Abstract
Macroautophagy was recently shown to regulate both lymphocyte biology and innate immunity. In this study we sought to determine whether a deregulation of autophagy was linked to the development of autoimmunity. Genome-wide association studies have pointed out nucleotide polymorphisms that can be associated with systemic lupus erythematosus, but the potential role of autophagy in the initiation and/or development of this syndrome is still unknown. Here, we provide first clues of macroautophagy deregulation in lupus. By the use of LC3 conversion assays and electron microscopy experiments, we observed that T cells from two distinct lupus-prone mouse models, i.e., MRLlpr/lpr and (NZB/NZW)F1, exhibit high loads of autophagic compartments compared with nonpathologic control CBA/J and BALB/c mice. Unlike normal mice, autophagy increases with age in murine lupus. In vivo lipopolysaccharide stimulation in CBA/J control mice efficiently activates T lymphocytes but fails to upregulate formation of autophagic compartments in these cells. This argues against a deregulation of autophagy in lupus T cells solely resulting from an acute inflammation injury. Autophagic vacuoles quantified by electron microscopy are also found to be significantly more frequent in T cells from lupus patients compared with healthy controls and patients with non-lupus autoimmune diseases. This elevated number of autophagic structures is not distributed homogeneously and appears to be more pronounced in certain T cells. These results suggest that autophagy could regulate the survival of autoreactive T cell during lupus, and could thus lead to design new therapeutic options for lupus.
Introduction
Macroautophagy is a catabolic process characterized by sequestration of cytoplasmic material in double-membrane vacuoles called autophagosomes, that ultimately fuse with lysosomes leading to degradation of their contents.Citation1 The role of this process was first described in yeast as a promoter of cell survival under nutrient starvation. Autophagy was later shown to play numerous vital roles in higher eukaryotes, notably in the immune system. Thus, autophagy was shown to regulate immune cell homeostasis and activation, induction of central tolerance and finally inflammation processes.Citation2
Autophagy in autoimmune and autoinflammatory disorders is suspected to be a key component of their etiology. Genetic polymorphisms on the gene coding for ATG16L1 was reported with strong association for patients suffering from Crohn disease leading to the identification of autophagy as a critical regulator of inflammation in the gut.Citation3,Citation4 In patients suffering from multiple sclerosis, ATG5 protein upregulation was described in T cells infiltrating inflammatory lesions, yet the precise impact of this expression pattern remains to be defined.Citation5 To date, no study investigated the possible involvement of autophagy in systemic lupus erythematosus (SLE). This prototypic systemic autoimmune disease is characterized by abnormal B and T cell activation, multiple organ inflammation, and production of autoantibodies (autoAbs) targeting mainly nuclear components. It was found from genome-wide association studies that two single nucleotide polymorphisms near and in the Atg5 locus are associated with SLE initiation and/or development.Citation6,Citation7 Moreover, drugs modulating autophagy such as hydroxychloroquine,Citation8 rapamycinCitation9 and the P140 peptideCitation10,Citation11 provide beneficial effects on the development of the pathology in lupus-prone mouse models as well as in patients with SLE.Citation12 To date, little information is available regarding the role of autophagic activity in lymphocytes under infectious or autoimmune events. Inflammation, cytokine environment and chronic antigenic stimulation characterizing autoimmune pathologies are eager to modulate autophagy in lymphocytes.
Autophagy was shown to be required for activation of T cells and for their survival after stimulationCitation13 and differentiation.Citation14 This survival seems highly related to quality control and turnover of mitochondria as shown with mouse models characterized by T cell-specific deletion of Atg5 or Atg7.Citation15,Citation16 Moreover, excess of autophagy that is detrimental to T cell survival under IFN-γ stimulation, is tightly regulated by immunity-related GTPase family M proteins in mice.Citation17 Finally autophagic flux was shown to degrade procaspases in T cell, thus antagonizing apoptosis.Citation18 This latter study shows a direct role for autophagy in survival of autoreactive T cells during experimental autoimmune encephalomyelitis. Thus regulation of autophagic activity is integral for T cell to decide between activation and death. Knowing that physiological survival-death equilibrium is deregulated in SLE, where autoreactive T cells are not clonally deleted,Citation19 we hypothesized that autophagy could play a role in the persistence of autoreactive cells.
This study is the first that aims at describing autophagic activity in lupus T cells. Intensity of autophagy was evaluated in central and peripheral lymphoid organs from two distinct lupus-prone mouse models, namely MRLlpr/lpr and (NZB/NZW)F1 (NZB/W) mice. Autophagic activity was also assessed in the human pathology by quantifying autophagic structures in peripheral blood T cells from SLE patients. These results were compared with those obtained in normal mice that received lipopolysaccharide (LPS) to define if autophagy deregulation was a direct consequence of an acute inflammation.
Results
Autophagic flux is increased in thymocytes from lupus-prone mice
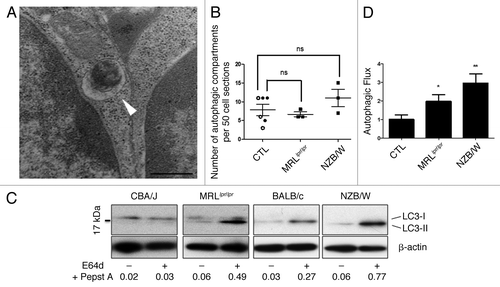
In order to evaluate autophagic activity in central T cells, we quantified autophagic compartments on thymus sections obtained from MRLlpr/lpr and NZB/W lupus-prone mice. Quantification was performed by transmission electron microscopy (TEM) in cells with lymphocyte morphology (diameter < 10 µM, high nuclear/cytoplasm ratio) to exclude other cell types, especially thymic epithelial cells known to exhibit high constitutive autophagic activity. An example of autophagic vacuole is depicted in . Quantification of autophagic compartments on 50 cell sections failed to reveal any significant difference between lupus mice (8 week-MRLlpr/lpr and 12-weeks-NZB/W lupus mice) and CBA/J and BALB/c control mice (). Microtubule-associated protein 1 light chain 3 (LC3) conversion assays were also performed (). No obvious difference in lupus mice vs. controls could be noticed in terms of LC3-II expression in nontreated cells, confirming the results obtained by TEM. However, when thymocytes were treated with inhibitors of lysosomal proteases E64d and pepstatin A, we could observe a significantly higher autophagic flux in MRLlpr/lpr and NZB/W mice compared with controls (). These results suggest that autophagic flux is increased in thymocytes from lupus-prone mice.Citation20
Figure 1. Increased autophagic flux in thymocytes from lupus-prone mice compared with controls (A) A representative autophagosome is indicated by the white arrow (black scale bar: 500 nm). (B) Quantification by TEM of autophagic vacuoles for 50 thymocyte sections randomly selected on thymus sections from 8 week-old CBA/J (open circles) and 12 week-old BALB/c (filled circles) control mice (CTL) and lupus-prone mice MRLlpr/lpr (8 week-old) and NZB/W (12 week-old). Each point represents measurement for an individual mouse. Central bars refer to the mean and vertical bars stand for standard deviation. ns = non-significant using unpaired t-test. (C) LC3 conversion assessed by western immunoblotting. Dissociated thymocytes obtained from 8 week-old control CBA/J and lupus MRLlpr/lpr mice or from 12 week-old control BALB/c and lupus NZB/W mice were cultured at 37°C for 16h. When indicated, cells were treated (+) or not (-) during the last 4 h of the culture with 5 µg/mL pepstatin A and 5 µg/mL E64d to block lysosomal degradation. Cell lysates were resolved by SDS-PAGE, transferred onto PVDF membranes before staining with anti-LC3 Ab. Loading controls were performed by staining actin β-chain. Each immunoblot is representative of three experiments with identical results. LC3-II/β-actin band intensity ratios are indicated as numbers under each immunoblot. (D) LC3-II levels were evaluated by densitometry and normalized to β-actin band intensities for at least three other independent experiments (right panel). Autophagic flux measurement consists on a ratio between the values with and without protease inhibitors (= autophagic flux). Histogram bars represent the means of individual experiments with standard errors. *p < 0.05 and **p < 0.01 using unpaired t-test between control and lupus conditions.
Autophagic activity is deregulated in peripheral T cells from lupus-prone mice
As autophagy is shown to be essential for peripheral T cell homeostasis, we sought to determine whether autophagic activity was deregulated in purified splenic T cells from lupus mice before the appearance of the first symptoms (8–12 week-old MRLlpr/lpr mice and 12–20 week-old NZB/W mice). LC3 conversion assays showed high levels of LC3-II expression in nonstimulated conditions (steady-state) for MRLlpr/lpr compared with CBA/J mice, with or without lysosomal protease inhibitors (). A small increase is also observed for NZB/W mice in comparison to BALB/c mice although statistical significance could not be reached (). Comparison of conditions without and with protease inhibitors under phorbol myristate acetate (PMA)/ionomycin stimulation showed LC3-II accumulation in both lupus and control mice reflecting effective autophagic flux in lupus and normal T cells. As LC3-II levels are proportional to the quantity of autophagic membranes, initial quantity of autophagic structures seemed increased in lupus conditions.
Figure 2. Autophagic activity in peripheral T cells from lupus-prone mice is raised compared with controls T cells sorted from spleens obtained from 8–12 week-old control CBA/J and lupus MRLlpr/lpr mice (A) or from 12–20 week-old control BALB/c and lupus NZB/W mice (B) were left unstimulated (steady-state) or stimulated by 50 ng/mL PMA and 1 µM Ionomycin (PMA/Iono) at 37°C for 16h. When indicated, cells were treated (+) or not (-) during the last 4 h of the culture with 5 µg/mL pepstatin A and 5 µg/mL E64d to block lysosomal degradation. Cell lysates were resolved by SDS-PAGE, transferred onto PVDF membranes before staining with anti-LC3 Ab. Loading controls were performed by staining actin-β chain. Each immunoblot is representative of at least five independent experiments with identical results. *Band corresponding to the Ig heavy chain retained in lysates obtained from oldest lupus mice. LC3-II/β-actin band intensity ratios are indicated as numbers under each immunoblot. (C and D) LC3-II levels were evaluated by densitometry and normalized to β-actin band intensities for at least five independent experiments. Histogram bars represent the means of individual experiments with standard errors. *p < 0.05, **p < 0.01, ***p < 0.001 using paired t-test between control and lupus conditions. Activation of sorted T cells from control and lupus mice was assessed by flow cytometry with CD69 staining (E). Dotted black lines and solid gray lines represent respectively control and lupus mice.
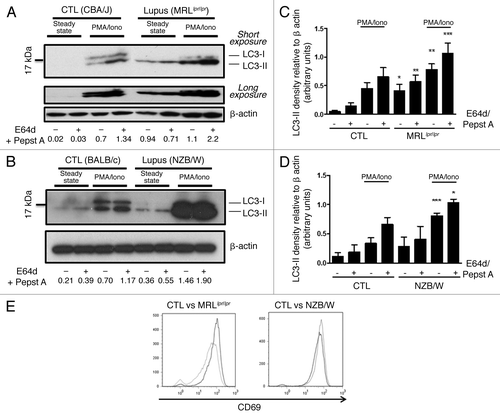
To determine if this difference was effectively related to lupus context or solely the consequence of in vivo activation of T cells in an inflammatory environment, we stimulated T cells in vitro with PMA/ionomycin (). Both in control and lupus T cells, LC3-II levels increased in the presence of protease inhibitors indicating that in both mice, autophagy was induced after T cell receptor (TCR)-like stimulation. It was also noticed that LC3-II expression was significantly higher in lupus-prone mice compared with controls, both with and without lysosomal protease inhibitors (). As accumulation of LC3-II in the presence of E64d and pepstatin A was still observed in lupus conditions, it could be concluded that the elevated LC3-II rates found in these mice are not the consequence of a total autophagic flux blockade. Moreover, LC3-II level rise observed in lupus mice under PMA/Ionomycin stimulation did not result from a higher sensitivity to these compounds because activation of lupus T cells compared with normal mice was at least equivalent as shown by CD69 staining after treatment (). Comparison of MRLlpr/lpr or NZB/W mice with three different control mouse strains (C57BL/6, BALB/c and CBA/J) in a single experiment (same immunoblot), confirms that relative levels of LC3-II are effectively raised in lupus conditions (Fig. S1).
We then checked if the difference shown in autophagic activity between MRLlpr/lpr and control mice was related to the lpr mutation. LC3 conversion assays performed in parallel with C57BL/6 and C57BL/6lpr/lpr T cells showed no significant difference between both strains even at the highest ages tested for comparison of MRLlpr/lpr and CBA/J (Fig. S2). We also performed preliminary experiments with the same settings, using B lymphocytes isolated from spleens collected from control and lupus mice (Fig. S3). In contrast to T cells, no difference of autophagic activity between normal and lupus conditions was observed in peripheral B cells.
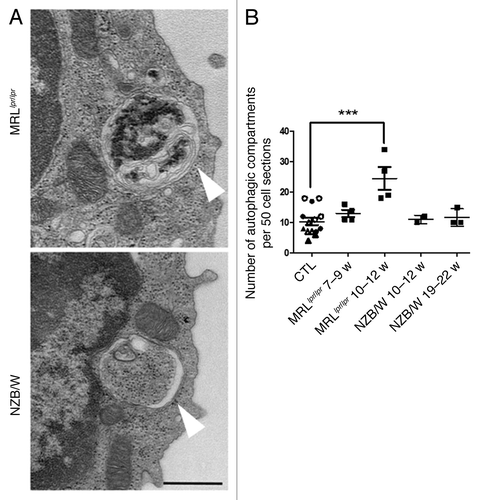
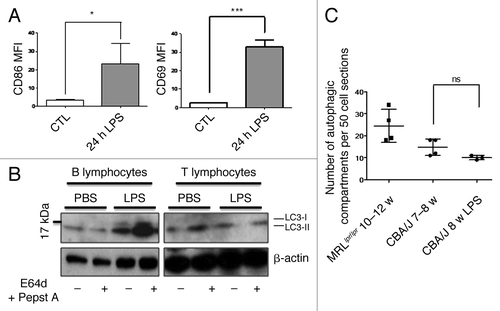
To understand if high levels of LC3-II measured in lupus-prone conditions reflected high numbers of autophagosomal structures, we performed TEM experiments on isolated T cells that were fixed just after isolation. Representative autophagic compartments in T cells sorted from spleens of MRLlpr/lpr and NZB/W mice are shown in . Quantification of autophagic compartments in 50 cell sections revealed a number of autophagic vacuoles that was higher in the oldest MRLlpr/lpr lupus mice tested (i.e., 10 to 12 weeks) compared with pooled controls (CBA/J and BALB/c mice at various ages, ). In contrast, NZB/W mice did not exhibit a significant increase in comparison to pooled controls. These results are reminiscent of LC3 conversion assays showing that differences in autophagic compartment load was mainly observed in NZB/W mice compared with controls under PMA/Ionomycin stimulations. It should be noticed that a strict comparison between T cells from 19–22 week-old NZB/W and control mice with corresponding ages, showed that the number of autophagic compartments was significantly higher in NZB/W mice (Fig. S4).
Figure 3. Increased number of autophagic vacuoles in peripheral T cells isolated from lupus mice compared with control mice. (A) Representative autophagic vacuoles indicated by white arrows, in peripheral T cells from lupus MRLlpr/lpr and NZB/W mice. Scale bar stands for both images (black scale bar: 500 nm). (B) Quantification by TEM of autophagosomes counted in 50 peripheral T lymphocyte sections sorted from spleens of control (7–8 week-old CBA/J mice (open circles), 10–12 week-old CBA/J mice (filled circles), 10–12 week-old BALB/c mice (open triangles) and 20–22 week-old BALB/c mice (filled triangles), MRLlpr/lpr and NZB/W lupus mice. Mice were sacrificed at the indicated ages. Each point represents measurement of an individual mouse. Central bars refer to the mean and vertical bars stand for standard deviation. ***p < 0.001 using unpaired t-test, w = week.
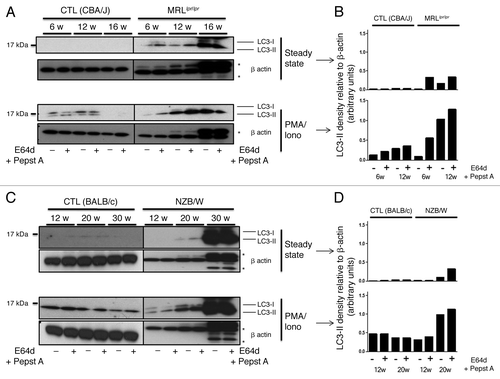
Additional LC3 conversion assays were performed to assess whether autophagic activity deregulation progressively takes place with the age of the mice (). LC3-II detection visualized in the presence or absence of pepstatin A and E64d, effectively increases with age in lupus-prone mice, both at steady-state and under PMA/ionomycin stimulation (as quantified for corresponding blots in ). In contrast, LC3-II conversion does not evolve with age in control mice and was even found to decrease in the oldest mice. LC3-II accumulation observed in MRLlpr/lpr mice was not found in C57BL/6lpr/lpr mice, even at higher ages (Fig. S2) revealing again that the lpr mutation per se is not responsible for autophagic deregulation found in MRLlpr/lpr mice. To understand if LC3-II accumulation was due to a decrease in lysosomal activity, we performed p62/SQSTM1 staining by immunoblots (Fig. S1). These results show that accumulation of p62 is observed in lupus-prone mice at ages where LC3-II levels are significantly higher in comparison to control mice (12 weeks for MRLlpr/lpr and 17 weeks for NZB/W). We can then rule out a total autophagic flux blockade as the only cause of LC3-II accumulation in lupus mice. Moreover, compared with control mice, LC3-II level increase in lupus mice is not related to higher LC3-encoding mRNA levels, as shown by Map1lc3a mRNA semi-quantified by RT-PCR (Fig. S5).
Figure 4. Autophagic activity in peripheral T cells increases with age in lupus-prone mice in contrast to control mice. T cells were sorted from spleens obtained from control (CBA/J) and lupus MRLlpr/lpr mice (A) or control (BALB/c) and lupus NZB/W mice (C) that were sacrificed at the indicated ages indicated in weeks. Cells were left unstimulated (steady-state) or stimulated with 50 ng/mL PMA and 1 µM Ionomycin (PMA/Iono) at 37°C for 16 h. When indicated, cells were treated (+) or not (-) during the last 4 h of culture with 5 µg/mL pepstatin A and 5 µg/mL E64d to block lysosomal degradation. Cell lysates were resolved by SDS-PAGE, transferred onto PVDF membranes before staining with anti-LC3 Ab. Loading controls were performed by staining actin-β chain. Each immunoblot is representative of three experiments with identical results. *Bands corresponding to Ig heavy and light chains retained in lysates obtained from oldest lupus mice. (B and D) LC3-II levels of immunoblots shown in (A and C) were evaluated by densitometry for the indicated ages and normalized to β-actin band intensities.
Autophagic activity is deregulated in peripheral T cells from SLE patients
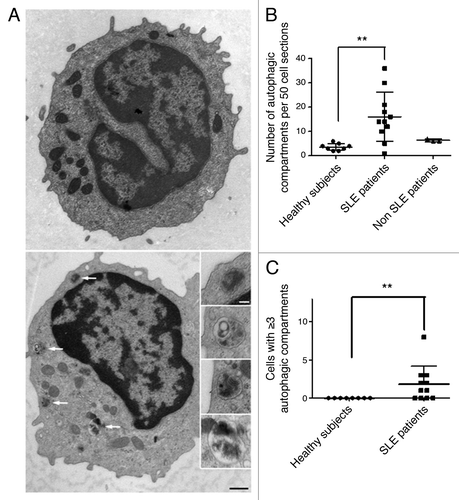
Although pathologies developed by MRLlpr/lpr and NZB/W mice display important similarities with human pathology, these models do not exhibit any quiescent phase and cannot fully compare with the clinical evolution of SLE. We thus decided to investigate if the deregulation of autophagy observed in lupus mice was also found in the human pathology. Lupus patients chosen for the study were mostly quiescent with nine patients out of 11 showing a SLEDAI ≤ 6, and 2 patients with higher SLEDAI (12 and 14, respectively) (). Autophagic vacuoles were counted in T lymphocytes isolated from the peripheral blood of healthy volunteers and SLE patients. One representative cell containing several autophagic compartments is shown in . The number of autophagic vacuoles was significantly higher in T cells from SLE patients compared with healthy volunteers (p = 0.0032, ). Moreover, autophagic activity does not seem to be related to disease activity. Of note, patient #7 (, 14 autophagic vacuoles for 50 lymphocyte sections, SLEDAI = 4) belongs to the same family (daughter) than two controls (father and mother) exhibiting 2 and 3 autophagic vacuoles per 50 T lymphocytes. This suggests that accumulation of autophagic compartments may be correlated with the development of the pathology and is not a common feature of a familial genetic background. We also tested three patients suffering from inflammatory pathologies unrelated to SLE (2 with vasculitis, 1 with primary Sjögren’s syndrome). None of these patients showed autophagic vacuole number as high as found in several SLE patients. Highly vacuolated autophagic T cells were found in lupus patients, a feature that was not visible in controls (p = 0.0086, ). These results led us to conclude that autophagic deregulation observed in lupus-prone mice is also found in T cells from SLE patients.
Table 1. Description of female patients suffering from SLE included in the study
Figure 5. Increased number of autophagic vacuoles in peripheral T cells from SLE patients compared with healthy subjects (A) An example of a cell with a high number of autophagic vacuoles (black scale bar: 500 nm). Magnifications of identified autophagic structures are shown on the right side. The white scale bar (100 nm) stands for all the magnified images (B) Double-blind quantification of autophagic vacuoles in peripheral T cells isolated from the PBMC fraction obtained from healthy donors, patients with SLE or with other autoimmune diseases (Sjögren’s syndrome and vasculitis). Autophagic vacuoles were counted in 50 T lymphocyte sections. (C) Cell sections containing three or more autophagic vacuoles were counted for each subject. Each point represents the result for one subject. Central bars refer to the means and vertical bars stand for standard deviation. **p < 0.01 using unpaired t-test (in B) or Mann-Whitney U test.
The increased number of autophagosomes in peripheral lupus T cells is not an immediate consequence of inflammatory event
Inflammatory conditions are known to stimulate autophagy in several cell types through pattern recognition receptor stimulation or in the presence of particular cytokines such as IFNγ. We then sought to determine if an in vivo stimulation by LPS of CBA/J control mice, known to generate a systemic inflammation, was sufficient to induce the accumulation of autophagosomes as the one observed in lupus T cells. We first checked the level of B and T lymphocyte activation in LPS-treated mice by measuring respectively CD86 and CD69 expression by flow cytometry (). Twenty-four hours after i.p. injection of 50 µg LPS, we noticed remarkable activation of splenic B and T cells. B cells are known to be directly stimulated by LPS through toll-like receptor 4 (TLR4) engagement. Moreover TLR4 is known to trigger autophagy in macrophages.Citation21,Citation22 LC3 conversion assays performed on B cells revealed that in vivo LPS stimulation activated autophagy in terms of basal LC3-II level but also in terms of flux (). In contrast, although activated as shown by CD69 expression, T cells failed to undergo autophagic flux under in vivo LPS stimulation. Moreover, no accumulation of autophagic vacuoles could be observed by TEM experiments (). Thus, short-term inflammatory cytokine environment is not sufficient to lead to the accumulation of autophagic compartments observed in lupus-prone mice.
Figure 6. No autophagic compartment accumulation in normal T cells during acute systemic inflammation induced by LPS. (A) CBA/J mice (8 week-old) were injected i.p. with 50 µg LPS or with PBS (Control). B cell (TCR-β/B220+) and T cell (TCR-β+/B220-) activation was checked by measuring CD86 and CD69 expression respectively (represented as mean fluorescence intensity, MFI) in treated (24h LPS) and nontreated mice (control). *p < 0.05 by paired t-test and ***p < 0.005 by paired t-test. (B) LC3 conversion was assessed for splenic B or T cells isolated 24 h after injection of PBS alone or LPS, and left in cultured at 37°C for 4 h with (+) or without (-) lysosomal proteases inhibitors E64d and pepstatin A (5 µg/mL). Each immunoblot is representative of three experiments with identical results. (C) Quantification by TEM of autophagic vacuoles in peripheral T cells from either 10–12 week-old MRLlpr/lpr lupus mice, control 7–8 week-old CBA/J mice, or LPS-treated mice (CBA/J, 8 week-old LPS). Autophagic vacuoles were counted in 50 peripheral T lymphocyte sections. Each point represents measurement for an individual mouse. Central bars refer to the means and vertical bars stand for standard deviation. ns = nonsignificant using unpaired t-test.
Discussion
This work showed for the first time a clear deregulation of autophagic activity in T cells from two lupus-prone mouse models of distinct haplotypes. Autophagic flux was affected in thymocytes isolated from these two murine strains. Compared with normal mice, a significant accumulation of autophagic vacuoles was observed in peripheral lupus T cells. Interestingly, this higher number of autophagic vacuoles was not detectable in T cells from the thymus, which is involved in central tolerance. These results are in good agreement with previous work indicating that autophagy is more crucial in peripheral T cells homeostasis than during earlier immature stages of T cell development.Citation16 The origin of this phenotype is not known and it would be important to define if the accumulation of autophagic compartments observed in lupus T cells is due to disequilibrium between autophagosome formation and capacity of lysosomal degradation. It is nevertheless possible to conclude that total flux blockade is not the initial consequence of such autophagosome accumulation according to the increase in p62 level in lupus mice under inhibition of lysosomal proteolytic activity. Higher levels of LC3-II in lupus mice do not seem either to be related to de novo transcription of Map1lc3 genes, at least of one isoform. The precise nature of the stimulus leading to increased autophagic activity in lupus conditions is thus not defined. From PMA/Ionomycin stimulation experiments showing that, in lupus mice, autophagic activity is higher under stimulation than in control mice, we can conclude that TCR signaling pathways that are known to be abnormal in lupus,Citation23 could be involved in this “hyperautophagic” phenotype.
Stimulations by innate immune receptors, like TLRs,Citation24 or by cytokines, like IFNγ17, are known to induce autophagy in different cell types. In vivo LPS injection experiments suggest that TLR4 stimulation induces autophagy in nonlupus B cells. We found further that the increased number of autophagic vacuoles in lupus T cells is not just the consequence of an in vivo inflammatory stimulus and certainly requires a particular genetic environment to occur. It remains to be defined if other inflammation stimuli than LPS can induce autophagy in T cells and more importantly if a chronicity of inflammatory events could be responsible for the increased autophagosomal load in lupus T cells.
Interestingly, the increased autophagic vacuole number in lupus T cells is not a feature that is restricted to mouse models. We show that some patients suffering from SLE also exhibit a high number of autophagic compartments in peripheral T cells. Despite the strong heterogeneity in the etiology and symptoms of lupus pathology, we identified several patients with an autophagosomal load that is much higher compared with controls. An important consideration is that most patients included in this study were quiescent. The number of samples studied is certainly insufficient to establish correlations between the autophagic compartment number, the nature of the symptoms and treatment. Nevertheless, we can argue that autophagic vacuole accumulation is not only a secondary effect of hydroxychloroquine (Plaquenil) treatment routinely used for SLE treatment, and known to block autophagic flux, since highly vacuolated cells were also found in cells collected from patients treated with other drugs (). It would be important to define if the increased autophagosome number observed in lupus T cells is a general feature of autoinflammatory and autoimmune diseases. Our results show that T cells from two patients suffering from vasculitis and one patient suffering from Sjögren’s syndrome do not exhibit a number of autophagic compartments as high as the ones found in lupus patients.
The precise impacts of autophagy deregulation on T cell fate and the overall progression of the pathology also have to be investigated. Autophagy can exert both pro- and antisurvival effects on T cells. It could be a survival mechanism allowing autoreactive cells to persist as recently shown in experimental autoimmune encephalomyelitis model.Citation18 Interestingly, we found that cells with high numbers of autophagic vacuoles (more than three per cell section) are more frequent in some SLE patients than in healthy donors. It is tempting to speculate that these cells are autoreactive cells and that autophagy promotes their survival and contributes to their persistence in autoimmune conditions. In certain settings e.g., during apoptosis blockadeCitation25 or viral infections,Citation26 autophagy can also lead to a “nonclassical” cell death that do not only rely on caspase activation.Citation27 The impact of this kind of cell death on immune response is not fully understood and could be relevant in lupus pathology where dead cells constitute a source of antigens and proinflammatory molecules. At this stage in vivo models deficient for autophagy in lupus development are necessary to determine how autophagy contributes to lupus disease. An important question will be to determine if certain autophagy-related genes or upstream regulators of the autophagy pathway are involved in the lupus disease in mice and patients.
An important aspect will also be to identify which T and B lymphocyte subsets are affected. Our first results do not show any significant deregulation of autophagy in the total B cell population. It is possible that only some B cell subtypes in secondary lymphoid organs or in other tissues exhibit deregulated autophagy. Additional studies must be done using sorted B cells, and especially B-1a cells, which are known to play an important role in lupus development. B-1a cells seem to be particularly dependent on autophagy for their survival compared with B-2 conventional B cells.Citation28 Moreover, autophagy in B cells could contribute to the systemic autoimmune disease by providing MHC-II molecules loaded with self-peptides from endogenous origin or by favoring BCR/TLR9 cosignaling as suggested by Chaturvedi et al.Citation29 In this context it is interesting to remind that hydoxychloroquine as well as P140 peptide are both able to modulate autophagic flux and affect peptide presentation by MHC-II molecules.Citation11,Citation30 These findings could lead to new therapeutic options, or to the improvement of existing ones, aiming at targeting more specifically pathogenic lymphocytes, avoiding a global inhibition of immune system that is harmful to SLE patients.
Patients, Materials and Methods
Patients and normal donors
Peripheral blood T cells were obtained from 11 female patients with SLE (mean age = 32, range 16 to 52), two male patients with vasculitis, one female patient with primary Sjögren’s syndrome and eight healthy donors (4 men, 4 women, mean age = 31, range 18 to 45). Autoimmune patients fulfilled the American College of Rheumatology criteria for their respective diseases. They were recruited from the Strasbourg University Hospital after informed consent was obtained. Clinical parameters and ongoing treatments of SLE patients at the time of sampling are listed in .
Mice
MRLlpr/lpr, NZB/W, BALB/c, CBA/J, C57BL/6 and C57BL/6lpr/lpr mice were purchased from Harlan. All mice were bred and maintained in accordance with guidelines of the local Institutional Animal Care and Use Committee (CREMEAS).
Flow cytometry
All Abs used for flow cytometry analyses were purchased from BD Biosciences: fluorescein isothyocyanate (FITC)-labeled anti-mouse TCR-β (clone H57-597, 553171), phycoerythrin (PE)-labeled anti-mouse CD69 (clone H1.2F3, ref. 553237), allophycocyanin (APC)-labeled anti-mouse B220 (clone R13-6B2, 553092), FITC-labeled anti-human CD3ε (clone UCHT1, 55916) and APC-labeled anti-human CD19 (clone HIB19, 555415). Cells were incubated with fluorochrome-conjugated antibodies and in the case of mouse cell staining, rat anti-mouse CD16/CD32 monoclonal Ab (mAb, clone 2.4G2, 553142) was used to block Fcγ receptors, for 30 min at 4°C in phosphate-buffered saline (PBS) pH = 7.4 containing 2% (v/v) fetal calf serum (FCS). Data were collected on a Gallios flow cytometer (Beckman Coulter) and analyzed using FlowJo software (Tree Star).
Cell culture and isolation
Thymocytes and splenic T cells were collected from MRLlpr/lpr, CBA/J, C57BL/6, C57BL/6lpr/lpr, NZB/W and BALB/c mice and immediately cultured at 37°C, 5% CO2 in RPMI 1640 medium (Lonza BioWhittaker) supplemented with 10% FCS, 10 µg/mL gentamycin (Lonza BioWhittaker), 10 mM HEPES (Lonza BioWhittaker) and 0.05 mM β-mercaptoethanol (Lonza BioWhittaker) at a concentration of 5 × 106 cellules/mL. Splenic T cells were purified by negative selection. Briefly, spleen cell suspensions were depleted from monocytes, granulocytes, B cells and NK cells using Dynal T cell Negative Isolation Kit (Dynal-Life Technologies, 114-13D) according to the manufacturer’s instructions. CD3+CD4-CD8- double-negative T cells that are frequent in MRLlpr/lpr mice, were discarded by anti-B220 Abs included in the commercial preparation. Human peripheral blood mononuclear cells (PBMC) were isolated on a Ficoll (PAA, J15-004) density gradient and T cells were then sorted by using the Pan T cell isolation Kit II, human (Miltenyi Biotech, 130-091-156). Resulting TCR-β+/B220- mouse T cell and CD3ε+/CD19- human T cell preparations were > 90% pure as determined by flow cytometry. Mouse B cells were isolated using Dynal magnetic beads coupled with Abs to rat immunoglobulins (Ig) after incubating splenocyte suspension with the following rat Abs (BD Biosciences): anti-CD11b (clone M1/70, 553308), anti-Gr1 (clone RB6-8C5, 550291), anti-CD49b (clone DX5, 553855) and anti-TCRβ (clone H57-597, 553167). Preparations were > 90% pure according to TCR-β/B220 staining.
In vivo LPS injections
CBA/J mice received 50 µg LPS (Sigma Aldrich, L2630) in PBS or PBS alone intraperitoneally (i.p.). Twenty-four hours later, spleens were harvested and splenocytes were gently dissociated. Activation of lymphocytes was assessed by flow cytometry with CD69 staining for T cells and CD86 staining for B cells. Purified B and T cells were sorted and cultured as described above. Autophagic flux was measured as described below by LC3 staining on western immunoblots.
Western immunoblotting
The Abs used for western immunoblotting were specific for β-actin (Santa Cruz Biotechnology, clone C4, sc-47778), LC3 (MBL, clone 51–11, ref M115–3) and p62/SQSTM1 (rabbit polyclonal antibody, Sigma-Aldrich, P0068). In some experiments, lysosomal protease inhibitors E64d and pepstatin A (Sigma-Aldrich, P5318 and E8640) were added at 5 µg/mL each. When indicated, cells were treated with 50 ng/mL PMA (Sigma-Aldrich, P8139) and 1 µM ionomycin (Sigma-Aldrich, I0634). To evaluate the autophagosomal membrane load and monitor autophagic flux, whole cell proteins were extracted from cultured cells using Laemmli buffer (TRIS-HCl 125 mM pH 6.8; 2% (w/v) sodium dodecyl sulfate (SDS); 10% (v/v) glycerol; 5% (v/v) β-mercaptoethanol). Cell lysates were separated on 8–16% Novex Tris-glycine SDS-PAGE gradient gels (Life Technologies, EC6078BOX) and then transferred onto a polyvinylidene difluoride (PVDF) membrane. Membranes were blocked with PBS containing 0.1% (v/v) Tween 20 (PBS-T) and 3% (w/v) non-fat dry milk for 1h and then incubated overnight at 4°C with 1 μg/mL anti-LC3 Ab in PBS-T containing 1% nonfat dry milk. After washing with PBS-T, membranes were incubated for 30 min at room temperature with horseradish peroxidase-conjugated goat anti-mouse IgG antibody (Southern Biotech, 1030-05). Signal was detected using enhanced chemiluminescence detection reagents (Immobilon Western Millipore, WBKLS0500). When indicated, LC3-II levels were normalized by densitometry to β-actin level using ImageJ Software. Autophagic flux was quantified by calculating a ratio between these values obtained with and without protease inhibitors.
Real-time PCR (RT-PCR)
Total RNA was isolated from 5 × 106 purified T cells using RNeasy Mini Kit (Qiagen, 74103) according to the manufacturer’s instructions. After treatment by DNase (Qiagen, 79254) to remove residual genomic DNA, mRNA was retro-transcribed with Improm-II reverse transcriptase (Promega, A3800). Twenty nanograms of cDNA was used for real-time PCR on StepOne apparatus (Applied Biosystems). Briefly, LC3 Map1lc3a, Actb and Hprt (respectively encoding for LC3, β-actin and HPRT1) cDNA were amplified using Taqman Gene Expression Assays provided by Applied Biosystems (Mm01249999-g1, Mm01249999_g1, Mm00458724_m1). Amplicons and probes were designed to span two exons, limiting the risk of amplifying residual genomic DNA. Relative Map1lc3a mRNA quantifications were made by defining ΔCT (ΔCT = CT β-actin–CT LC3 and CT hprt1–CT LC3 where CT is “Cycle Threshold”) and ΔΔCT (ΔΔCT = ΔCT sample-ΔCT of one control mouse per plate) using StepOne software (Applied Biosystems). Results shown represent 2-ΔΔCT values where one control mouse per plate is arbitrarily equal to 1.
Electron microscopy and quantification of autophagic structures
Mouse and human T cells as well as entire mouse thymus were fixed using 2.5% (v/v) glutaraldehyde in phosphate buffer 0.1 M, pH 7.4 overnight at 4°C. They were post-fixed using 1% (v/v) osmium tetroxide for 90 min and dehydrated in ascending series of ethanol dilutions. They were then treated with propylene oxide, impregnated in ascending dilutions of resin in propylene oxide, left in pure resin overnight (Epon, Inland Europe), embedded and polymerized at 60°C for 48 h. Ultra-thin sections (70 nm) performed on ultramicrotome (Leica ultracut R) were collected on butvar-coated single-slot copper grids, stained with 1% (v/v) uranyl acetate for 30 min and with lead citrate for 2 min. Grids were examined by TEM (Hitachi H600) and the images were acquired using a digital Hamamatsu camera. Fifty cell sections per condition were examined and autophagic compartments were quantified. Only grids prepared from different resin blocks were considered for quantification, avoiding counting the same cell several times. Vesicles were considered as autophagosomes when meeting at least two of the following criteria:Citation31 double membrane, absence of ribosome in cytosolic side of the vacuole, similar density of the luminal side of the vesicle compared with cytosol, complete or remains of organelles inside the vesicle. Single-membrane vesicles containing dense or clear amorphous material were considered as autolysosomes. Autophagosomes and autolysosomes counts were pooled and termed “autophagic vacuoles” or “autophagic compartments.” Statistical analysis comparing either normal vs. lupus-prone mice, or SLE patients vs. healthy donors were performed using unpaired t-test when samples followed a normal distribution and by Mann-Whitney U test when Gaussian distribution of values could not be assumed. Both tests were considered significant when p < 0.05
| Abbreviations: | ||
| APC | = | allophycocyanin |
| FITC | = | fluorescein isothyocyanate |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| NZB/W | = | (NZB/NZW)F1 |
| LPS | = | lipopolysaccharide |
| PBMC | = | peripheral blood mononuclear cells |
| PBS | = | phosphate-buffered saline |
| PE | = | phycoerythrin |
| SLE | = | systemic lupus erythematosus |
Additional material
Download Zip (921.2 KB)Acknowledgments
We thank Jean-Louis Pasquali and Jean-Paul Briand for carefully reading the manuscript. We also thank Monique Duval for technical assistance. This work was funded by the French Centre National de la Recherche Scientifique, Région Alsace and University of Strasbourg.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/20275
References
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124 - 31; http://dx.doi.org/10.1016/j.ceb.2009.11.014; PMID: 20034776
- Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol 2010; 189:925 - 35; http://dx.doi.org/10.1083/jcb.201002021; PMID: 20548099
- Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 2007; 39:596 - 604; http://dx.doi.org/10.1038/ng2032; PMID: 17435756
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264 - 8; http://dx.doi.org/10.1038/nature07383; PMID: 18849965
- Alirezaei M, Fox HS, Flynn CT, Moore CS, Hebb AL, Frausto RF, et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 2009; 5:152 - 8; http://dx.doi.org/10.4161/auto.5.2.7348; PMID: 19066443
- Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, et al, International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN). Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008; 40:204 - 10; http://dx.doi.org/10.1038/ng.81; PMID: 18204446
- Orozco G, Eyre S, Hinks A, Bowes J, Morgan AW, Wilson AG, et al, UKRAG consortium. Study of the common genetic background for rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis 2011; 70:463 - 8; http://dx.doi.org/10.1136/ard.2010.137174; PMID: 21068098
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 2010; 69:20 - 8; http://dx.doi.org/10.1136/ard.2008.101766; PMID: 19103632
- Perl A. Emerging new pathways of pathogenesis and targets for treatment in systemic lupus erythematosus and Sjogren’s syndrome. Curr Opin Rheumatol 2009; 21:443 - 7; http://dx.doi.org/10.1097/BOR.0b013e32832efe6b; PMID: 19584730
- Page N, Gros F, Schall N, Décossas M, Bagnard D, Briand J-P, et al. HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann Rheum Dis 2011; 70:837 - 43; http://dx.doi.org/10.1136/ard.2010.139832; PMID: 21173017
- Page N, Gros F, Schall N, Briand JP, Muller S. A therapeutic peptide in lupus alters autophagic processes and stability of MHCII molecules in MRL/lpr B cells. Autophagy 2011; 7:539 - 40; http://dx.doi.org/10.4161/auto.7.5.14845; PMID: 21282971
- Monneaux F, Muller S. Molecular therapies for systemic lupus erythematosus: clinical trials and future prospects. Arthritis Res Ther 2009; 11:234; http://dx.doi.org/10.1186/ar2711; PMID: 19591653
- Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med 2007; 204:25 - 31; http://dx.doi.org/10.1084/jem.20061303; PMID: 17190837
- Arsov I, Adebayo A, Kucerova-Levisohn M, Haye J, MacNeil M, Papavasiliou FN, et al. A role for autophagic protein beclin 1 early in lymphocyte development. J Immunol 2011; 186:2201 - 9; http://dx.doi.org/10.4049/jimmunol.1002223; PMID: 21239722
- Stephenson LM, Miller BC, Ng A, Eisenberg J, Zhao Z, Cadwell K, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy 2009; 5:625 - 35; http://dx.doi.org/10.4161/auto.5.5.8133; PMID: 19276668
- Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol 2009; 182:4046 - 55; http://dx.doi.org/10.4049/jimmunol.0801143; PMID: 19299702
- Feng CG, Zheng L, Jankovic D, Báfica A, Cannons JL, Watford WT, et al. The immunity-related GTPase Irgm1 promotes the expansion of activated CD4+ T cell populations by preventing interferon-gamma-induced cell death. Nat Immunol 2008; 9:1279 - 87; http://dx.doi.org/10.1038/ni.1653; PMID: 18806793
- Kovacs JR, Li C, Yang Q, Li G, Garcia IG, Ju S, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ 2012; 19:144 - 52; http://dx.doi.org/10.1038/cdd.2011.78; PMID: 21660048
- Tsokos GC, Mitchell JP, Juang YT. T cell abnormalities in human and mouse lupus: intrinsic and extrinsic. Curr Opin Rheumatol 2003; 15:542 - 7; http://dx.doi.org/10.1097/00002281-200309000-00004; PMID: 12960478
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J 2008; 27:1110 - 21; http://dx.doi.org/10.1038/emboj.2008.31; PMID: 18337753
- Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007; 27:135 - 44; http://dx.doi.org/10.1016/j.immuni.2007.05.022; PMID: 17658277
- Nambiar MP, Juang YT, Krishnan S, Tsokos GC. Dissecting the molecular mechanisms of TCR zeta chain downregulation and T cell signaling abnormalities in human systemic lupus erythematosus. Int Rev Immunol 2004; 23:245 - 63; http://dx.doi.org/10.1080/08830180490452602; PMID: 15204087
- Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A 2011; 108:1427 - 32; http://dx.doi.org/10.1073/pnas.1014156108; PMID: 21220332
- Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci U S A 2008; 105:16677 - 82; http://dx.doi.org/10.1073/pnas.0808597105; PMID: 18946037
- Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest 2006; 116:2161 - 72; http://dx.doi.org/10.1172/JCI26185; PMID: 16886061
- Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev 2010; 24:2592 - 602; http://dx.doi.org/10.1101/gad.1984410; PMID: 21123646
- Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 2008; 4:309 - 14; PMID: 18188005
- Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity 2008; 28:799 - 809; http://dx.doi.org/10.1016/j.immuni.2008.03.019; PMID: 18513998
- Katz SJ, Russell AS. Re-evaluation of antimalarials in treating rheumatic diseases: re-appreciation and insights into new mechanisms of action. Curr Opin Rheumatol 2011; 23:278 - 81; http://dx.doi.org/10.1097/BOR.0b013e32834456bf; PMID: 21448012
- Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F. Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol 2010; 185:7349 - 57; http://dx.doi.org/10.4049/jimmunol.1000576; PMID: 21059894