Abstract
Protein folding stress is a salient feature of the most frequent neurodegenerative diseases. Although the accumulation of abnormally folded proteins is a well-characterized event underlying the pathology, the way cells respond to this phenomenon is not well understood. Signs of endoplasmic reticulum (ER) stress are a common marker of neurodegeneration in many diseases, which may represent two contrasting processes: cell protection events due to activation of adaptive programs, or a chronic stress state that culminates in apoptosis to eliminate irreversibly injured cells. Autophagy has been proposed as a protective mechanism to overcome neurodegeneration that is also modulated by ER stress. In this issue of autophagy Bertrand Mollereau’s group provides novel evidence indicating that engagement of nonharmful levels of ER stress protects against experimental Parkinson disease. At the mechanistic level, a homeostatic crosstalk between ER stress signaling and the autophagy pathway was proposed to mediate the therapeutic effects. This study, together with recent findings, supports the involvement of a “hormesis mechanism” to handle degeneration through preconditioning mediated by a dynamic balance between ER stress and autophagy. The implications for aging and future therapeutic development are discussed.
The accumulation of diffusible oligomers or large aggregates of specific misfolded proteins is a salient characteristic of many neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), Alzheimer disease, Parkinson disease (PD), and Huntington disease (HD). Perturbation in protein homeostasis is currently proposed as a key factor underlying the pathological effects of most of the well-characterized brain disease-linked proteins.Citation1,Citation2 Strategies to either decrease protein aggregation or attenuate proximal downstream signaling are thought to be relevant targets for disease intervention. In this context, autophagy is emerging as an interesting candidate for therapeutic development because it operates as an efficient and selective mechanism for the degradation of abnormally folded proteins.Citation3
Although the molecular mechanisms underlying neurodegeneration in general are poorly understood, accumulating evidence suggests that perturbation in ER function is a common event in many neurodegenerative diseases.Citation1 ER stress is mitigated through a series of complementary mechanisms that as a whole are known as the unfolded protein response (UPR).Citation4 The UPR is a complex signal transduction pathway that selectively modulates the expression of proteins involved in diverse processes including folding, quality control, protein entry into the ER, and organelle biogenesis among others.Citation4 Conversely, chronic or irreversible ER stress triggers cell death by apoptosis. The UPR is governed by several stress sensors located at the ER membrane. These sensors transduce information about the protein folding status at the ER lumen to the nucleus and cytosol by controlling expression of specific transcription factors and other proximal signaling components.Citation4 The ER stress sensor inositol requiring kinase 1 (ERN1/IRE1α) induces the expression of the transcription factor XBP1, which modulates many genes related to folding and quality control mechanisms. ERN1/IRE1α also controls the activation of c-Jun-N-terminal kinase (MAPK8/JNK), which has been linked to the upregulation of autophagy under ER stress.Citation5 Other mechanisms also link ER stress signaling with autophagy (see review in ref. Citation6). Autophagy is proposed to operate as a mechanism able to eliminate damaged organelles and aggregated proteins under conditions of ER stress.
ER Stress in Neurodegenerative Diseases
Although many reports have linked the occurrence of ER stress with neurodegeneration, genetic and pharmacological manipulations of the pathway have revealed a complex scenario, where the exact contribution of the ER stress to the pathology is difficult to predict a priori. The occurrence of ER stress may represent: (1) a chronic damaging process triggering neuronal loss, (2) the active engagement of prosurvival responses to adapt to the stress and reestablish protein homeostasis, or (3) a late epiphenomenon due to the dramatic alterations in homeostasis. Several studies have reported the expected result where attenuation of ER stress levels protects against neurodegeneration (reviewed in ref. Citation1). Unexpectedly, genetic inactivation of the UPR transcription factor XBP1 in the nervous system delays degeneration in mouse models of ALSCitation7 and HD.Citation8 These protective effects were attributed to the induction of autophagy, associated with the clearance of mutant SOD1 or HTT/huntingtin, respectively.Citation7,Citation8 Nonetheless, although most reports have shown a positive effect of ERN1/MAPK8 signaling in the activation of autophagy, a recent study suggested that this pathway may also inhibit autophagy flux in the context of HD.Citation9 Other alternative theoretical mechanisms have been proposed to explain these unexpected results, including the idea that targeting specific UPR components may actually generate low levels (nonlethal) of stress that could then engage a compensatory adaptive response (i.e., autophagy, see below).Citation10 These observations suggest an interesting concept where the autophagy and UPR pathways operate as supra-autoregulatory networks that integrate information to handle perturbations of protein homeostasis in disease.
Hormesis: What Doesn’t Kill You Makes You Stronger
The idea that low levels of stress may actually protect against a subsequent injury has been around for decades in the toxicology area. In the context of ER stress, this concept has been applied also to the field of neurodegeneration and brain ischemia-reperfusion. Mild or low levels of ER stress selectively engages a subset of UPR signaling events, highlighting the specific induction of XBP1 and not of pro-apoptotic components such as DDIT3/CHOP.Citation11-Citation13 ER stress preconditioning (i.e., exposure to nonlethal doses of pharmacological ER stressors) protects against brain ischemia.Citation14,Citation15 Low levels of ER stress also reduce retinal endothelial inflammationCitation16 and attenuate heart ischemia/reperfusion possibly due to expression of XBP1.Citation17 The genetic induction of low ER stress also alleviates retinal degeneration in Drosophila models.Citation13
Hormesis (from Greek hórmēsis “rapid motion, eagerness,” from ancient Greek hormáein “to set in motion, impel, urge on”) refers to a biological favorable response triggered by subjecting an organism/cell to a low exposure of toxins and other stressors.Citation18 Conditions that stimulate hormesis can engage adaptive stress signaling events rendering cells resistant against a high dose of the same stimuli. In terms of toxicological definitions, Calabrese and Baldwin considered hormesis as “an adaptive response characterized by biphasic dose responses of generally similar quantitative features with respect to amplitude and range of the stimulatory response that are either directly induced or the result of compensatory biological processes following an initial disruption in homeostasis.”Citation19 It is becoming evident that mild perturbations of ER function may trigger a hormetic mechanism of protection possibly due to the activation of a prosurvival UPR response that bursters the folding and quality control capacity of the ER.Citation4 This phenomenon is actually observed in most physiological systems that requires the UPR for their proper function, such as specialized secretory cells.Citation4 New evidence provided in this issue of Autophagy by Fouillet et al., support the concept that one of the main hormetic mechanisms triggered by exposure to nonlethal ER stress is the engagement of autophagy.Citation20
Interplay Between Autophagy and ER Stress in Parkinson Disease: A Complex Hormetic Rheostat
PD is the second most frequent neurodegenerative disease involving the loss of dopaminergic neurons in the substantia nigra pars compacta. One of the most studied PD-related gene products is SNCA/α-synuclein, which is the main component of intracellular inclusions termed Lewy bodies, a key histopathological feature of the disease. Many studies have demonstrated the occurrence of ER stress in animal and cellular models of PD, in addition to postmortem brain tissue from PD patients (see examples in refs. Citation21–Citation24). The functional contribution of ER stress to PD in vivo has been only recently addressed. It was reported that the accumulation of SΝCΑ occurs inside the ER lumen during early stages of PD,Citation22 associated with a later and increasing chronic ER stress response, that correlates with the occurrence of neurodegeneration. Consistent with this idea, attenuation of ER stress levels with pharmacological or gene therapy approaches delays disease progression.Citation23,Citation24 Thus, chronic levels of irreversible ER stress were proposed as an important factor underlying neuronal loss in PD. Many other studies have linked ER-related injuries with PD (review in ref. Citation1).
Mollereau's study indicates that sublethal levels of ER stress generate a preconditioning effect that provides strong neuroprotection against experimental PD in Drosophila and mouse models.Citation20 The neuroprotective effects of nonlethal ER stress are functionally linked to the engagement of autophagy. For example, feeding transgenic flies overexpressing SNCA with tunicamycin (a pharmacological ER stress agent) triggers low levels of stress in the brain reflected in the selective activation of XBP1 splicing and upregulation of autophagy. In this model, genetic inactivation of essential autophagy genes reverses the protective effects of ER stress. Surprisingly, the authors were able to treat mice with tunicamycin through intraperitoneal injections and protect dopaminergic neurons against a PD-inducing neurotoxin, improving motor performance. Experiments in mammalian cells corroborated the role of autophagy in the beneficial effects of mild ER stress preconditioning against experimental PD. This study demonstrates a hormetic mechanism in the control of degeneration in PD where a direct interplay between the UPR and the autophagy pathways generates efficient and long-term neuroprotection (). The molecular link mediating the crosstalk between these two homeostatic pathways in PD remains to be elucidated. A few reports have also suggested that the homeostatic balance between autophagy and ER stress can operate in the opposite direction. For example, in models of ischemia/preconditioning, autophagy was recently shown to regulate ER stress levels.Citation25 MTOR (an upstream regulator of autophagy) signaling also modulates ER stress signaling.Citation26 In addition, autophagy-deficient tumors show a dramatic upregulation of ER chaperones, which may contribute to tumor survival and growth under hypoxic and nutrient starvation conditions.Citation27 Finally, deficiency of ATG7 in the liver triggers ER stress, contributing to insulin resistance in obesity models.Citation28 Thus, the autophagy and UPR signaling network may operate as a delicate rheostat that balances physiological fluctuations and pathological perturbations in protein homeostasis in both directions: UPR signaling controlling autophagy levels, and autophagy signaling/activity controlling ER stress levels.
Figure 1. Hormesis mechanism to avoid degeneration in PD. During presymptomatic stages of PD, low levels of stress (i.e., oxidative and ER stress) may trigger the activation of autophagy as a survival pathway to reduce stress levels. This preconditioning may effect protect against a subsequent toxic stimuli that prevents or delays degeneration of dopaminergic neurons through a hormesis mechanism. The appearance of the clinical manifestation of PD may be due in part to a gradual increase in stress levels to an irreversible point where hormesis mechanisms (i.e., UPR prosurvival/adaptive signaling and upregulation of autophagy) are not sufficient to recover cellular homeostasis.
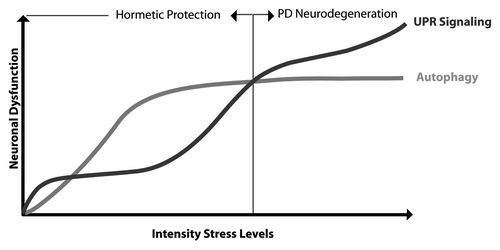
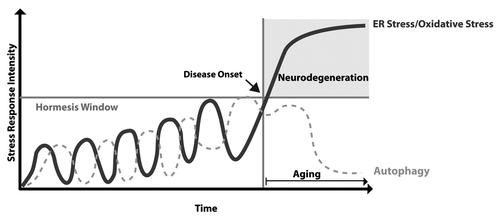
We also speculate that a hormesis-related mechanism may underlie the notion that most neurodegenerative diseases are clinically manifested during aging despite the fact the pathological events are operating since very early presymptomatic stages. This is especially evident in familial forms of the disease where the patient is born with a genetic mutation. In this scenario, low or fluctuating perturbations of ER function during early presymptomatic phases of the disease may actually promote adaptive programs that in the long-term protect neurons against subsequent and stronger pathological stimuli (). Then, only when stress levels reach an irreversible point, neurological impairment is expressed, which is in agreement with recent finding by Lee’s group.Citation22,Citation23 In this context, aging-related events (the major risk factor to developing neurodegenerative diseases) may constitute a crucial factor in ablating hormetic mechanisms of survival due to the known attenuation of the adaptive capacity of the cell against protein homeostasis alterations ().Citation3 In fact, chronic treatment with low doses of rapamycin (an autophagy inducer) or exposure to organismal stress (i.e., nonlethal heat shock or oxidative stress) have a tremendous effect on prolonging life span in animal models.Citation3 The impairment of adaptive mechanisms during aging may dramatically burster ER and oxidative stress levels in a vicious cycle, leading to the irreversible damage of the cell.Citation2 Based on increasing evidence suggesting a causative role of autophagy defects and ER perturbations in neurodegenerative diseases, these new studies set an interesting scenario where targeting one particular component of a stress pathway may compensate or alleviate defects in other points of the protein homeostasis network through a hormetic mechanism.
Figure 2. The aging and hormesis rehostat in neurodegenerative diseases. During presymptomatic stages of disease, low or fluctuating perturbations of ER function trigger adaptive programs that protect neurons against subsequent stress episodes. When stress levels reach an irreversible point, neurological impairment is expressed. The gradual decrease of autophagy activity and increased oxidative stress during aging ablates the hormetic capacity of the cell, further increasing stress/injury levels in a vicious cycle, culminating in irreversible neuronal dysfunction and possibly neuronal loss.
| Abbreviations: | ||
| ALS | = | amyotrophic lateral sclerosis |
| PD | = | Parkinson disease |
| HD | = | Huntington disease |
| ER | = | endoplasmic reticulum |
| ERN1/IRE1α | = | inositol requiring kinase 1 |
| UPR | = | unfolded protein response |
| MAPK8/JNK | = | c-Jun-N terminal kinase |
| XBP1 | = | X-Box Binding protein-1 |
| SNCA | = | α-synuclein |
Acknowledgments
This article was funded by the Michael J. Fox Foundation for Parkinson’s Research (C.H.). In addition we received support from FONDECYT no. 1100176, FONDAP grant no. 15010006, Millennium Institute no. P09-015-F, Muscular Dystrophy Association, ALS Therapy Alliance, North American Spine Society and Alzheimer’s Disease Foundation (C.H.), Insercion de Capital Humano Avanzado CONICYT no. 79100007 (S.M.) and FONDECYT no. 3100112 (K.C.).
References
- Matus S, Glimcher LH, Hetz C. Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr Opin Cell Biol 2011; 23:239 - 52; http://dx.doi.org/10.1016/j.ceb.2011.01.003; PMID: 21288706
- Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 2011; 71:35 - 48; http://dx.doi.org/10.1016/j.neuron.2011.06.031; PMID: 21745636
- Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell 2011; 146:682 - 95; http://dx.doi.org/10.1016/j.cell.2011.07.030; PMID: 21884931
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012; 13:89 - 102; PMID: 22251901
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26:9220 - 31; http://dx.doi.org/10.1128/MCB.01453-06; PMID: 17030611
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280 - 93; http://dx.doi.org/10.1016/j.molcel.2010.09.023; PMID: 20965422
- Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 2009; 23:2294 - 306; http://dx.doi.org/10.1101/gad.1830709; PMID: 19762508
- Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum Mol Genet 2012; 21:2245 - 62; http://dx.doi.org/10.1093/hmg/dds040; PMID: 22337954
- Lee H, Noh JY, Oh Y, Kim Y, Chang JW, Chung CW, et al. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum Mol Genet 2012; 21:101 - 14; http://dx.doi.org/10.1093/hmg/ddr445; PMID: 21954231
- Matus S, Nassif M, Glimcher LH, Hetz C. XBP-1 deficiency in the nervous system reveals a homeostatic switch to activate autophagy. Autophagy 2009; 5:1226 - 8; http://dx.doi.org/10.4161/auto.5.8.10247; PMID: 19855189
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol 2006; 4:e374; http://dx.doi.org/10.1371/journal.pbio.0040374; PMID: 17090218
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007; 318:944 - 9; http://dx.doi.org/10.1126/science.1146361; PMID: 17991856
- Mendes CS, Levet C, Chatelain G, Dourlen P, Fouillet A, Dichtel-Danjoy ML, et al. ER stress protects from retinal degeneration. EMBO J 2009; 28:1296 - 307; http://dx.doi.org/10.1038/emboj.2009.76; PMID: 19339992
- Mao XR, Crowder CM. Protein misfolding induces hypoxic preconditioning via a subset of the unfolded protein response machinery. Mol Cell Biol 2010; 30:5033 - 42; http://dx.doi.org/10.1128/MCB.00922-10; PMID: 20733002
- Calabrese V, Cornelius C, Dinkova-Kostova AT, Calabrese EJ, Mattson MP. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid Redox Signal 2010; 13:1763 - 811; http://dx.doi.org/10.1089/ars.2009.3074; PMID: 20446769
- Petrovski G, Das S, Juhasz B, Kertesz A, Tosaki A, Das DK. Cardioprotection by endoplasmic reticulum stress-induced autophagy. Antioxid Redox Signal 2011; 14:2191 - 200; http://dx.doi.org/10.1089/ars.2010.3486; PMID: 20726815
- Li J, Wang JJ, Zhang SX. Preconditioning with endoplasmic reticulum stress mitigates retinal endothelial inflammation via activation of X-box binding protein 1. J Biol Chem 2011; 286:4912 - 21; http://dx.doi.org/10.1074/jbc.M110.199729; PMID: 21138840
- Martins I, Galluzzi L, Kroemer G. Hormesis, cell death and aging. Aging (Albany NY) 2011; 3:821 - 8; PMID: 21931183
- Calabrese EJ, Baldwin LA. Defining hormesis. Hum Exp Toxicol 2002; 21:91 - 7; http://dx.doi.org/10.1191/0960327102ht217oa; PMID: 12102503
- Fouillet A, Levet C, Virgone A, Robin M, Dourlen P, Rieusset J, et al. ER stress inhibits neuronal death by promoting autophagy. Autophagy 2012; 8:915 - 26; http://dx.doi.org/10.4161/auto.19716; PMID: 22660271
- Wang HQ, Takahashi R. Expanding insights on the involvement of endoplasmic reticulum stress in Parkinson’s disease. Antioxid Redox Signal 2007; 9:553 - 61; http://dx.doi.org/10.1089/ars.2006.1524; PMID: 17465880
- Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK. Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J Neurosci 2012; 32:3301 - 5; http://dx.doi.org/10.1523/JNEUROSCI.5368-11.2012; PMID: 22399752
- Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, et al. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J Neurosci 2012; 32:3306 - 20; http://dx.doi.org/10.1523/JNEUROSCI.5367-11.2012; PMID: 22399753
- Gorbatyuk MS, Shabashvili A, Chen W, Meyers C, Sullivan LF, Salganik M, et al. Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of Parkinson disease. Mol Ther 2012; In press http://dx.doi.org/10.1038/mt.2012.28; PMID: 22434142
- Sheng R, Liu X-Q, Zhang L-S, Gao B, Han R, Wu Y-Q, et al. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy 2012; 8:310 - 25; http://dx.doi.org/10.4161/auto.18673; PMID: 22361585
- Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ 2012; 19:310 - 20; http://dx.doi.org/10.1038/cdd.2011.98; PMID: 21779001
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062 - 75; http://dx.doi.org/10.1016/j.cell.2009.03.048; PMID: 19524509
- Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010; 11:467 - 78; http://dx.doi.org/10.1016/j.cmet.2010.04.005; PMID: 20519119