses the paucity of knowledge regarding the regulation of autophagy in podocytes. Resolving the relationship between the MTOR pathway and autophagy has been identified in this review as another crucial area for future research.
Elucidating this area of uncertainty may also help us understand a dilemma regarding the potential role of MTOR inhibition as a novel therapeutic strategy in DN and other renal diseases in humans. While an abundance of published data convincingly shows that rapamycin-type MTOR inhibitors substantially ameliorate the glomerular pathology and proteinuria in a variety of different animal models of DN, there is also now clear evidence that rapamycin may cause or exacerbate proteinuria in some situations in humans. The fact that rapamycin can cause proteinuria in humans has, understandably, markedly dampened any enthusiasm for the initiation of clinical trials of rapamycin-type inhibitors for the treatment of DN. This review points out how research to further elucidate the role of the MTOR pathway in the control of autophagy in podocytes and other glomerular cells, may help guide us in deciding if and when it is safe to initiate trials of MTOR inhibitors in patients with DN and other glomerular diseases.
The review also deals with the role played by autophagy in PKD (). The fact that MTOR inhibits autophagy, that MTOR is hyperactive in cystic epithelial cells, and that inhibition of MTOR with rapamycin, (an intervention that increases autophagy) ameliorates cyst progression in animal models of PKD, taken together, suggests that autophagy is likely suppressed in this disease. However, our current understanding of role of autophagy in PKD in animal models of the disease remains largely descriptive. In addition, results of trials in humans with PKD using rapamycin have been disappointing. Therefore, more research in humans and in animal models elucidating the relationship between autophagy, apoptosis and MTOR in PKD is recommended, before deciding whether additional studies regarding the use of MTOR inhibitors in humans is appropriate in this disease.
Available evidence regarding a role for autophagy in renal aging is also considered in this review (). Glomerulosclerosis and tubulo-interstitial damage are typical histological changes in the aging kidney. It is therefore of interest that podocyte-specific deletion of the Atg5 gene results in renal changes very similar to those of the aging kidney, including loss of podocytes and glomerulosclerosis.Citation66 Furthermore, proximal tubule-specific deletion of Atg5 leads to significant increases in age-dependent accumulation of abnormal mitochondria and ubiquitinated proteins, events that eventually lead to to apoptosis of these cells.Citation113 These findings clearly show that deficiency of basal autophagy in both podocytes and proximal tubular cells are likely partly responsible for the changes and decline in renal function associated with kidney aging.
It is also important to point out the complexity regarding the interpretation of the array of techniques developed to evaluate autophagy. One area of potential confusion, that deserves particular mention in this review, regards the interpretation of techniques that determine the conversion of LC3-I to LC3-II. It is important to appreciate that increased formation of LC3-II is not a measure of autophagic flux, a term which refers to the complete process of autophagy, including the delivery of cargo by autophagosomes to lysosomes, and its subsequent breakdown and recycling. Instead, the accumulation of LC3-II generally indicates either an increase in the induction of autophagy (i.e., an increase in the formation of autophagosomes) and/or a blockade of autophagosome clearance. Additional methods (discussed in detail in other reviewsCitation187,Citation268) are needed to assess the extent to which conversion of LC3-I to LC3-II indicates the induction vs. a blockade of autophagy.
In conclusion, there has been a rapid acceleration during the past few years in the development of a variety of knockout mice that have had a substantial impact on our understanding of the role of autophagy in the kidney. This trend is bound to continue. We anticipate that exciting new developments regarding the role of autophagy in the kidney in many different physiological and pathological situations will occur at a rapid pace over the next few years. These developments are likely to change and improve our therapeutic approaches to many renal problems.
Acknowledgments
We thank members of our laboratories for critical reading of the manuscript. This work was supported in part by Deutsche Forschungsgemeinschaft grant KFO 201 (to T.B.H.), by the Excellence Initiative of the German Federal and State Governments EXC 294 (to T.B.H.), by GerontosysII?NephAge (031589GA) (to T.B.H.), by the U.S.National Institutes of Health (RO1DK056851 and DK074835 to C.E., DK058831 and DK087843 to Z.D., DK083491 to K.I.), by the U.S.Veterans Administration (to Z.D. and to W.L.), by Grant-in-Aid For Diabetic Nephropathy Research from the Ministry of Health, Labour and Welfare of Japan (to D.K.), and the KAKENHI 21591148 (to D.K.) and 00452235 (to S.K.).
References
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443:780 - 6; http://dx.doi.org/10.1038/nature05291; PMID: 17051204
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814 - 22; http://dx.doi.org/10.1038/ncb0910-814; PMID: 20811353
- Weide T, Huber TB. Implications of autophagy for glomerular aging and disease. Cell Tissue Res 2011; 343:467 - 73; http://dx.doi.org/10.1007/s00441-010-1115-0; PMID: 21286756
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823 - 30; http://dx.doi.org/10.1038/ncb0910-823; PMID: 20811354
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004; 6:1122 - 8; http://dx.doi.org/10.1038/ncb1183; PMID: 15467718
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 2002; 10:457 - 68; http://dx.doi.org/10.1016/S1097-2765(02)00636-6; PMID: 12408816
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 2004; 14:1296 - 302; http://dx.doi.org/10.1016/j.cub.2004.06.054; PMID: 15268862
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002; 110:177 - 89; http://dx.doi.org/10.1016/S0092-8674(02)00833-4; PMID: 12150926
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002; 110:163 - 75; http://dx.doi.org/10.1016/S0092-8674(02)00808-5; PMID: 12150925
- Kim D-H, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell 2003; 11:895 - 904; http://dx.doi.org/10.1016/S1097-2765(03)00114-X; PMID: 12718876
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009; 137:873 - 86; http://dx.doi.org/10.1016/j.cell.2009.03.046; PMID: 19446321
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 2007; 25:903 - 15; http://dx.doi.org/10.1016/j.molcel.2007.03.003; PMID: 17386266
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 2007; 9:316 - 23; http://dx.doi.org/10.1038/ncb1547; PMID: 17277771
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004; 18:1926 - 45; http://dx.doi.org/10.1101/gad.1212704; PMID: 15314020
- Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell 2010; 40:310 - 22; http://dx.doi.org/10.1016/j.molcel.2010.09.026; PMID: 20965424
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell 2006; 124:471 - 84; http://dx.doi.org/10.1016/j.cell.2006.01.016; PMID: 16469695
- Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci U S A 1998; 95:1432 - 7; http://dx.doi.org/10.1073/pnas.95.4.1432; PMID: 9465032
- Ganley IG, Lam H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297 - 305; http://dx.doi.org/10.1074/jbc.M900573200; PMID: 19258318
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981 - 91; http://dx.doi.org/10.1091/mbc.E08-12-1248; PMID: 19211835
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011; 332:1317 - 22; http://dx.doi.org/10.1126/science.1199498; PMID: 21659604
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992 - 2003; http://dx.doi.org/10.1091/mbc.E08-12-1249; PMID: 19225151
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011; 146:408 - 20; http://dx.doi.org/10.1016/j.cell.2011.06.034; PMID: 21816276
- Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villén J, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011; 332:1322 - 6; http://dx.doi.org/10.1126/science.1199484; PMID: 21659605
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12:21 - 35; http://dx.doi.org/10.1038/nrm3025; PMID: 21157483
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 2004; 166:213 - 23; http://dx.doi.org/10.1083/jcb.200403069; PMID: 15249583
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol 2004; 14:1650 - 6; http://dx.doi.org/10.1016/j.cub.2004.08.026; PMID: 15380067
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004; 431:200 - 5; http://dx.doi.org/10.1038/nature02866; PMID: 15306821
- Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol 2006; 16:1865 - 70; http://dx.doi.org/10.1016/j.cub.2006.08.001; PMID: 16919458
- Thedieck K, Polak P, Kim ML, Molle KD, Cohen A, Jenö P, et al. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One 2007; 2:e1217; http://dx.doi.org/10.1371/journal.pone.0001217; PMID: 18030348
- Woo SY, Kim DH, Jun CB, Kim YM, Haar EV, Lee SI, et al. PRR5, a novel component of mTOR complex 2, regulates platelet-derived growth factor receptor beta expression and signaling. J Biol Chem 2007; 282:25604 - 12; http://dx.doi.org/10.1074/jbc.M704343200; PMID: 17599906
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098 - 101; http://dx.doi.org/10.1126/science.1106148; PMID: 15718470
- Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell 2011; 144:757 - 68; http://dx.doi.org/10.1016/j.cell.2011.02.014; PMID: 21376236
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 2003; 11:1457 - 66; http://dx.doi.org/10.1016/S1097-2765(03)00220-X; PMID: 12820960
- Inoki K, Li Y, Xu T, Guan K-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003; 17:1829 - 34; http://dx.doi.org/10.1101/gad.1110003; PMID: 12869586
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 2008; 10:935 - 45; http://dx.doi.org/10.1038/ncb1753; PMID: 18604198
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008; 320:1496 - 501; http://dx.doi.org/10.1126/science.1157535; PMID: 18497260
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 2003; 13:1259 - 68; http://dx.doi.org/10.1016/S0960-9822(03)00506-2; PMID: 12906785
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 2003; 5:578 - 81; http://dx.doi.org/10.1038/ncb999; PMID: 12771962
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010; 141:290 - 303; http://dx.doi.org/10.1016/j.cell.2010.02.024; PMID: 20381137
- Sekiguchi T, Hirose E, Nakashima N, Ii M, Nishimoto T. Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J Biol Chem 2001; 276:7246 - 57; http://dx.doi.org/10.1074/jbc.M004389200; PMID: 11073942
- Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol 2005; 15:702 - 13; http://dx.doi.org/10.1016/j.cub.2005.02.053; PMID: 15854902
- Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, Eguchi S, et al. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem 2007; 282:20329 - 39; http://dx.doi.org/10.1074/jbc.M702636200; PMID: 17517883
- Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem 2003; 278:32493 - 6; http://dx.doi.org/10.1074/jbc.C300226200; PMID: 12842888
- Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol 2003; 5:566 - 71; http://dx.doi.org/10.1038/ncb996; PMID: 12766776
- Inoki K, Li Y, Zhu T, Wu J, Guan K-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002; 4:648 - 57; http://dx.doi.org/10.1038/ncb839; PMID: 12172553
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005; 121:179 - 93; http://dx.doi.org/10.1016/j.cell.2005.02.031; PMID: 15851026
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 2002; 10:151 - 62; http://dx.doi.org/10.1016/S1097-2765(02)00568-3; PMID: 12150915
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010; 584:1287 - 95; http://dx.doi.org/10.1016/j.febslet.2010.01.017; PMID: 20083114
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000; 150:1507 - 13; http://dx.doi.org/10.1083/jcb.150.6.1507; PMID: 10995454
- Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 1997; 192:245 - 50; http://dx.doi.org/10.1016/S0378-1119(97)00084-X; PMID: 9224897
- Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell 2005; 16:2544 - 53; http://dx.doi.org/10.1091/mbc.E04-08-0669; PMID: 15743910
- Kawamata T, Kamada Y, Kabeya Y, Sekito T, Ohsumi Y. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol Biol Cell 2008; 19:2039 - 50; http://dx.doi.org/10.1091/mbc.E07-10-1048; PMID: 18287526
- Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol 2011; 23:744 - 55; http://dx.doi.org/10.1016/j.ceb.2011.09.003; PMID: 21963299
- Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol 2009; 20:2493 - 502; http://dx.doi.org/10.1681/ASN.2008111186; PMID: 19875810
- 2009 Annual Data Report Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. United States Renal Data System http://www.usrds.org, 2009:231-9.
- Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest 1997; 99:342 - 8; http://dx.doi.org/10.1172/JCI119163; PMID: 9006003
- Lemley KV, Lafayette RA, Safai M, Derby G, Blouch K, Squarer A, et al. Podocytopenia and disease severity in IgA nephropathy. Kidney Int 2002; 61:1475 - 85; http://dx.doi.org/10.1046/j.1523-1755.2002.00269.x; PMID: 11918755
- White KE, Bilous RW, Marshall SM, El Nahas M, Remuzzi G, Piras G, et al. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes 2002; 51:3083 - 9; http://dx.doi.org/10.2337/diabetes.51.10.3083; PMID: 12351451
- Huber TB, Walz G, Kuehn EW. mTOR and rapamycin in the kidney: signaling and therapeutic implications beyond immunosuppression. Kidney Int 2011; 79:502 - 11; http://dx.doi.org/10.1038/ki.2010.457; PMID: 21085109
- Lloberas N, Cruzado JM, Franquesa M, Herrero-Fresneda I, Torras J, Alperovich G, et al. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J Am Soc Nephrol 2006; 17:1395 - 404; http://dx.doi.org/10.1681/ASN.2005050549; PMID: 16597691
- Nagai K, Matsubara T, Mima A, Sumi E, Kanamori H, Iehara N, et al. Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int 2005; 68:552 - 61; http://dx.doi.org/10.1111/j.1523-1755.2005.00433.x; PMID: 16014032
- Sataranatarajan K, Mariappan MM, Lee MJ, Feliers D, Choudhury GG, Barnes JL, et al. Regulation of elongation phase of mRNA translation in diabetic nephropathy: amelioration by rapamycin. Am J Pathol 2007; 171:1733 - 42; http://dx.doi.org/10.2353/ajpath.2007.070412; PMID: 17991718
- Yang Y, Wang J, Qin L, Shou Z, Zhao J, Wang H, et al. Rapamycin prevents early steps of the development of diabetic nephropathy in rats. Am J Nephrol 2007; 27:495 - 502; http://dx.doi.org/10.1159/000106782; PMID: 17671379
- Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest 2011; 121:2197 - 209; http://dx.doi.org/10.1172/JCI44774; PMID: 21606591
- Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest 2010; 120:1084 - 96; http://dx.doi.org/10.1172/JCI39492; PMID: 20200449
- Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest 2011; 121:2181 - 96; http://dx.doi.org/10.1172/JCI44771; PMID: 21606597
- Asanuma K, Tanida I, Shirato I, Ueno T, Takahara H, Nishitani T, et al. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J 2003; 17:1165 - 7; PMID: 12709412
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101 - 11; http://dx.doi.org/10.1091/mbc.E03-09-0704; PMID: 14699058
- Sato S, Kitamura H, Adachi A, Sasaki Y, Ghazizadeh M. Two types of autophagy in the podocytes in renal biopsy specimens: ultrastructural study. J Submicrosc Cytol Pathol 2006; 38:167 - 74; PMID: 17784646
- Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011; 332:966 - 70; http://dx.doi.org/10.1126/science.1205407; PMID: 21512002
- Young AR, Narita M, Narita M. Spatio-temporal association between mTOR and autophagy during cellular senescence. Autophagy 2011; 7:1387 - 8; http://dx.doi.org/10.4161/auto.7.11.17348; PMID: 21799306
- Eremina V, Baelde HJ, Quaggin SE. Role of the VEGF–a signaling pathway in the glomerulus: evidence for crosstalk between components of the glomerular filtration barrier. Nephron, Physiol 2007; 106:32 - 7; http://dx.doi.org/10.1159/000101798
- Simons M, Saffrich R, Reiser J, Mundel P. Directed membrane transport is involved in process formation in cultured podocytes. J Am Soc Nephrol 1999; 10:1633 - 9; PMID: 10446930
- Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem 2004; 263:55 - 72; http://dx.doi.org/10.1023/B:MCBI.0000041848.57020.57; PMID: 15524167
- Kon M, Cuervo AM. Chaperone-mediated autophagy in health and disease. FEBS Lett 2010; 584:1399 - 404; http://dx.doi.org/10.1016/j.febslet.2009.12.025; PMID: 20026330
- Dice JF. Chaperone-mediated autophagy. Autophagy 2007; 3:295 - 9; PMID: 17404494
- Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 2006; 103:5805 - 10; http://dx.doi.org/10.1073/pnas.0507436103; PMID: 16585521
- Kaushik S, Massey AC, Mizushima N, Cuervo AM. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 2008; 19:2179 - 92; http://dx.doi.org/10.1091/mbc.E07-11-1155; PMID: 18337468
- Cuervo AM, Knecht E, Terlecky SR, Dice JF. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol 1995; 269:C1200 - 8; PMID: 7491910
- Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 2004; 15:4829 - 40; http://dx.doi.org/10.1091/mbc.E04-06-0477; PMID: 15331765
- Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 2005; 1:131 - 40; http://dx.doi.org/10.4161/auto.1.3.2017; PMID: 16874025
- Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989; 246:382 - 5; http://dx.doi.org/10.1126/science.2799391; PMID: 2799391
- Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 2008; 28:5747 - 63; http://dx.doi.org/10.1128/MCB.02070-07; PMID: 18644871
- Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, et al. Chaperone-mediated autophagy at a glance. J Cell Sci 2011; 124:495 - 9; http://dx.doi.org/10.1242/jcs.073874; PMID: 21282471
- Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem 2000; 275:27447 - 56; PMID: 10862611
- Wing SS, Chiang HL, Goldberg AL, Dice JF. Proteins containing peptide sequences related to Lys-Phe-Glu-Arg-Gln are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem J 1991; 275:165 - 9; PMID: 2018472
- Cuervo AM, Terlecky SR, Dice JF, Knecht E. Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. J Biol Chem 1994; 269:26374 - 80; PMID: 7929357
- Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, et al. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009; 323:124 - 7; http://dx.doi.org/10.1126/science.1166088; PMID: 19119233
- Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of α-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 2004; 24:1888 - 96; http://dx.doi.org/10.1523/JNEUROSCI.3809-03.2004; PMID: 14985429
- Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 2011; 42:719 - 30; http://dx.doi.org/10.1016/j.molcel.2011.04.025; PMID: 21700219
- Cuervo AM, Dice JF. Regulation of lamp2a levels in the lysosomal membrane. Traffic 2000; 1:570 - 83; http://dx.doi.org/10.1034/j.1600-0854.2000.010707.x; PMID: 11208145
- Sooparb S, Price SR, Shaoguang J, Franch HA. Suppression of chaperone-mediated autophagy in the renal cortex during acute diabetes mellitus. Kidney Int 2004; 65:2135 - 44; http://dx.doi.org/10.1111/j.1523-1755.2004.00639.x; PMID: 15149326
- Han K, Zhou H, Pfeifer U. Inhibition and restimulation by insulin of cellular autophagy in distal tubular cells of the kidney in early diabetic rats. Kidney Blood Press Res 1997; 20:258 - 63; http://dx.doi.org/10.1159/000174155; PMID: 9398032
- Cuervo AM, Hildebrand H, Bomhard EM, Dice JF. Direct lysosomal uptake of α 2-microglobulin contributes to chemically induced nephropathy. Kidney Int 1999; 55:529 - 45; http://dx.doi.org/10.1046/j.1523-1755.1999.00268.x; PMID: 9987077
- Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol 2003; 14:2199 - 210; http://dx.doi.org/10.1097/01.ASN.0000079785.13922.F6; PMID: 12874476
- Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest 2004; 114:5 - 14; PMID: 15232604
- Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol 2011; 7:189 - 200; http://dx.doi.org/10.1038/nrneph.2011.16; PMID: 21364518
- Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol 2006; 17:1503 - 20; http://dx.doi.org/10.1681/ASN.2006010017; PMID: 16707563
- American Society of Nephrology. American Society of Nephrology Renal Research Report. J Am Soc Nephrol 2005; 16:1886 - 903; http://dx.doi.org/10.1681/ASN.2005030285; PMID: 15888557
- Xue JL, Daniels F, Star RA, Kimmel PL, Eggers PW, Molitoris BA, et al. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol 2006; 17:1135 - 42; http://dx.doi.org/10.1681/ASN.2005060668; PMID: 16495381
- Waikar SS, Curhan GC, Wald R, McCarthy EP, Chertow GM. Declining mortality in patients with acute renal failure, 1988 to 2002. J Am Soc Nephrol 2006; 17:1143 - 50; http://dx.doi.org/10.1681/ASN.2005091017; PMID: 16495376
- Vaidya VS, Ferguson MA, Bonventre JV. Biomarkers of acute kidney injury. Annu Rev Pharmacol Toxicol 2008; 48:463 - 93; http://dx.doi.org/10.1146/annurev.pharmtox.48.113006.094615; PMID: 17937594
- Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 2008; 73:994 - 1007; http://dx.doi.org/10.1038/sj.ki.5002786; PMID: 18272962
- Lieberthal W, Nigam SK. Acute renal failure. I. Relative importance of proximal vs. distal tubular injury. Am J Physiol 1998; 275:F623 - 31; PMID: 9815122
- Lieberthal W, Nigam SK. Acute renal failure. II. Experimental models of acute renal failure: imperfect but indispensable. Am J Physiol Renal Physiol 2000; 278:F1 - 12; PMID: 10644651
- Chien CT, Shyue SK, Lai MK. Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation 2007; 84:1183 - 90; http://dx.doi.org/10.1097/01.tp.0000287334.38933.e3; PMID: 17998875
- Wu HH, Hsiao TY, Chien CT, Lai MK. Ischemic conditioning by short periods of reperfusion attenuates renal ischemia/reperfusion induced apoptosis and autophagy in the rat. J Biomed Sci 2009; 16:19; http://dx.doi.org/10.1186/1423-0127-16-19; PMID: 19272187
- Suzuki C, Isaka Y, Takabatake Y, Tanaka H, Koike M, Shibata M, et al. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun 2008; 368:100 - 6; http://dx.doi.org/10.1016/j.bbrc.2008.01.059; PMID: 18222169
- Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol 2010; 176:1181 - 92; http://dx.doi.org/10.2353/ajpath.2010.090594; PMID: 20075199
- Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem 2006; 281:29776 - 87; http://dx.doi.org/10.1074/jbc.M603783200; PMID: 16882669
- Inoue K, Kuwana H, Shimamura Y, Ogata K, Taniguchi Y, Kagawa T, et al. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin Exp Nephrol 2010; 14:112 - 22; http://dx.doi.org/10.1007/s10157-009-0254-7; PMID: 20013139
- Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 2011; 22:902 - 13; http://dx.doi.org/10.1681/ASN.2010070705; PMID: 21493778
- Liu S, Hartleben B, Kretz O, Wiech T, Igarashi T, Mizushima N, et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012; 8:826 - 37; http://dx.doi.org/10.4161/auto.19419; PMID: 22617445
- Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 2008; 74:631 - 40; http://dx.doi.org/10.1038/ki.2008.214; PMID: 18509315
- Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol 2008; 294:F777 - 87; http://dx.doi.org/10.1152/ajprenal.00590.2007; PMID: 18256309
- Pallet N, Bouvier N, Legendre C, Gilleron J, Codogno P, Beaune P, et al. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy 2008; 4:783 - 91; PMID: 18628650
- Chargui A, Zekri S, Jacquillet G, Rubera I, Ilie M, Belaid A, et al. Cadmium-induced autophagy in rat kidney: an early biomarker of subtoxic exposure. Toxicol Sci 2011; 121:31 - 42; http://dx.doi.org/10.1093/toxsci/kfr031; PMID: 21325019
- Kimura A, Ishida Y, Wada T, Hisaoka T, Morikawa Y, Sugaya T, et al. The absence of interleukin-6 enhanced arsenite-induced renal injury by promoting autophagy of tubular epithelial cells with aberrant extracellular signal-regulated kinase activation. Am J Pathol 2010; 176:40 - 50; http://dx.doi.org/10.2353/ajpath.2010.090146; PMID: 20008137
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124 - 31; http://dx.doi.org/10.1016/j.ceb.2009.11.014; PMID: 20034776
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res 2010; 20:748 - 62; http://dx.doi.org/10.1038/cr.2010.82; PMID: 20548331
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280 - 93; http://dx.doi.org/10.1016/j.molcel.2010.09.023; PMID: 20965422
- Bolisetty S, Traylor AM, Kim J, Joseph R, Ricart K, Landar A, et al. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J Am Soc Nephrol 2010; 21:1702 - 12; http://dx.doi.org/10.1681/ASN.2010030238; PMID: 20705711
- Høyer-Hansen M, Jäättelä M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 2007; 14:1576 - 82; http://dx.doi.org/10.1038/sj.cdd.4402200; PMID: 17612585
- Gozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H, et al. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ 2008; 15:1875 - 86; http://dx.doi.org/10.1038/cdd.2008.121; PMID: 18806755
- Kawakami T, Inagi R, Takano H, Sato S, Ingelfinger JR, Fujita T, et al. Endoplasmic reticulum stress induces autophagy in renal proximal tubular cells. Nephrol Dial Transplant 2009; 24:2665 - 72; http://dx.doi.org/10.1093/ndt/gfp215; PMID: 19454529
- Kitamura M. Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am J Physiol Renal Physiol 2008; 295:F323 - 34; http://dx.doi.org/10.1152/ajprenal.00050.2008; PMID: 18367660
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 2005; 102:8204 - 9; http://dx.doi.org/10.1073/pnas.0502857102; PMID: 15928081
- Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126:121 - 34; http://dx.doi.org/10.1016/j.cell.2006.05.034; PMID: 16839881
- Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 2009; 28:3015 - 26; http://dx.doi.org/10.1038/emboj.2009.242; PMID: 19713938
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10:676 - 87; http://dx.doi.org/10.1038/ncb1730; PMID: 18454141
- Isaka Y, Suzuki C, Abe T, Okumi M, Ichimaru N, Imamura R, et al. Bcl-2 protects tubular epithelial cells from ischemia/reperfusion injury by dual mechanisms. Transplant Proc 2009; 41:52 - 4; http://dx.doi.org/10.1016/j.transproceed.2008.10.026; PMID: 19249473
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927 - 39; http://dx.doi.org/10.1016/j.cell.2005.07.002; PMID: 16179260
- Maiuri MC, Le Toumelin G, Criollo A, Rain J-C, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007; 26:2527 - 39; http://dx.doi.org/10.1038/sj.emboj.7601689; PMID: 17446862
- Chang NC, Nguyen M, Germain M, Shore GC. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J 2010; 29:606 - 18; http://dx.doi.org/10.1038/emboj.2009.369; PMID: 20010695
- Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 2007; 25:193 - 205; http://dx.doi.org/10.1016/j.molcel.2006.12.009; PMID: 17244528
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22:132 - 9; http://dx.doi.org/10.1016/j.ceb.2009.12.004; PMID: 20056399
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456 - 61; http://dx.doi.org/10.1126/science.1196371; PMID: 21205641
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132 - 41; http://dx.doi.org/10.1038/ncb2152; PMID: 21258367
- Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One 2010; 5:e15394; http://dx.doi.org/10.1371/journal.pone.0015394; PMID: 21072212
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A 2011; 108:4788 - 93; http://dx.doi.org/10.1073/pnas.1100844108; PMID: 21383122
- Löffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, et al. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011; 7:696 - 706; http://dx.doi.org/10.4161/auto.7.7.15451; PMID: 21460634
- Dunlop EA, Hunt DK, Acosta-Jaquez HA, Fingar DC, Tee AR. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy 2011; 7:737 - 47; http://dx.doi.org/10.4161/auto.7.7.15491; PMID: 21460630
- Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ 2011; 18:1598 - 607; http://dx.doi.org/10.1038/cdd.2011.33; PMID: 21475306
- Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol 2010; 191:155 - 68; http://dx.doi.org/10.1083/jcb.201002100; PMID: 20921139
- Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, et al. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol 2012; 180:517 - 25; http://dx.doi.org/10.1016/j.ajpath.2011.11.001; PMID: 22265049
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741 - 52; http://dx.doi.org/10.1038/nrm2239; PMID: 17717517
- Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol 2009; 11:1355 - 62; http://dx.doi.org/10.1038/ncb1980; PMID: 19838173
- Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy 2010; 6:891 - 900; http://dx.doi.org/10.4161/auto.6.7.13038; PMID: 20724831
- Zhou X, Zhou J, Li X, Guo C, Fang T, Chen Z. GSK-3β inhibitors suppressed neuroinflammation in rat cortex by activating autophagy in ischemic brain injury. Biochem Biophys Res Commun 2011; 411:271 - 5; http://dx.doi.org/10.1016/j.bbrc.2011.06.117; PMID: 21723251
- Fujishima Y, Nishiumi S, Masuda A, Inoue J, Nguyen NM, Irino Y, et al. Autophagy in the intestinal epithelium reduces endotoxin-induced inflammatory responses by inhibiting NF-κB activation. Arch Biochem Biophys 2011; 506:223 - 35; http://dx.doi.org/10.1016/j.abb.2010.12.009; PMID: 21156154
- Scarlatti F, Granata R, Meijer AJ, Codogno P. Does autophagy have a license to kill mammalian cells?. Cell Death Differ 2009; 16:12 - 20; http://dx.doi.org/10.1038/cdd.2008.101; PMID: 18600232
- Chen Y, Klionsky DJ. The regulation of autophagy—unanswered questions. J Cell Sci 2011; 124:161 - 70; http://dx.doi.org/10.1242/jcs.064576; PMID: 21187343
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004 - 10; http://dx.doi.org/10.1038/nrm2529; PMID: 18971948
- Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ 2009; 16:966 - 75; http://dx.doi.org/10.1038/cdd.2009.33; PMID: 19325568
- Belibi F, Zafar I, Ravichandran K, Segvic AB, Jani A, Ljubanovic DG, et al. Hypoxia-inducible factor-1α (HIF-1α) and autophagy in polycystic kidney disease (PKD). Am J Physiol Renal Physiol 2011; 300:F1235 - 43; http://dx.doi.org/10.1152/ajprenal.00348.2010; PMID: 21270095
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Edelstein CL. What is the role of tubular epithelial cell apoptosis in polycystic kidney disease (PKD)?. Cell Cycle 2005; 4:1550 - 4; http://dx.doi.org/10.4161/cc.4.11.2185; PMID: 16258272
- Tao Y, Kim J, Faubel S, Wu JC, Falk SA, Schrier RW, et al. Caspase inhibition reduces tubular apoptosis and proliferation and slows disease progression in polycystic kidney disease. Proc Natl Acad Sci U S A 2005; 102:6954 - 9; http://dx.doi.org/10.1073/pnas.0408518102; PMID: 15863619
- Tao Y, Zafar I, Kim J, Schrier RW, Edelstein CL. Caspase-3 gene deletion prolongs survival in polycystic kidney disease. J Am Soc Nephrol 2008; 19:749 - 55; http://dx.doi.org/10.1681/ASN.2006121378; PMID: 18272845
- Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 1993; 75:229 - 40; http://dx.doi.org/10.1016/0092-8674(93)80065-M; PMID: 8402909
- Germain M, Slack RS. Dining in with BCL-2: new guests at the autophagy table. Clin Sci (Lond) 2010; 118:173 - 81; http://dx.doi.org/10.1042/CS20090310; PMID: 19845510
- Saeki K, Yuo A, Okuma E, Yazaki Y, Susin SA, Kroemer G, et al. Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ 2000; 7:1263 - 9; http://dx.doi.org/10.1038/sj.cdd.4400759; PMID: 11175264
- Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ 2005; 12:Suppl 2 1528 - 34; http://dx.doi.org/10.1038/sj.cdd.4401777; PMID: 16247500
- Lin HH, Yang TP, Jiang ST, Yang HY, Tang MJ. Bcl-2 overexpression prevents apoptosis-induced Madin-Darby canine kidney simple epithelial cyst formation. Kidney Int 1999; 55:168 - 78; http://dx.doi.org/10.1046/j.1523-1755.1999.00249.x; PMID: 9893125
- Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature 2006; 444:949 - 52; http://dx.doi.org/10.1038/nature05348; PMID: 17122773
- Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J 2011; 278:403 - 13; http://dx.doi.org/10.1111/j.1742-4658.2010.07965.x; PMID: 21182587
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004; 304:1500 - 2; http://dx.doi.org/10.1126/science.1096645; PMID: 15131264
- Giansanti V, Torriglia A, Scovassi AI. Conversation between apoptosis and autophagy: “Is it your turn or mine?”. Apoptosis 2011; 16:321 - 33; http://dx.doi.org/10.1007/s10495-011-0589-x; PMID: 21404107
- Kaushal GP, Kaushal V, Herzog C, Yang C. Autophagy delays apoptosis in renal tubular epithelial cells in cisplatin cytotoxicity. Autophagy 2008; 4:710 - 2; PMID: 18497570
- Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med 2009; 361:1570 - 83; http://dx.doi.org/10.1056/NEJMra0901217; PMID: 19828534
- Belibi FA, Edelstein CL. Novel targets for the treatment of autosomal dominant polycystic kidney disease. Expert Opin Investig Drugs 2010; 19:315 - 28; http://dx.doi.org/10.1517/13543781003588491; PMID: 20141351
- Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 2006; 103:5466 - 71; http://dx.doi.org/10.1073/pnas.0509694103; PMID: 16567633
- Weimbs T. Regulation of mTOR by polycystin-1: is polycystic kidney disease a case of futile repair?. Cell Cycle 2006; 5:2425 - 9; http://dx.doi.org/10.4161/cc.5.21.3408; PMID: 17102641
- Wu M, Wahl PR, Le Hir M, Wackerle-Men Y, Wuthrich RP, Serra AL. Everolimus retards cyst growth and preserves kidney function in a rodent model for polycystic kidney disease. Kidney Blood Press Res 2007; 30:253 - 9; http://dx.doi.org/10.1159/000104818; PMID: 17596700
- Tao Y, Kim J, Schrier RW, Edelstein CL. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol 2005; 16:46 - 51; http://dx.doi.org/10.1681/ASN.2004080660; PMID: 15563559
- Zafar I, Ravichandran K, Belibi FA, Doctor RB, Edelstein CL. Sirolimus attenuates disease progression in an orthologous mouse model of human autosomal dominant polycystic kidney disease. Kidney Int 2010; 78:754 - 61; http://dx.doi.org/10.1038/ki.2010.250; PMID: 20686448
- Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol 2010; 21:489 - 97; http://dx.doi.org/10.1681/ASN.2009040421; PMID: 20075061
- Korolchuk VI, Rubinsztein DC. Regulation of autophagy by lysosomal positioning. Autophagy 2011; 7:927 - 8; http://dx.doi.org/10.4161/auto.7.8.15862; PMID: 21521941
- Walz G, Budde K, Mannaa M, Nürnberger J, Wanner C, Sommerer C, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363:830 - 40; http://dx.doi.org/10.1056/NEJMoa1003491; PMID: 20581392
- Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363:820 - 9; http://dx.doi.org/10.1056/NEJMoa0907419; PMID: 20581391
- Natoli TA, Smith LA, Rogers KA, Wang B, Komarnitsky S, Budman Y, et al. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat Med 2010; 16:788 - 92; http://dx.doi.org/10.1038/nm.2171; PMID: 20562878
- Belibi F, Ravichandran K, Zafar I, He Z, Edelstein CL. mTORC1/2 and rapamycin in female Han:SPRD rats with polycystic kidney disease. Am J Physiol Renal Physiol 2011; 300:F236 - 44; http://dx.doi.org/10.1152/ajprenal.00129.2010; PMID: 20943770
- Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 2010; 70:288 - 98; http://dx.doi.org/10.1158/0008-5472.CAN-09-1751; PMID: 20028854
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res 2007; 17:839 - 49; http://dx.doi.org/10.1038/cr.2007.78; PMID: 17893711
- Ecder T, Melnikov VY, Stanley M, Korular D, Lucia MS, Schrier RW, et al. Caspases, Bcl-2 proteins and apoptosis in autosomal-dominant polycystic kidney disease. Kidney Int 2002; 61:1220 - 30; http://dx.doi.org/10.1046/j.1523-1755.2002.00250.x; PMID: 11918728
- Takakura A, Nelson EA, Haque N, Humphreys BD, Zandi-Nejad K, Frank DA, et al. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum Mol Genet 2011; 20:4143 - 54; http://dx.doi.org/10.1093/hmg/ddr338; PMID: 21821671
- Yu W, Kong T, Beaudry S, Tran M, Negoro H, Yanamadala V, et al. Polycystin-1 protein level determines activity of the Galpha12/JNK apoptosis pathway. J Biol Chem 2010; 285:10243 - 51; http://dx.doi.org/10.1074/jbc.M109.070821; PMID: 20106977
- Høyer-Hansen M, Jäättelä M. AMP-activated protein kinase: a universal regulator of autophagy?. Autophagy 2007; 3:381 - 3; PMID: 17457036
- Grimaldi C, Chiarini F, Tabellini G, Ricci F, Tazzari PL, Battistelli M, et al. AMP-dependent kinase/mammalian target of rapamycin complex 1 signaling in T-cell acute lymphoblastic leukemia: therapeutic implications. Leukemia 2011; PMID: 21968881
- Xie Z, He C, Zou MH. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy 2011; 7:1254 - 5; http://dx.doi.org/10.4161/auto.7.10.16740; PMID: 21685727
- Takiar V, Nishio S, Seo-Mayer P, King JD Jr., Li H, Zhang L, et al. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci U S A 2011; 108:2462 - 7; http://dx.doi.org/10.1073/pnas.1011498108; PMID: 21262823
- Parkhitko A, Myachina F, Morrison TA, Hindi KM, Auricchio N, Karbowniczek M, et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci U S A 2011; 108:12455 - 60; http://dx.doi.org/10.1073/pnas.1104361108; PMID: 21746920
- Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell 2008; 14:90 - 102; http://dx.doi.org/10.1016/j.ccr.2008.06.004; PMID: 18598947
- El-Zoghby ZM, Stegall MD, Lager DJ, Kremers WK, Amer H, Gloor JM, et al. Identifying specific causes of kidney allograft loss. Am J Transplant 2009; 9:527 - 35; http://dx.doi.org/10.1111/j.1600-6143.2008.02519.x; PMID: 19191769
- Nankivell BJ, Borrows RJ, Fung CL, O’Connell PJ, Allen RD, Chapman JR. The natural history of chronic allograft nephropathy. N Engl J Med 2003; 349:2326 - 33; http://dx.doi.org/10.1056/NEJMoa020009; PMID: 14668458
- Nankivell BJ, Chapman JR. Chronic allograft nephropathy: current concepts and future directions. Transplantation 2006; 81:643 - 54; http://dx.doi.org/10.1097/01.tp.0000190423.82154.01; PMID: 16534463
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 2010; 40:294 - 309; http://dx.doi.org/10.1016/j.molcel.2010.09.022; PMID: 20965423
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323 - 35; http://dx.doi.org/10.1038/nature09782; PMID: 21248839
- Matzinger P. The danger model: a renewed sense of self. Science 2002; 296:301 - 5; http://dx.doi.org/10.1126/science.1071059; PMID: 11951032
- Pallet N, Bouvier N, Bendjallabah A, Rabant M, Flinois JP, Hertig A, et al. Cyclosporine-induced endoplasmic reticulum stress triggers tubular phenotypic changes and death. Am J Transplant 2008; 8:2283 - 96; http://dx.doi.org/10.1111/j.1600-6143.2008.02396.x; PMID: 18785955
- Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci U S A 2011; 108:10562 - 7; http://dx.doi.org/10.1073/pnas.1107559108; PMID: 21670280
- Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol 2006; 8:1053 - 63; http://dx.doi.org/10.1038/ncb1471; PMID: 16964246
- Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int 2008; 74:867 - 72; http://dx.doi.org/10.1038/ki.2008.350; PMID: 18633339
- Perico N, Cattaneo D, Sayegh MH, Remuzzi G. Delayed graft function in kidney transplantation. Lancet 2004; 364:1814 - 27; http://dx.doi.org/10.1016/S0140-6736(04)17406-0; PMID: 15541456
- Turkmen K, Martin J, Akcay A, Nguyen Q, Ravichandran K, Faubel S, et al. Apoptosis and autophagy in cold preservation ischemia. Transplantation 2011; 91:1192 - 7; http://dx.doi.org/10.1097/TP.0b013e31821ab9c8; PMID: 21577181
- Zhu C, Wang X, Xu F, Bahr BA, Shibata M, Uchiyama Y, et al. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ 2005; 12:162 - 76; http://dx.doi.org/10.1038/sj.cdd.4401545; PMID: 15592434
- Gotoh K, Lu Z, Morita M, Shibata M, Koike M, Waguri S, et al. Participation of autophagy in the initiation of graft dysfunction after rat liver transplantation. Autophagy 2009; 5:351 - 60; http://dx.doi.org/10.4161/auto.5.3.7650; PMID: 19158494
- Lu Z, Dono K, Gotoh K, Shibata M, Koike M, Marubashi S, et al. Participation of autophagy in the degeneration process of rat hepatocytes after transplantation following prolonged cold preservation. Arch Histol Cytol 2005; 68:71 - 80; http://dx.doi.org/10.1679/aohc.68.71; PMID: 15827380
- Pallet N, Thervet E, Legendre C, Anglicheau D. Sirolimus early graft nephrotoxicity: clinical and experimental data. Curr Drug Saf 2006; 1:179 - 87; http://dx.doi.org/10.2174/157488606776930580; PMID: 18690929
- Thaunat O, Beaumont C, Chatenoud L, Lechaton S, Mamzer-Bruneel MF, Varet B, et al. Anemia after late introduction of sirolimus may correlate with biochemical evidence of a chronic inflammatory state. Transplantation 2005; 80:1212 - 9; http://dx.doi.org/10.1097/01.tp.0000179106.07382.6a; PMID: 16314788
- Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity 2010; 33:301 - 11; http://dx.doi.org/10.1016/j.immuni.2010.09.002; PMID: 20870173
- Säemann MD, Haidinger M, Hecking M, Hörl WH, Weichhart T. The multifunctional role of mTOR in innate immunity: implications for transplant immunity. Am J Transplant 2009; 9:2655 - 61; http://dx.doi.org/10.1111/j.1600-6143.2009.02832.x; PMID: 19788500
- Beljanski V, Knaak C, Smith CD. A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J Pharmacol Exp Ther 2010; 333:454 - 64; http://dx.doi.org/10.1124/jpet.109.163337; PMID: 20179157
- Chaigne-Delalande B, Guidicelli G, Couzi L, Merville P, Mahfouf W, Bouchet S, et al. The immunosuppressor mycophenolic acid kills activated lymphocytes by inducing a nonclassical actin-dependent necrotic signal. J Immunol 2008; 181:7630 - 8; PMID: 19017951
- Simons M, Hartleben B, Huber TB. Podocyte polarity signalling. Curr Opin Nephrol Hypertens 2009; 18:324 - 30; http://dx.doi.org/10.1097/MNH.0b013e32832e316d; PMID: 19542980
- Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 2007; 71:1205 - 14; http://dx.doi.org/10.1038/sj.ki.5002222; PMID: 17410103
- Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 2005; 16:2941 - 52; http://dx.doi.org/10.1681/ASN.2005010055; PMID: 16107576
- Pavenstädt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev 2003; 83:253 - 307; PMID: 12506131
- Sato S, Adachi A, Sasaki Y, Dai W. Autophagy by podocytes in renal biopsy specimens. J Nihon Med Sch 2006; 73:52 - 3; http://dx.doi.org/10.1272/jnms.73.52; PMID: 16641527
- Sato S, Yanagihara T, Ghazizadeh M, Ishizaki M, Adachi A, Sasaki Y, et al. Correlation of autophagy type in podocytes with histopathological diagnosis of IgA nephropathy. Pathobiology 2009; 76:221 - 6; http://dx.doi.org/10.1159/000228897; PMID: 19816081
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383 - 435; http://dx.doi.org/10.1152/physrev.00030.2009; PMID: 20959619
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007; 13:619 - 24; http://dx.doi.org/10.1038/nm1574; PMID: 17450150
- Anderson S, Brenner BM. The aging kidney: structure, function, mechanisms, and therapeutic implications. J Am Geriatr Soc 1987; 35:590 - 3; PMID: 3553292
- Floege J, Hackmann B, Kliem V, Kriz W, Alpers CE, Johnson RJ, et al. Age-related glomerulosclerosis and interstitial fibrosis in Milan normotensive rats: a podocyte disease. Kidney Int 1997; 51:230 - 43; http://dx.doi.org/10.1038/ki.1997.28; PMID: 8995738
- Kaplan C, Pasternack B, Shah H, Gallo G. Age-related incidence of sclerotic glomeruli in human kidneys. Am J Pathol 1975; 80:227 - 34; PMID: 51591
- McCray BA, Taylor JP. The role of autophagy in age-related neurodegeneration. Neurosignals 2008; 16:75 - 84; http://dx.doi.org/10.1159/000109761; PMID: 18097162
- Riediger F, Quack I, Qadri F, Hartleben B, Park JK, Potthoff SA, et al. Prorenin receptor is essential for podocyte autophagy and survival. J Am Soc Nephrol 2011; 22:2193 - 202; http://dx.doi.org/10.1681/ASN.2011020200; PMID: 22034640
- Fischer EG, Moore MJ, Lager DJ. Fabry disease: a morphologic study of 11 cases. Mod Pathol 2006; 19:1295 - 301; http://dx.doi.org/10.1038/modpathol.3800634; PMID: 16799480
- Kobayashi H, Takahashi-Fujigasaki J, Fukuda T, Sakurai K, Shimada Y, Nomura K, et al. Pathology of the first autopsy case diagnosed as mucolipidosis type III α/β suggesting autophagic dysfunction. Mol Genet Metab 2011; 102:170 - 5; http://dx.doi.org/10.1016/j.ymgme.2010.09.014; PMID: 21051253
- Servais A, Morinière V, Grünfeld JP, Noël LH, Goujon JM, Chadefaux-Vekemans B, et al. Late-onset nephropathic cystinosis: clinical presentation, outcome, and genotyping. Clin J Am Soc Nephrol 2008; 3:27 - 35; http://dx.doi.org/10.2215/CJN.01740407; PMID: 18178779
- Kashtan CE, Nevins TE, Posalaky Z, Vernier RL, Fish AJ. Proteinuria in a child with sialidosis: case report and histological studies. Pediatr Nephrol 1989; 3:166 - 74; http://dx.doi.org/10.1007/BF00852901; PMID: 2701867
- Santoro D, Rosenbloom BE, Cohen AH. Gaucher disease with nephrotic syndrome: response to enzyme replacement therapy. Am J Kidney Dis 2002; 40:E4; http://dx.doi.org/10.1053/ajkd.2002.33935; PMID: 12087590
- Schiffmann R. Fabry disease. Pharmacol Ther 2009; 122:65 - 77; http://dx.doi.org/10.1016/j.pharmthera.2009.01.003; PMID: 19318041
- Chévrier M, Brakch N, Céline L, Genty D, Ramdani Y, Moll S, et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010; 6:589 - 99; http://dx.doi.org/10.4161/auto.6.5.11943; PMID: 20431343
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517 - 27; http://dx.doi.org/10.1016/j.molcel.2009.01.021; PMID: 19250912
- Sever S, Altintas MM, Nankoe SR, Möller CC, Ko D, Wei C, et al. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest 2007; 117:2095 - 104; http://dx.doi.org/10.1172/JCI32022; PMID: 17671649
- Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 2008; 14:931 - 8; http://dx.doi.org/10.1038/nm.1857; PMID: 18724379
- Yaddanapudi S, Altintas MM, Kistler AD, Fernandez I, Möller CC, Wei C, et al. CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J Clin Invest 2011; 121:3965 - 80; http://dx.doi.org/10.1172/JCI58552; PMID: 21911934
- Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329:841 - 5; http://dx.doi.org/10.1126/science.1193032; PMID: 20647424
- Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem 2008; 283:21540 - 9; http://dx.doi.org/10.1074/jbc.M800214200; PMID: 18505729
- Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet 2008; 24:604 - 12; http://dx.doi.org/10.1016/j.tig.2008.10.002; PMID: 18992957
- Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem 2000; 275:31505 - 13; http://dx.doi.org/10.1074/jbc.M002102200; PMID: 10806201
- Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, et al. Prevalence of chronic kidney disease in the United States. JAMA 2007; 298:2038 - 47; http://dx.doi.org/10.1001/jama.298.17.2038; PMID: 17986697
- Collins AJ, Foley RN, Herzog C, Chavers B, Gilbertson D, Ishani A, et al. US Renal Data System 2010 Annual Data Report. Am J Kidney Dis 2011; 57:Suppl 1 A8; http://dx.doi.org/10.1053/j.ajkd.2010.10.007; PMID: 21184928
- Chronopoulos A, Cruz DN, Ronco C. Hospital-acquired acute kidney injury in the elderly. Nat Rev Nephrol 2010; 6:141 - 9; http://dx.doi.org/10.1038/nrneph.2009.234; PMID: 20125094
- Imai E, Horio M, Watanabe T, Iseki K, Yamagata K, Hara S, et al. Prevalence of chronic kidney disease in the Japanese general population. Clin Exp Nephrol 2009; 13:621 - 30; http://dx.doi.org/10.1007/s10157-009-0199-x; PMID: 19513802
- Schmitt R, Cantley LG. The impact of aging on kidney repair. Am J Physiol Renal Physiol 2008; 294:F1265 - 72; http://dx.doi.org/10.1152/ajprenal.00543.2007; PMID: 18287400
- Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol 2002; 13:3005 - 15; http://dx.doi.org/10.1097/01.ASN.0000039661.06947.FD; PMID: 12444221
- Christensen EI, Nielsen S. Structural and functional features of protein handling in the kidney proximal tubule. Semin Nephrol 1991; 11:414 - 39; PMID: 1947495
- Kume S, Uzu T, Horiike K, Chin-Kanasaki M, Isshiki K, Araki S, et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest 2010; 120:1043 - 55; http://dx.doi.org/10.1172/JCI41376; PMID: 20335657
- Cui J, Bai X-Y, Shi S, Cui S, Hong Q, Cai G, et al. Age-related changes in the function of autophagy in rat kidneys. Age (Omaha) 2011; 34:329 - 39; http://dx.doi.org/10.1007/s11357-011-9237-1
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009; 325:201 - 4; http://dx.doi.org/10.1126/science.1173635; PMID: 19590001
- Guarente L, Picard F. Calorie restriction--the SIR2 connection. Cell 2005; 120:473 - 82; http://dx.doi.org/10.1016/j.cell.2005.01.029; PMID: 15734680
- McKiernan SH, Tuen VC, Baldwin K, Wanagat J, Djamali A, Aiken JM. Adult-onset calorie restriction delays the accumulation of mitochondrial enzyme abnormalities in aging rat kidney tubular epithelial cells. Am J Physiol Renal Physiol 2007; 292:F1751 - 60; http://dx.doi.org/10.1152/ajprenal.00307.2006; PMID: 17344189
- Wiggins JE, Goyal M, Sanden SK, Wharram BL, Shedden KA, Misek DE, et al. Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: prevention by calorie restriction. J Am Soc Nephrol 2005; 16:2953 - 66; http://dx.doi.org/10.1681/ASN.2005050488; PMID: 16120818
- Mimura I, Nangaku M. The suffocating kidney: tubulointerstitial hypoxia in end-stage renal disease. Nat Rev Nephrol 2010; 6:667 - 78; http://dx.doi.org/10.1038/nrneph.2010.124; PMID: 20877304
- Samokhvalov V, Scott BA, Crowder CM. Autophagy protects against hypoxic injury in C. elegans. Autophagy 2008; 4:1034 - 41; PMID: 18849662
- Hars ES, Qi H, Ryazanov AG, Jin S, Cai L, Hu C, et al. Autophagy regulates ageing in C. elegans. Autophagy 2007; 3:93 - 5; PMID: 17204841
- Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 2007; 3:597 - 9; PMID: 17912023
- Meléndez A, Tallóczy Z, Seaman M, Eskelinen E-L, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003; 301:1387 - 91; http://dx.doi.org/10.1126/science.1087782; PMID: 12958363
- De Meyer GR, De Keulenaer GW, Martinet W. Role of autophagy in heart failure associated with aging. Heart Fail Rev 2010; 15:423 - 30; http://dx.doi.org/10.1007/s10741-010-9166-6; PMID: 20383579
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab 2009; 10:507 - 15; http://dx.doi.org/10.1016/j.cmet.2009.10.008; PMID: 19945408
- Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 2008; 14:959 - 65; http://dx.doi.org/10.1038/nm.1851; PMID: 18690243
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496