Abstract
Activation of hepatic stellate cells (HSC), a resident pericytic cell in liver, into a proliferative and fibrogenic cell type, is the principal event underlying hepatic fibrosis following injury. Release of lipid droplets (LD) containing retinyl esters and triglyceride is a defining feature of HSC activation, yet the basis for this release has remained mysterious. Here we offer a surprising discovery that autophagy is the missing link underlying LD release, by stimulating metabolism of their contents to provide the energy vital to fuel HSC activation. By specifically inhibiting the autophagic pathway in activated HSC, LD release is impaired and cellular ATP levels are decreased. Moreover, animals with HSC-specific deletion of Atg7 display attenuated activation following liver injury, leading to reduced fibrosis in vivo. We further demonstrate that fibrogenic cells from other organs, including kidney and lung, also rely on autophagy as a core pathway driving the scarring response. Our results provide a novel framework for understanding pathways underlying fibrogenic cell responses to tissue injury.
Liver injury of any type provokes a brisk wound healing response that leads to the deposition of extracellular matrix, which, over time, can culminate in cirrhosis. Hepatic stellate cell (HSC), a remarkably protean resident non-parenchymal cell type, are essential for the liver’s response to injury. These pericyte-like star-shaped cells within the subendothelial space of Disse, are normally filled with perinuclear lipid droplets (LD) containing retinyl esters. During injury, however, the cells acquire a highly proliferative, fibrogenic, and contractile phenotype, while losing their LD, leading to the release of free retinol into the extracellular space. The fate of the fatty acid component of the retinyl esters has been mysterious, however.
Based on recent findings, hepatocytes, the resident parenchymal cell in liver, exploit autophagy to digest intracellular lipids to maintain energy homeostasis. The contribution of autophagy to intracellular lipid degradation in hepatocytes led us to hypothesize that autophagic signaling might also drive the release of LD during HSC activation.
Consistent with our hypothesis, HSC autophagy induction was documented during cellular activation in culture and in vivo in mice following either carbon tetrachloride (CCl4)-or thioacetamide (TAA)-induced liver injury, as well as in human HSCs from hepatitis B-infected liver. Autophagy was also upregulated in lung fibroblasts from patients with idiopathic pulmonary fibrosis, compared with cells from normal human lung.
In culture-activated mouse HSCs, pharmacological antagonism of autophagy using either 3-methyladenine or chloroquine leads to reduced expression of critical fibrogenic genes (Col1a1/collagen α1(I) and Col1a2/α2(I), Pdgfrb, Acta2/α-sma and Mmp2) and their corresponding proteins. Using a more specific approach, knockdown of the essential autophagy genes Atg7 or Atg5 using a lentiviral vector expressing small hairpin RNAs, also inhibited fibrogenic gene expression in HSCs.
Based on these findings, we next established that autophagy contributes to HSC activation during liver injury in vivo using a mouse line with HSC-specific deletion of Atg7, which was generated by crossing GFAP-Cre transgenic mice with Atg7loxP/loxP animals (HSCs typically express neural crest markers, including GFAP). Following liver injury due to either CCl4 or TAA, the number of HSCs and liver architecture in these mice were not altered, yet tissue fibrosis was markedly reduced. Consistent with attenuated HSC activation, oil-red-O positive droplets were preserved in the cells in culture and in vivo when autophagy was suppressed.
In order to determine whether engagement of autophagy is a generalized feature of the fibrotic response or is restricted to fibrogenic cells in liver, we examined whether fibrogenic cells from kidney and lung also rely on autophagy. Indeed, inhibition of autophagy in renal mesangial cells and pulmonary fibroblasts also attenuated fibrogenesis, indicating that autophagy is likely to be a core pathway of fibrogenesis common to most, if not all tissues.
We further postulated that in HSCs, and probably in other fibrogenic cells, metabolism of LD by autophagy provides cellular energy critical to fueling the catabolic pathways of cellular activation. As predicted, inhibition of autophagy in HSCs leads to an increase in triglyceride-containing LD, associated with a decrease in total ATP levels, which could be partially rescued by the addition of the free fatty acid oleate. Moreover, inhibition of fatty acid oxidation by etomoxir blocks HSC activation in a manner parallel to that conferred by inhibiting autophagy.
In summary, autophagy provides the energy required for initiating and perpetuating the activated phenotype of hepatic fibrogenic cells (HSCs) following injury (). This new perspective indicates that while in epithelial cells autophagy may be a critical pathway to attenuate injury and maintain short-term homeostasis, its consequences in fibrogenic cells may ultimately worsen long-term disease outcomes by promoting tissue fibrosis. Selectively blocking autophagy in fibrogenic cells therefore emerges as an attractive candidate antifibrotic strategy. Nonetheless, cell-specific inhibition of autophagy in a complex tissue remains a challenging prospect that is currently not possible. The findings also offer yet another indication that autophagy controls an enlarging range of responses critical to cellular and tissue homeostasis.
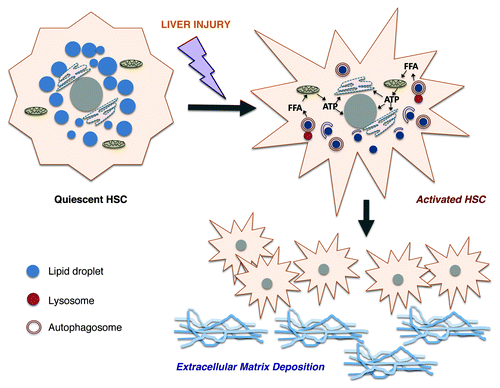
Figure 1. Role of autophagy in hepatic stellate cell (HSC) activation. Upon injury, quiescent HSCs upregulate autophagy, which leads to hydrolysis of retinyl esters within their perinuclear droplets. This process liberates free fatty acids (FFA) that can undergo mitochondrial β-oxidation. As a result, autophagy generates ATP energy to maintain the activated phenotype. Activated HSCs proliferate and produce large quantities of extracellular matrix molecules that contribute to hepatic fibrosis.