Abstract
Although reactive oxygen species (ROS) have been reported to evoke different autophagic pathways, how ROS or their secondary products modulate the selective clearance of oxidatively damaged organelles is less explored. To investigate the signaling role of ROS and the impact of their compartmentalization in autophagy pathways, we used murine fibrosarcoma L929 cells overexpressing different antioxidant enzymes targeted to the cytosol or mitochondria and subjected them to photodynamic (PD) stress with the endoplasmic reticulum (ER)-associated photosensitizer hypericin. We show that following apical ROS-mediated damage to the ER, predominantly cells overexpressing mitochondria-associated glutathione peroxidase 4 (GPX4) and manganese superoxide dismutase (SOD2) displayed attenuated kinetics of autophagosome formation and overall cell death, as detected by computerized time-lapse microscopy. Consistent with a primary ER photodamage, kinetics and colocalization studies revealed that photogenerated ROS induced an initial reticulophagy, followed by morphological changes in the mitochondrial network that preceded clearance of mitochondria by mitophagy. Overexpression of cytosolic and mitochondria-associated GPX4 retained the tubular mitochondrial network in response to PD stress and concomitantly blocked the progression toward mitophagy. Preventing the formation of phospholipid hydroperoxides and H2O2 in the cytosol as well as in the mitochondria significantly reduced cardiolipin peroxidation and apoptosis. All together, these results show that in response to apical ER photodamage ROS propagate to mitochondria, which in turn amplify ROS production, thereby contributing to two antagonizing processes, mitophagy and apoptosis.
Introduction
Accumulating evidence indicates that loss of cellular redox homeostasis is associated to cancer initiation and progression and is crucial in mediating the cellular response to various ROS-based therapeutic agents, including chemo-, radio- and photodynamic therapy (PDT).Citation1-Citation4 These anticancer modalities impose cancer cell death by generating lethal levels of ROS that alter cancer cell’s resistance mechanisms of adaptation to oxidative-injury. While it is well established that excessive ROS can instigate apoptosis, emerging data have also revealed a signaling role for ROS in the activation of macroautophagy (hereafter referred to as autophagy), a major lysosomal pathway for the degradation and recycling of proteins and organelles with prosurvival function.Citation5,Citation6 During autophagy, cytoplasmic material is sequestered into a double-membrane structure called the autophagosome for subsequent delivery to lysosomes for degradation.Citation7,Citation8 Activation of autophagy in response to the increased status of oxidative stress during cancer progression and in response to anticancer therapy plays a role in the modulation of cellular redox homeostasis with consequences that depend on the genetic background of the cancer cells, their ability to undergo apoptosis, progression stage and type of ROS insults.Citation9-Citation11
At the molecular level, both hydrogen peroxide (H2O2) and superoxide radical (O2•−) have been proposed as signaling mediators in autophagy.Citation11 Scherz-Shouval et al.Citation12 showed that H2O2 accumulation in the mitochondria as result of nutrient starvation was essential for the activation of autophagy. Other studies showed that increased levels of O2•− produced by inhibition of superoxide dismutase (SOD), addition of exogeneous H2O2, starvation or blockade of the electron transport chain, were involved in the regulation of autophagy.Citation13-Citation16 Jiang et al.,Citation17 however, have reported that although mitochondrial ROS may act as a cause and indirect initiators of autophagy, they are not required in the execution of this process.
The systematic characterization of ROS as molecular mediators of autophagy and apoptosis pathways is complex since these species can be rapidly neutralized or interconverted from one to another by the action of, e.g., endogenous antioxidants. For example, O2•−, a membrane impermeable and thus more spatially confined type of ROS, can be converted by SOD into H2O2, a less reactive molecule with ability to diffuse and cross membranes. In turn, H2O2 can be neutralized to H2O and O2 by catalase or peroxidases or can be further converted into the highly reactive •OH, by metal ions.Citation18
It is largely accepted that the primary ROS produced in PDT by the combination of a photosensitizer, molecular oxygen and visible light, is singlet oxygen (O2, 1Δg). This species has an inherent lifetime of few microseconds which limits the primary photo-oxidation reactions of 1O2 with biomolecules in the close vicinity to the site of the photosensitizer accumulation.Citation19 Photodynamic (PD) stress has been the subject of intense research in recent decades due to its therapeutic applications in PDT of cancer, age-related macular degeneration, microbial infections or dermatological diseases, such as actinic keratosis or acne vulgaris.Citation20-Citation25 Several of the cellular events that occur in the phase comprised between the primary photo-oxidative damage and the final therapeutic effect, which is cell death and eradication of diseased tissue, are modulated by secondary ROS. Secondary ROS can extend the initial photodamage intracellularly, as seen by Ouedrago et al.Citation26 where lipid hydroperoxides produced in the cell membrane after photooxidation using deuteroporphyrin can spread the oxidative damage within the cells and produce nuclear DNA damage. Furthermore, secondary ROS such as H2O2 produced via NADPH-oxidase activation after PD stress using DP and benzoporphyrin derivative, membrane- and mitochondrial-localized photosensitizers, respectively, had a signaling role in the amplification of the oxidative damage and cell death within the treated cell population.Citation27 This ROS was found to extend the levels of oxidative stress to greater distances reaching neighboring nontreated cells, by a so-called “bystander effect.”Citation28,Citation29
In our previous studies, we showed that light activation of the endoplasmic reticulum (ER)-associated hypericin (Hyp) induces a rapid drop in sarco(endo)plasmic-reticulum Ca2+-ATPase (SERCA) pump protein levels, as the primary event mediated by locally generated 1O2.Citation30 SERCA photodamage causes disruption of Ca2+ homeostasis, ER stress and eventual mitochondrial apoptosis.Citation30 However, Hyp-mediated photodynamic (Hyp-PD) stress also activates autophagy and the more selective chaperone-mediated autophagy, whereby oxidized soluble proteins are targeted for degradation in a protein-by-protein fashion,Citation30,Citation31 and both pathways have been shown to increase the resistance against PD stress.Citation31 The alleviation of both autophagic and apoptotic processes by overexpression of the cytosolic membrane-associated antioxidant enzyme GPX4 (phospholipid glutathione peroxidase) already revealed a role for ROS originated at the ER in the further propagation of oxidative damage. Nevertheless, the nature of the different intracellular ROS produced in response to the initial 1O2-mediated insult at the ER is still unknown. In addition, the way these ROS further modulate the autophagy-apoptosis crosstalk is still unclear.
In this work, we addressed these questions by using L929 murine fibrosarcoma cells overexpressing different antioxidant enzymes (phospholipid glutathione peroxidase (GPX4), catalase (CAT) and superoxide dismutase (SOD) targeted at different locations (cytosol and mitochondria). We found that while lipid hydroperoxides or their metabolites produced following the initial ROS insult at the ER stimulate ER-selective autophagy (reticulophagy), this oxidative damage is being rapidly conveyed to the mitochondrial phospholipid cardiolipin. The subsequent generation of ROS by the mitochondria is a key event which coordinates both the selective removal of mitochondria through mitophagy and the initiation of apoptotic cell death.
Results
Hyp-PD mediated ER photodamage propagates ROS and autophagy in L929 cells
Hyp is a photosensitizer that localizes preferentially in the ER of different cell lines including L929 cells,Citation30 while is excluded from mitochondria, thereby ensuring that primary Hyp-mediated photodamage is confined to the ER (Fig. S1A and S1B). This is also confirmed by the rapid SERCA photo-oxidation (Fig. S1C) following Hyp-PD exposure of L929 cells, in line with previous results.Citation30 In order to study the cellular responses to Hyp-PD stress, we first monitored the levels of ROS produced as a function of time using computerized time-lapse microscopy (CTLM). L929 cells continuously produced ROS long after the initial PD insult, with a peak at around 9–10 h (), as also reflected by a corresponding time-dependent accumulation of carbonylated (oxidized) proteins ().
Figure 1. Localized ER photodamage promotes ROS production and stimulation of autophagy in L929 cells. (A) Background-subtracted intracellular DCF fluorescence measured as a function of time for a population after photodynamic treatment with Hyp (25 nM) and (●) 7.8 J•cm−2 or (○) 0 J•cm−2 (control) of 572 ± 12-nm light. In the absence of photosensitizer or initial energy dose, negligible changes in DFC fluorescence were observed. The values plotted correspond to the intracellular DCF fluorescence values corrected by the background, since the oxidized probe gradually leaks out the cell with time. (B) Protein carbonylation quantification (nmol of carbonylated protein/mg protein) as a function of time after Hyp-PD stress with an energy dose of 1.1 J•cm−2 (white light) compared with control. (C) LC3-I-to-LC3-II conversion determined by western blot analysis of whole cell lysates as a function of the initial energy dose (white light), detected 6 h after initial PD insult and quantification of the western blot analysis. Quantifications are performed as the ratio of LC3-II over actin. A representative western blot (n = 3) is shown. (Inset) Levels of ROS detected in the empty-vector transfected cells (i.e., Neo cells), measured by DCF-fluorescence using FACS, at different energy doses (white light). (D) Kinetics of GFP-LC3 puncta formation in a L929 Neo population challenged with 4.5 J•cm−2 (●) of 572 ± 12-nm light delivered by microscopic PDT, monitored by CTLM, compared with the control (○), i.e., cells with Hyp, but with no initial excitation. (E) Confocal microscopy images corresponding to the formation of GFP-LC3 labeled autophagosomes in L929 Neo(NaSe) cells taken at 6 and 16 h after PD stress (1.1 J•cm−2, white light) ([CQ] = 5 μM). Scale bar: 10 μm.
![Figure 1. Localized ER photodamage promotes ROS production and stimulation of autophagy in L929 cells. (A) Background-subtracted intracellular DCF fluorescence measured as a function of time for a population after photodynamic treatment with Hyp (25 nM) and (●) 7.8 J•cm−2 or (○) 0 J•cm−2 (control) of 572 ± 12-nm light. In the absence of photosensitizer or initial energy dose, negligible changes in DFC fluorescence were observed. The values plotted correspond to the intracellular DCF fluorescence values corrected by the background, since the oxidized probe gradually leaks out the cell with time. (B) Protein carbonylation quantification (nmol of carbonylated protein/mg protein) as a function of time after Hyp-PD stress with an energy dose of 1.1 J•cm−2 (white light) compared with control. (C) LC3-I-to-LC3-II conversion determined by western blot analysis of whole cell lysates as a function of the initial energy dose (white light), detected 6 h after initial PD insult and quantification of the western blot analysis. Quantifications are performed as the ratio of LC3-II over actin. A representative western blot (n = 3) is shown. (Inset) Levels of ROS detected in the empty-vector transfected cells (i.e., Neo cells), measured by DCF-fluorescence using FACS, at different energy doses (white light). (D) Kinetics of GFP-LC3 puncta formation in a L929 Neo population challenged with 4.5 J•cm−2 (●) of 572 ± 12-nm light delivered by microscopic PDT, monitored by CTLM, compared with the control (○), i.e., cells with Hyp, but with no initial excitation. (E) Confocal microscopy images corresponding to the formation of GFP-LC3 labeled autophagosomes in L929 Neo(NaSe) cells taken at 6 and 16 h after PD stress (1.1 J•cm−2, white light) ([CQ] = 5 μM). Scale bar: 10 μm.](/cms/asset/96c3eea8-f967-47b4-9916-b94bc60dc806/kaup_a_10920763_f0001.gif)
We recently reported that ROS generated by Hyp-PD stress stimulate autophagy pathways in both fibroblasts and HeLa cells, in the attempt to limit the propagation of oxidative damage and protecting the cells from mitochondrial apoptosis.Citation31 Likewise in L929 cells Hyp-PD stress led to a dose- and time-dependent accumulation of lipidated LC3-II as measured by western blotting and as quantified through analysis of LC3 puncta formation by CTLM in GFP-LC3 stably transfected L929 cells (). Moreover, consistent with previous works,Citation31 the presence of 3-methyladenine (3-MA), an inhibitor of the class III PtdIns 3-kinase complex required for autophagic vacuole formation,Citation32 or the siRNA mediated knockdown of the essential autophagy gene ATG5, increased the cytotoxic effects of Hyp-PD stress, suggesting that autophagy stimulation has a similar cytoprotective role in L929 cells (Fig. S1D–G).
Unirradiated control cells containing Hyp did not display changes in autophagosome number over time, measured by CTLM, therefore ruling out the contribution of the imaging process to the detection of this cellular response. PD-treated cells overexpressing GFP-only did not show puncta formation (data not shown), confirming that the puncta observed in GFP-LC3 treated cells are due to autophagosome formation rather than unspecific GFP aggregation.
The accumulation of LC3-II () or GFP-LC3 puncta () correlated well with the increased ROS production detected at different energy doses (, inset) and times () and was increased by the addition of the lysosomotric agent chloroquine (CQ), which inhibits the fusion between autophagosomes and lysosomes.Citation33 Presence of CQ increased the number of GFP-labeled autophagosomes (; see for quantification), which is more evident at 16 h due to the higher accumulation of oxidative damage, thereby confirming that Hyp-PD stress also stimulates autophagic flux in L929 cells.
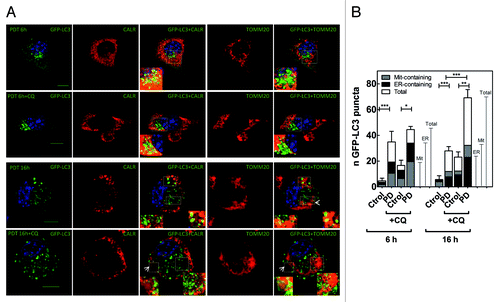
Figure 2. Spatio-temporal evolution of autophagy (A) Confocal microscopy images corresponding to the analysis of the autophagic flux for Neo(Nase) cells, taken at 6 and 16 h after PD stress (1.1 J•cm−2, white light), and for the colocalization of the autophagosomes with ER- and mitochondria-fragments. Calreticulin (CALR) was labeled with the Texas Red fluorophore, whereas TOMM20 with AlexaFluor 647; for clarity of the merged images, images corresponding to TOMM20 are shown in false color red. Arrows indicate ring-shaped autophagosomes. Scale bar: 10 μm. (B) Fluorescence microscopy analysis of the autophagic flux for Neo(NaSe) cells. The graph plots the average of GFP-labeled autophagosomes per cell. Each bar includes the number of autophagosomes that contain/colocalize with ER and mitochondria fragments. Data represent mean ± SD of 4 independent experiments; in each experiment 30 cells were scored per condition.
All together these results point to a close correlation between the progression/propagation of the oxidative damage after the initial Hyp-PD stress at the ER and autophagy stimulation in L929 cells.
Hyp-PD stress activates reticulophagy and mitophagy in a spatiotemporal manner
Our previous studies indicated that although oxidative damage is initiated at the ER and induces immediate loss of ER-Ca2+ homeostasis, mitochondrial apoptosis is rapidly engaged.Citation30 This suggests the existence of a close crosstalk between ER and mitochondria, which uses ROS as second messengers.
Since autophagy can be used for the removal/clearance of defective/damaged organelles, we next investigated whether activation of autophagy by Hyp-PD stress could involve the selective clearance of the ER (reticulophagy) and/or mitochondria (mitophagy). To this end, we used immunofluorescence techniques to monitor whether autophagosomes colocalized or contained fragments of ER (immunolabeled with an anti-calreticulin antibody) or mitochondria (immunolabeled with anti-TOMM20 antibody), thus revealing the presence of reticulophagy and mitophagy. Confocal microscopy analysis confirmed that after Hyp-PD stress the number of autophagosomes co-localizing with the ER luminal chaperone calreticulin, relative to the total number of autophagosomes accumulating in the cell, was initially higher than those containing mitochondrial fragments, but decreased at later time points (i.e., 16 h). Intriguingly, at longer time points (i.e., 16 h) this situation was reversed and the amount of autophagosomes containing mitochondria fragments relative to the total number of autophagosomes became predominant (). Organelle-containing autophagosomes, which appear as rings-containing mitochondrial or ER fragments, were also clearly detected (; Fig. S2). Moreover, blockage of the autophagic flux by CQ further evidenced the temporal transition between these two forms of organelle-selective autophagy, since blockage of lysosomal degradation by CQ addition at the different phases of the autophagic process, caused the accumulation of undigested autophagosomes with different cargo (). Interestingly, the transition between these two types of selected autophagy was accompanied by a noticeable change in mitochondrial morphology (). At shorter times, i.e., 6 h after PD stress when reticulophagy was predominant, mitochondria appeared elongated forming a reticular network. On the other hand, at the time when mitophagy was prevalently stimulated (i.e., 16 h after irradiation) mitochondria appeared more fragmented. These observations are consistent with recent works reporting that during starvation elongated mitochondria are protected from autophagic elimination, whereas mitochondrial fragmentation precedes mitophagy.Citation34-Citation36
All together these results suggest that the ER localized photogeneration of ROS by Hyp-PD stimulates a spatio-temporal and organelle-specific autophagic degradation, which involves first reticulophagy and then mitophagy.
Hyp-PD mediated apoptosis is blocked by protecting mitochondria from oxidative damage
To further identify the nature, localization and signaling role of the ROS produced following Hyp-PD stress, we overexpressed different antioxidant enzymes (Fig. S3) in L929 cells at different subcellular locations: cytosolic catalase (CAT), cytosolic membranes-associated phospholipid glutathione peroxidase (GPX4),Citation37 and mitochondrial superoxide dismutase (SOD2). Cytosolic GPX4 is present in the cytosol where it is associated with plasma, nuclear or other organelle membranes, including the ER.Citation38-Citation40 Moreover, we engineered mutants of CAT and GPx4 to enable their forced expression (mt-CAT and mt-GPX4, respectively) in the mitochondria. Since oxidative stress is the initial trigger of all cellular responses in our paradigm, we first analyzed the impact of these antioxidant enzymes on different cell death parameters. While all cells accumulated Hyp to similar levels within the ER (Fig. S4A), the increased level of cell death observed at 24 h with increasing PD doses in the empty-vector transfected cells (i.e., Neo cells), was attenuated by the overexpression of the antioxidant enzymes, although clearly to a different extent (). Interestingly, while the protection provided by cytosolic CAT and GPX4 vanished after 24 h at higher PD doses, overexpression of mitochondrial antioxidants, predominantly SOD2 and mt-GPX4, was visibly more cytoprotective within the range of PD dose used. Moreover, CTLM analysis showed that overexpression of SOD2 slowed down the rate of cell death in a dose-dependent way (; Fig. S4B). It should be noted that the Neo-L929 cells that were cultured in medium containing sodium selenite (NaSe) required for the functions of GPX4 (i.e., NeoNaSe), displayed a reduced sensitivity to PD-mediated stress, likely due to an enhancement of the antioxidant activity of the basal levels of the seleno-dependent enzymes GPX within the range of concentration used.Citation41
Figure 3. Protecting mitochondria from oxidative damage blocks Hyp-PD apoptosis. (A) Hyp-induced PD cell death (trypan-blue-positive cells) in L929 Neo or L929 cells overexpressing different antioxidants as a function of the energy dose (white light), 24 after the initial PD insult. (*p < 0.05, **p < 0.01 and ***p < 0.001). (B) Effect of overexpression of SOD2 on the kinetics of cell death, measured by PI uptake using CTLM, as a function of energy dose (572 ± 12-nm light), compared with Neo cells. (C) Effect of antioxidants on cardiolipin peroxidation detected at 6 h after an energy dose of 1.1 J•cm−2 (white light), measured as percentage of cell population showing loss of NAO fluorescence. (D) Measurement of caspase-3 activity detected at 6 h after an energy dose of 1.1 J•cm−2 (white light), measured as fluorescence AMC released after caspase-3-fluorescent substrate cleavage. (*p < 0.05, **p < 0.01 and ***p < 0.001). L929 cells overexpressing the selenium dependent enzyme GPX4 were grown in media supplemented with sodium selenite (NaSe) required for the enzyme activity.
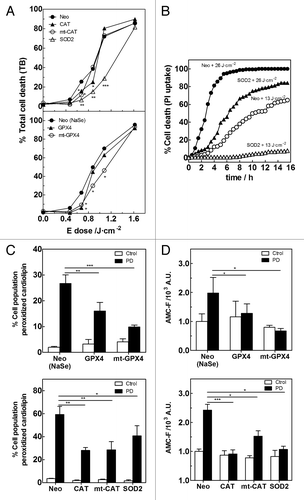
To evaluate the propagation of the oxidative damage from the ER to the mitochondria, we next examined the levels of cardiolipin (CL) peroxidation produced after PD stress in the different L929 cell lines. CL is a mitochondrial-specific anionic phospholipid that is located in the inner and outer leaflets of the mitochondrial inner membrane where it interacts with cytochrome C (Cyt C) forming a complex with peroxidase activity.Citation42,Citation43 CL-peroxidation is one of the earliest apoptotic events occurring in response to different pro-oxidant insults, thereby providing a marker for redox-regulated mitochondrial events linked to cell death.Citation44,Citation45 Photodamage at the ER yields CL peroxidation in L929 Neo cells, indicative of mitochondrial damage (). Overexpression of mitochondrial antioxidant enzymes, like SOD2 and mt-CAT attenuated CL peroxidation following Hyp-PD stress suggesting that production of O2•− and H2O2 by mitochondria contributes to this event. Interestingly, overexpression of mt-GPX4, which reduces CL-peroxides to the corresponding alcohols in situ, yielded the predominant protection (). Moreover, the strong protection displayed by the cytosolic membranes-associated GPX4 and CAT suggests that ER-photogenerated phospholipid hydroperoxides and H2O2 can propagate oxidative damage to the mitochondrial CL (). Consistent with a role of CL peroxidation in Cyt C release,Citation44,Citation45 immunocytochemistry analysis showed that the redistribution of Cyt C in the cytosol after PD stress and loss of transmembrane potential, were prevented by the overexpression of cytosolic GPX4 or SOD2 (Fig. S4C and S4D). Moreover, caspase-3 activation after PD stress was significantly blocked by both cytosolic GPX4 and CAT, but also by mitochondrial antioxidants (). Nevertheless, because overexpression of SOD2 and mt-GPX4 was overall more cytoprotective (), these data suggest that through a 1O2-induced ROS-release, mitochondrial ROS and mitochondria lipid hydroperoxides amplify cell death pathways.
All together, these results reveal that the delocalization of oxidative damage from ER to the mitochondria via phospholipid hydroperoxides and H2O2 generates secondary mitochondrial ROS, which amplify cell death.
Organelle-specific autophagy is modulated by secondary ROS
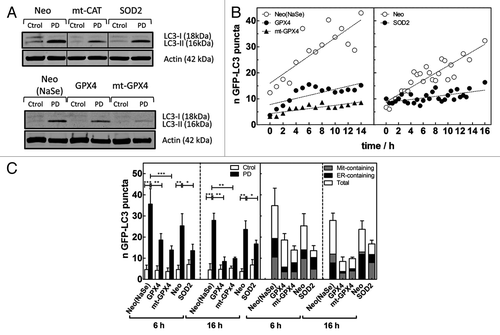
We next studied the effect of the overexpression of antioxidant enzymes on different autophagic parameters. As expected, overexpression of different antioxidants alleviated the oxidative burden and significantly reduced LC3-II accumulation and the number of GFP-labeled autophagosomes detected after Hyp-PD stress (Fig. 4). Furthermore, using CTLM after microscopic PD stress, we were able to compare the kinetics of autophagosome formation in the absence and presence of the antioxidant enzymes (). The increased expression of cytosolic GPX4 or mitochondrial SOD2 slowed down the kinetics of autophagosome formation compared with the PD-treated Neo population, consistent with the attenuation of LC3-II levels detected by western blot (). The CTLM analysis confirmed the higher efficiency of mt-GPX4 in mitigating autophagy induction by Hyp-PD stress (), therefore confirming a major role for mitochondrial PhLOOH in the amplification of this ROS-mediated autophagy.
Figure 4A–C. Secondary ROS modulate organelle-specific autophagy. (A) Effect of overexpression of antioxidant enzymes on LC3-I-to-LC3-II conversion determined by western blot analysis of whole cell lysates detected 6 h after initial PD insult (1.1 J•cm−2, white light). A representative western blot (n = 3) is shown. The upper panel is a composite of 3 different lanes (same exposure) of the same gel. (B) Effect of cytosolic membranes-associated and mitochondrial GPX4 (left) and SOD2 (right) on the kinetics of GFP-LC3 puncta formation, compared with the Neo(NaSe) population, in populations challenged with 4.5 J•cm−2 of 572 ± 12-nm light delivered by microscopic PDT, monitored by CTLM. (C) (Left) Quantification of the number of GFP-labeled autophagosomes detected in the images (D), represented as the average of GFP-LC3 puncta per cell (*p < 0.05, **p < 0.01 and ***p < 0.001) and (right) quantification of the number of autophagosomes that contain/colocalize with ER and mitochondria fragments. Data represent mean ± SD of 4 independent experiments; in each experiment 30 cells were scored per condition.
Overexpression of antioxidants not only reduced the kinetics of autophagosome formation, but it also affected the levels of reticulophagy and mitophagy detected after Hyp-PD stress, thus providing information on the predominant ROS contributing to these processes.
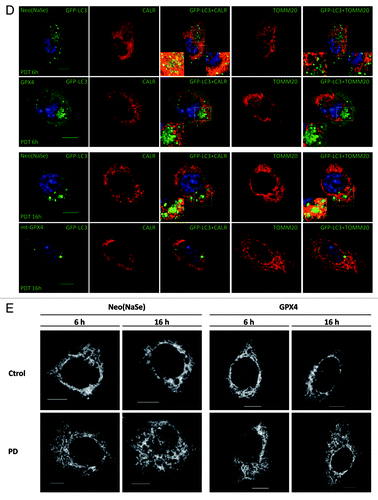
Confocal microscopy analysis () indicated that overexpression of cytosolic GPX4 curbed mostly the selective reticulophagy (relative to the total number of autophagosomes) occurring 6 h after Hyp-PD of Neo cells and which preceded the clearance of mitochondria at later time points (see also ). Mitigation of reticulophagy resulted in a reduction of the number of mitochondria colocalizing with the autophagosomes over time (i.e., 16 h), thus suggesting that following the apical ER photodamage, PhLOOH propagate the oxidative wave to mitochondria. In contrast, while overexpression of mitochondrial antioxidants reduced the total number of autophagosomes formed after Hyp-PD (), it did not affect the relative fraction of ER-containing autophagosomes at 6 h after irradiation, when compared with Neo(NaSe) cells. As expected, the fraction of autophagosomes colocalizing with mitochondria at 16 h was significantly reduced by mitochondrial antioxidants.
Intriguingly, the overexpression of cytosolic GPX4 and mitochondria-associated enzymes, mt-GPX4 and SOD2, also prevented the Hyp-PD induced morphological changes in the mitochondrial network ().
Figure 4D and E. Secondary ROS modulate organelle-specific autophagy. (D) Confocal microscopy images corresponding to the effect of antioxidants on the number of GFP-labeled autophagosomes detected at 6 and 16 h after Hyp-PD stress as well as the colocalization of the autophagosomes with ER- and mitochondria fragments. Calreticulin (CALR) antibody was labeled with the Texas Red fluorophore, whereas TOMM20 antibody with AlexaFluor 647; for clarity of the merged images, images corresponding to TOMM20 are shown in false color red. (E) Confocal microscopy images corresponding to the changes in mitochondrial morphology that accompany transition from reticulophagy to mitophagy in Neo(NaSe) cells and protection displayed by GPX4. Scale bar: 10 μm.
These results strongly suggest that after the initial photo-oxidative stress to the ER, PhLOOHs propagate to the adjacent mitochondria and trigger the disruption of the reticular mitochondrial network, which favors subsequent mitochondria ROS generation and mitophagy.
Discussion
Recent studies have reported that the local photogeneration of ROS (i.e., photodynamic or PD stress) initiated by photosensitization is a trigger of autophagy pathways.Citation30,Citation46-Citation49 In particular, we recently reported that PD stress with hypericin activates (macro)autophagy, as well as chaperone-mediated autophagy and found that both pathways are functionally involved in mitigating ROS-damage and apoptotic cell death.Citation31 Although autophagy stimulation was associated with the rapid ROS-mediated downregulation of the Akt-mTOR pathway and not with the loss of function of BCL2 by direct photodamage,Citation50,Citation51 the effects of PD stress on organelle clearance by (macro)autophagy have not been investigated thus far. Since PD stress induces a primary boost of ROS that is initially confined to the subcellular sites or the organelle where the photosensitizer preferentially accumulates,Citation19,Citation52 it provides an interesting paradigm to study the impact of ROS-mediated organelle damage and the involvement of selective organelle degradation by autophagy.
In this study, we used L929 cells overexpressing antioxidant enzymes located at the cytosol/cytosolic membranes or targeted to the mitochondria to evaluate the type of ROS and the impact of their compartmentalization on the clearance of the ER and mitochondria in response to Hyp-PD stress. Since initially Hyp-photosensitized 1O2 generation is spatially confined in the ER membranes, but secondary ROS are ultimately conveyed to the mitochondria, we evaluated the kinetics of selective autophagy (i.e., reticulophagy and mitophagy) as well as apoptotic/cell death parameters in these different cell lines.
This study highlights that the major contribution to cellular oxidative damage, which is the trigger of both autophagy and cell death mechanisms, is due to secondary mitochondrial ROS generation, rather than direct ER-photo-oxidation. Indeed, cytosolic antioxidants, and especially GPX4, can attenuate initial ER-photodamage, which is the event triggering the selective and apical clearance of this organelle (reticulophagy), but are less efficient in preventing overall cell death, when compared with the more long-lasting cytoprotective effects conferred by the mitochondria-associated antioxidant enzymes.
Because Hyp is excluded from mitochondria, mitochondrial ROS production can only be a downstream process occurring after primary ER-photodamage and for which a signaling mediator is required. We hypothesized that hydroperoxides (or their secondary metabolites) initially photogenerated at the ER or H2O2 could have this signaling role. In fact, overexpression of cytosolic GPX4 and CAT reduced both autophagy (reticulophagy first and later on mitophagy) and mitochondrial apoptotic responses, thus assigning to PhLOOH (or PhLOOH-derived metabolites) and H2O2 a signaling role as oxidative mediators in the propagation of the initial photo-oxidative damage. Interestingly, 4-hydroxynonenal (4-HNE), an end product of lipid peroxidation that quickly forms adducts with proteins, has been shown to participate in the activation of both processes in other cellular systems.Citation53-Citation55
Different studies have shown that PhLOOHs can translocate/transfer between membranes,Citation26,Citation56-Citation59 through the cellular aqueous compartments, to expand their signaling oxidative range throughout the cell, at locations remote from the initial site of photo-oxidation. The occurrence of these events is consistent with the generation of long-lived species such as hydroperoxides, including H2O2. Moreover, PhLOOH intermembrane transfer is in some cases facilitated by proteins. Under oxidative stress conditions, the sterol carrier protein-2 (SCP-2), which has been implicated in the cholesterol (Ch) movement from the ER to mitochondria,Citation60,Citation61 was involved in transferring ChOOHs and PhLOOHs to the mitochondria, which exacerbated oxidative damage resulting in a significant loss of ΔΨm. PhLOOH translocation between ER- and mitochondria could be mediated by the mitochondrial-associated membranes (MAMs). MAMs are the proteinaceous structures tethering the ER and mitochondria, which allow regulation of Ca2+ fluxes and phospholipids transfer between these organelles (10–25 nm).Citation62 Interestingly, we have recently observed that MAMs disruption prevents the propagation of ROS to the mitochondrial apoptotic machinery after Hyp-PD stress (Verfaillie et al., unpublished data), therefore suggesting that MAMs could facilitate hydroperoxides translocation/dissemination between these organelles after PD stress.
This study also shows that as a result of this ROS event, damaged mitochondria amplify ROS generation in the form of O2•−, which easily dismutates into H2O2. H2O2 is thought to be the ROS mediating CL-peroxidation, which is catalyzed by the peroxidase activity of the complex CL-Cyt C.Citation42,Citation43 A recent work by Belikova et al.,Citation63 however, revealed that fatty acid hydroperoxides can also provide the oxidizing equivalents to the peroxidase complex that results in CL-peroxidation. Therefore, it is possible that fatty acid hydroperoxides hydrolyzed by phospholipase-mediated hydrolysis once translocated from the ER to the mitochondria, can directly mediate CL-peroxidation. Our CTLM experiments show that deactivation of O2•− by SOD2 decreases drastically the rate of cell death, thus confirming the contribution of these mitochondrially-generated secondary ROS in Hyp-PD mediated photokilling. Moreover, the observation that SOD2 (and also mt-CAT) expression results in a major attenuation of Hyp-PD mediated autophagy by reducing predominantly mitophagy, reveals the contribution of mitochondrial O2•− (and H2O2) production to autophagy stimulation, as reported in other ROS paradigms or after starvation.Citation12-Citation16 Recent studies using photosensitizers that accumulate preferentially in the mitochondria and which, therefore, initiate photo-oxidation in these organelles, report the clearance of photodamaged mitochondria by mitophagy.Citation64,Citation65
In our study, since the initial ROS damage occurs at the ER, we could evaluate the spatio-temporal transition from reticulophagy to mitophagy, and show that this process is accompanied by changes in mitochondrial morphology. The reticular mitochondrial network observed 6 h after Hyp-PD stress, where selective reticulophagy is the dominant process, becomes fragmented at longer times, (i.e., 16 h) when mitophagy takes over. Overexpression of GPX4 and mt-GPX4, both reducing the levels of mitophagy after PD stress, keeps the highly fused mitochondrial network. These observations are consistent with recent works showing that induction of autophagy by starvation leads to mitochondrial elongation to maintain mitochondrial mass,Citation34-Citation36 although, when mitochondrial integrity is compromised and these organelles become autophagosomal targets, their fragmentations precedes their engulfment by selective mitophagy. Furthermore, our observations are consistent with those of Twigg et al.Citation34 reporting that mitochondrial fission is linked to mitophagy, but not to reticulophagy.
On the other hand, our study further identifies in the downstream products of mitochondrial ROS, mainly in the form of CLOOH, the major ROS-derived molecules produced in the mitochondria are still able to have an effect on cellular responses induced by PD-mediated oxidative damage. Indeed, detoxification of CLOOH by mt-GPX4 reduces the oxidative damage amplified by mitochondria ROS production and consequently also slows down the kinetics of autophagosome formation, especially by reducing mitophagy.
These results are consistent with the study of Kissová et al.Citation66 which reported that inhibition of the mitochondrial lipids oxidation slows down autophagy in yeasts. Likewise, reduction of CLOOH to the corresponding alcohol decreases caspase-3 activation and eventual cell death (), thus confirming previous studies defining the anti-apoptotic role of mt-GPX4.Citation40,Citation67
CLOOH has been reported to be an upstream event in apoptosis activation when initial 1O2 mediated photo-oxidation is targeted/localized in the mitochondria by using ALA- or photofrin-photosensitized PD stress.Citation68,Citation69 Although the protective effect shown by mt-GPX4 is expected to be against CLOOH, it cannot be excluded that PhLOOH produced immediately after initial photodamage at the ER is able to translocate into the mitochondria and results in further peroxidation of mitochondrial components.
Processes downstream of CL-peroxidation lead to Cyt C- and other pro-apoptotic factors- release, which is considered as the point of no return in many paradigms of cell death.Citation70 Therefore, our results support the hypothesis proposed by Kagan et al.Citation45 that CLOOH could act as a ‘molecular switch’ that initiates the development of pre-apoptotic events when oxidative pressure inside the mitochondria is too high and mitophagy fails to effectively eliminate oxidatively damaged cellular components. In line with this, Korytowski et al.Citation71 have recently published that CLOOHs and other CL oxidation products can translocate via the aqueous intermembrane space from the inner mitochondrial membrane to the outer mitochondrial membrane of oxidatively stressed mitochondria in order to sensitize the outer membrane for pro-apoptotic recruitment of tBID and BAX. This is an interesting possibility which deserves to be explored in further studies.
Materials and Methods
Chemicals
Hyp was synthesized and purified as described previously.Citation72 Antibodies against LC3 (3868) and SERCA2 (4388) were from Cell Signaling Technology. Anti-Cyt C antibody (556433) was from BD PharMingen. Antibodies against TOMM20 (ab56783) and calreticulin (ab92516) were from Abcam. DMEM medium (21969), phenol red-free DMEM medium (31053-028), DMEM/F-12 (1:1) medium (11039-021), geneticin (10131-019), puromycin (A11138-03), 3,3′-dihexyloxacarbocyanine iodide (DioC6(3), D-273), 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCF-DA, C6827), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, D1306), ER-tracker Blue-WhiteDPX (E12353), MitoTracker Deep Red (M-22426), ProLong Gold antifade reagent (P36934), Texas Red goat anti-rabbit IgG(H+L) (T2767) and Alexa Fluor 647 goat anti-mouse IgG(H+L) (A21235) were purchased from Invitrogen. Fetal bovine serum (FBS, Hyclone, triple 0.1 μm sterile filtered, SV30160.03), DyLight 800 conjugated goat anti-rabbit IgG(H+L) (35571) and DyLight 680B conjugated goat anti-mouse IgG(H+L) (35525) were from Thermo Scientific. Penicillin/streptomycin (P0781), l-glutamine (G7513), trypan blue (TB, T-0776), 10-nonylacridine orange bromide (NAO, A-1372), 2,4-dinitrophenylhydrazine (97%, reagent grade, D199303), 3-MA (M9281), chloroquine diphosphate salt (CQ, C6628), dimethylsulfoxide (DMSO, ≥ 99.9%, ACS, spectrophotometric grade, 154938), ethyl acetate (≥ 99.5%, ACS, spectrophotometric grade, 154857), Dulbecco’s phosphate buffered saline (PBS, D8537), bovine serum albumin (BSA, ≥ 96%, essentially fatty acid free, A6003), goat serum (G9023), NaSe (S5261), propidium iodide (PI, 4170), anti-actin antibody (A5441) and thiazolyl blue tetrazolium bromide (MTT reagent) (M5655) were purchased from Sigma-Aldrich. ON-TARGET plus SMARTpool for Atg5 siRNA (L-064838-00), the ON-TARGET plus CONTROL POOL nontargeting siRNA (D001810-10-20) and Dharmafect Transfection Reagent 1 (T-2001-02) were from Dharmacon (Thermo Scientific).
Cell culture
L929 murine fibrosarcoma cells overexpressing antioxidants were maintained in DMEM containing 4.5 g•L−1 glucose and 0.11 g•L−1 sodium pyruvate and supplemented with 2 mM l-glutamine, 100 units•mL−1 penicillin, 100 μg•mL−1 streptomycin, 0.5 mg•mL−1 geneticin and 10% FBS. Media for GPX4- and mt-GPX4-overexpressing cells was supplemented with NaSe (43 nM). Stably GFP-LC3 expressing cells were grown in the previous media supplemented with puromycin (12 μg•mL−1).
Cloning of the antioxidant enzyme cDNA sequences in the pCAGGS expression vector and transfection of L929 cells
All antioxidant enzyme cDNA sequences were cloned into the pCAGGS expression vector under control of a eukaryotic hybrid β-actin/β-globulin promotor preceded by a human cytomegalovirus immediate early promotor. Following constructs were made: pCAGGS_human-SOD2 (pCAGGS_h-SOD2), _h-SOD1, rat-CAT (r-CAT), mitochondrial CAT_(rh-mt-CAT), pig-cytosolic GPX4 (_p-cGPX4) and mitochondrial GPX4_(ph-mt-GPX4). The rh-mt-CAT-construct is the genetically modified r-CAT gene wherein the mitochondrial targeting sequence of h-SOD2 was inserted N-terminally and the peroxisomal targeting sequence of r-CAT was deleted. Similarly to rh-mt-CAT, the ph-mt-GPX4 construct was obtained by inserting the mitochondrial targeting sequence of h-SOD2 N-terminally of the p-GPX4 construct. Specificity of the primers was established by verifying the length of the amplicon sequence along with the absence of amplification in the empty vector-transfected L929 cells (i.e., Neo cells) and cells overexpressing the antioxidant enzymes. However, as mt-CAT- and mt-GPX4-constructs were designed by modifying CAT and GPX4 respectively, amplification also occurs when r_CAT and p_GPX4 primers are used. Conversely, rh_mt-CAT and ph_mt-GPX4 primers were designed to specifically recognize the mitochondrial targeting sequence and therefore amplification only occurs with the mitochondrial enzymes. Transfection of the antioxidant enzyme constructs was performed through cotransfection with a geneticin-resistance construct in a 10:1 ratio using lipofectamine (Invitrogen) according to the manufacturer’s protocol. Stable overexpression of the antioxidant enzymes was obtained through culturing of the cells in the presence of geneticin.
Transduction of L929 cells with GFP-LC3 and GFP
Phoenix-Eco packaging cells (ATCC product SD-3443) were transfected with pBabe-EGFP-LC3 (obtained from Dr. Debnath; Department of Pathology and Biomedical Sciences Graduate Program) using the calcium phosphate precipitation method.Citation73 Twenty-four to 72 h after transfection viral supernatant was collected, filtered through a 0.45-µm filter and subsequently used to transduce L929 cells. Stable cell lines expressing GFP-LC3 were selected with puromycin (12 μg•mL−1).
Photosensitization
Photodynamic experiments were performed 24 h after plating cells in 10-cm plates. Cells were incubated with Hyp [200 nM, diluted from the stock solution (750 μM in DMSO)] in serum-free DMEM for 2 h. Prior to irradiation, Hyp solution was removed and cells were washed with PBS. Irradiation was performed in PBS. Cell culture plates were placed on a plastic diffuser sheet which received the light of a set of seven L18W30 fluorescent lamps (Osram) (white light). The irradiance of the lamps was 4.5 mW•cm−2 and was measured using an IL 1400 radiometer (detector SEL 033) (International Light). The fluence delivered (J•cm−2) to the cells was determined from the product of the irradiance and the duration of irradiation. After irradiation, cells were incubated in DMEM media containing 10% FBS until the time of the experiment. For autophagy monitoring experiments, CQ (5 μM) and 3-MA (5 mM) were added immediately after irradiation.
Computerized time-lapse microscopy (CTLM) and microscopic PDT
For microscopic PDT, cells were plated on Labtek 8-well chambered coverglasses (German borosilicate, Nunc). Prior to incubation of the photosensitizer, a cell population (field of view) was selected per well. After being washed in plain DMEM/F-12 (1:1), cells were incubated with Hyp (25 nM) for 1 h at 37°C in the buffered media in the stage of the microscope. In these experiments, Hyp concentration is lowered to ensure that there is not significant PD effect deriving from the imaging process that would perturb the study. After removing the excess of photosensitizer and washing cells with plain media, “live-imaging buffered media” [DMEM/F-12 (1:1) media containing 10% FBS, 100 units•mL−1 penicillin and 100 μg•mL−1streptomycin] was added to each well. The preselected cell population was illuminated through the objective of an Olympus XSi cell fluorescence microscope using a 572 ± 12 nm bandpass filter (Fig. S1A). Imaging started just after irradiation. The light irradiance was measured with an IL 1400 radiometer (detector SEL 033) (International Light). The detection area of this device is 0.33 cm2 and all light was incident into the detector surface. The measured irradiance was then corrected to the irradiance at the focal spot produced by the objective used in each experiment.
CTLM was used to follow the responses of the targeted population after PD treatment. Images were acquired for a given field at 30 min intervals over the duration of the experiment. Cell viability experiments were performed using the PI exclusion method, where a PI solution (2.24 μM in “live-imaging buffered media”) was added to the well prior to imaging. Fluorescence of PI-positive cells was measured using the Texas Red filter set (Δλexc: 572 ± 12 nm; Δλem: 627 ± 27 nm). The kinetics of autophagosome formation was determined by following the formation of GFP-LC3 puncta as a function of time. ROS levels were determined by measuring the oxidized fluorescent form of CM-H2DCF-DA, DCF. Immediately after irradiation, CM-H2DCF-DA (was added to each well to a final concentration of 1.6 μM (in this case the “live-imaging buffered media” only contains 3% FBS).The green fluorescence of the oxidized product DCF was imaged over time using a FITC filter set (Δλexc: 492 ± 9 nm; Δλem: 530 ± 20 nm).
Immunofluorescence
Cells were grown and treated on Labtek 2-well chambered coverglasses (German borosilicate, Nunc). For reticulophagy and mitophagy experiments, cells overexpressing GFP-LC3 were fixed at 6 and 16 h after treatment with 4% p-formaldehyde at 37°C, permeabilized with 0.1% TritonX-100 at room T for 20 min, blocked (PBS containing 5% BSA and 1% goat serum) and incubated with anti-calreticulin and anti-TOMM20 antibodies, which were labeled with Texas Red and AlexaFluor 647, respectively. For Cyt C release immunodetection, cells were fixed at 6 h after treatment and permeabilized with 0.1% TritonX-100 at 4°C. After blocking, cells were incubated with anti-Cyt C antibody and labeled with secondary antibody (DyLight680) in blocking buffer. In all experiments, cells were counterstained with DAPI (1 μg•mL−1), mounted using Prolong Gold antifade reagent and cured overnight. Fluorescence images were acquired using a Nikon A1R confocal unit mounted on a Ti2000 inverted microscope controlled by NIS elements acquisition software (Nikon Instruments Inc.) (reticulophagy and mitophagy experiments) using a Plan APO VC 60× 1.4NA oil immersion objective and an Olympus FluoView FV1000 confocal microscope using a UPlansAPO 60× 1.35NA oil immersion objective (Cyt C experiments).
Image analysis
Images were analyzed using ImageJ software from NIH.Citation74 For cell viability experiments, a total of 200–300 cells per field were analyzed where the total number of cells and the number of cells showing red (PI) fluorescence were counted to determine the percentage of cell death as a function of time. The kinetics of autophagosome formation was determined by counting the number of GFP-LC3 puncta produced as a function of time. The levels of intracellular ROS were determined by integration of intracellular DCF fluorescence using ImageJ. The background fluorescence of the medium proximal to each cell was also integrated and substracted from the intracellular intensity in order to obtain the corrected intracellular fluorescence. Colocalization analysis of GFP-labeled autophagosomes with ER and mitochondria was done with the help of the colocalization color map plugging of ImageJ.
Cell death assay
Cell death was quantified using the TB exclusion method, as described previously.Citation30
Cell viability assay (MTT assay)
At the specific times after treatment, cells were incubated with MTT solution (0.5 mg•mL−1) in DMEM media containing 10% FBS for 2 h. Afterwards, the formazan crystals were dissolved in a mixture ethanol/DMSO (1:1) and the absorbance of the obtained solutions was determined at 550 nm.
Caspase-3 activity
Cells were washed twice with ice-cold PBS, scrapped-off the plate and centrifuged to get a cell pellet that was resuspended in 60 μL of lysis buffer (100 mM HEPES pH7.4, 10% sucrose, 1% Triton-X100, 2.5 mM EDTA, 5mM DTT, 1 mM PMSF, 2 μg•mL−1 pepstatin and 2 μg•mL−1 leupeptin), 6 h after PD treatment. Samples containing 50 μg of protein were incubated with Ac-DEVD-AMC 50μM (Bachem, I-1660) in 250 μL lysis buffer and incubated at 37°C for 30 min. Fluorescence (λexc = 360 nm; λem = 460 nm) was measured in a Flexstation 3 Plate Reader (Molecular Devices) using a black 96-well (μ-clear) plate (Greiner Bio-One).
Cardiolipin peroxidation and mitochondria membrane potential (ΔΨm)
Cardiolipin peroxidation and loss of ΔΨm were measured by following the loss of NAO and DioC6(3) fluorescence, respectively, using fluorescence-activated cell sorting (FACS). At the specific times after treatment, cells were washed twice with pre-heated PBS at 37°C, trypsinized, neutralized with DMEM medium containing 10% FBS and centrifuged. The cell pellet was then resuspended in either NAO (5 nM) or DioC6(3) (10 nM) solutions in phenol red-free DMEM medium and incubated for 20 min. Afterwards, cell suspension was centrifuged and cell pellet resuspended in fresh phenol red-free DMEM medium.
Western blots
Preparation of cell lysates, determination of protein concentration and sample preparation forwestern blotting were described in a previous work.Citation75 Samples were processed on the Criterion TM system (Bio-Rad Laboratories) on 4–12% Bis-Tris gels and Protan 2 μm-pored nitrocellulose paper (Perkin-Elmer). Li-Cor Odissey IR imager was used as western blot detection system for scanning and quantification of immunoblots.
Activity assays
SOD-activity was analyzed on native 10% PAGE and staining with nitro-blue tetrazolium chloride according to Beauchamps and Fridovich.Citation76 CAT-activity was determined by analyzing the H2O2-mediated oxidation of CM-H2DCF-DA through FACS analysis. CM-H2DCF-DA (final concentration of 5 µM) and H2O2 (final concentration of 1 mM) were added simultaneously to the cell suspension and incubated for 30 min. Oxidation of the probe by H2O2 was determined by measuring the green fluorescence of the oxidized form DCF. GPx4-activity was measured by the coupled test procedure developed for measuring GPx-activity but with phosphatidylcholine hydroperoxide (PCOOH) as substrate.Citation77 PCOOH was prepared as described earlier.Citation78 One unit of activity is the amount of enzyme catalyzing the reduction of 1 µmol of PCOOH/min at 37°C and pH 7.4.
Protein carbonylation
Protein carbonylation was detected spectrophotometrically by following the absorption band (370 nm) of the hydrazone generated by reaction of the protein carbonyls with 2,4-dinitrophenylhydrazine, according to the method described by Hawkins et al.Citation79
siRNA transfection
Cells were transfected by adding 485 μL serum-free DMEM media with 10 μL Dharmafect 1 transfection reagent and a final concentration of 40 nM SiRNA to 10-cm dishes with 2 mL of DMEM media containing 1% FBS. Four hours after transfection 2.5 mL DMEM containing 10% FBS were added. A second transfection was performed 24 h later. Cells were re-plated and photosensitization experiments were performed 24 and 48 h, respectively, after the second transfection.
Statistical analysis
Results represent the mean ± SD of at least three independent experiments. Statistical analysis was performed using GraphPad Prism 5.03 and p values were calculated using the unpaired t-test with p < 0.05 considered statistically significant.
| Abbreviations: | ||
| CAT | = | catalase |
| CL | = | cardiolipin |
| CTLM | = | computerized time-lapse microscopy |
| Cyt C | = | cytochrome C |
| ER | = | endoplasmic reticulum |
| GPX4 | = | phospholipid glutathione peroxidase |
| Hyp | = | hypericin |
| mt | = | mitochondrial |
| PD | = | photodynamic |
| PDT | = | photodynamic therapy |
| PhLOOH | = | phospholipid hydroperoxide |
| ROS | = | reactive oxygen species |
| SERCA | = | sarco(endo)plasmic-reticulum Ca2+-ATPase |
| SOD | = | superoxide dismutase |
| TB | = | trypan blue |
| 1O2 | = | singlet oxygen |
| ΔΨm | = | mitochondria membrane potential |
Additional material
Download Zip (10.3 MB)Acknowledgments
This work is supported by GOA/11/2009 grant of the K.U.Leuven to P. Agostinis. This paper presents research results of the IAP6/18 funded by the Interuniversity Attraction Poles Programme, initiated by the Belgian State, Science Policy Office. N.Rubio thanks F.R.S.-FNRS for a postdoctoral grant (grant F/5/4/5-MCF/KP). Nikon confocal microscope was acquired through a Hercules Type 1 AKUL/09/037 to W. Annaert. GFP-LC3 L929 cell lines were generated by the GIGA Viral Vector platform.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/20768
References
- Dreher D, Junod AF. Role of oxygen free radicals in cancer development. Eur J Cancer 1996; 32A:30 - 8; http://dx.doi.org/10.1016/0959-8049(95)00531-5; PMID: 8695238
- Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer 2007; 121:2381 - 6; http://dx.doi.org/10.1002/ijc.23192; PMID: 17893868
- Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat 2004; 7:97 - 110; http://dx.doi.org/10.1016/j.drup.2004.01.004; PMID: 15158766
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach?. Nat Rev Drug Discov 2009; 8:579 - 91; http://dx.doi.org/10.1038/nrd2803; PMID: 19478820
- Ozben T. Oxidative stress and apoptosis: impact on cancer therapy. J Pharm Sci 2007; 96:2181 - 96; http://dx.doi.org/10.1002/jps.20874; PMID: 17593552
- Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 2010; 48:749 - 62; http://dx.doi.org/10.1016/j.freeradbiomed.2009.12.022; PMID: 20045723
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 77; http://dx.doi.org/10.1016/S1534-5807(04)00099-1; PMID: 15068787
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Azad MB, Chen YQ, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal 2009; 11:777 - 90; http://dx.doi.org/10.1089/ars.2008.2270; PMID: 18828708
- Gibson SB. A matter of balance between life and death: targeting reactive oxygen species (ROS)-induced autophagy for cancer therapy. Autophagy 2010; 6:835 - 7; http://dx.doi.org/10.4161/auto.6.7.13335; PMID: 20818163
- Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 2010; 6:838 - 54; http://dx.doi.org/10.4161/auto.6.7.12113; PMID: 20505317
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749 - 60; http://dx.doi.org/10.1038/sj.emboj.7601623; PMID: 17347651
- Chen YQ, McMillan-Ward E, Kong JM, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci 2007; 120:4155 - 66; http://dx.doi.org/10.1242/jcs.011163; PMID: 18032788
- Chen YQ, Gibson SB. Is mitochondrial generation of reactive oxygen species a trigger for autophagy?. Autophagy 2008; 4:246 - 8; PMID: 18094624
- Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 2008; 15:171 - 82; http://dx.doi.org/10.1038/sj.cdd.4402233; PMID: 17917680
- Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 2009; 16:1040 - 52; http://dx.doi.org/10.1038/cdd.2009.49; PMID: 19407826
- Jiang J, Maeda A, Ji J, Baty CJ, Watkins SC, Greenberger JS, et al. Are mitochondrial reactive oxygen species required for autophagy?. Biochem Biophys Res Commun 2011; 412:55 - 60; http://dx.doi.org/10.1016/j.bbrc.2011.07.036; PMID: 21806968
- Halliwell B, Gutteridge JMC. Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts. Arch Biochem Biophys 1986; 246:501 - 14; http://dx.doi.org/10.1016/0003-9861(86)90305-X; PMID: 3010861
- Redmond RW, Kochevar IE. Spatially resolved cellular responses to singlet oxygen. Photochem Photobiol 2006; 82:1178 - 86; http://dx.doi.org/10.1562/2006-04-14-IR-874; PMID: 16740059
- Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin 2011; 61:250 - 81; http://dx.doi.org/10.3322/caac.20114; PMID: 21617154
- Hamblin MR, Hasan T. Photodynamic therapy: a new antimicrobial approach to infectious disease?. Photochem Photobiol Sci 2004; 3:436 - 50; http://dx.doi.org/10.1039/b311900a; PMID: 15122361
- Dai T, Huang YY, Hamblin MR. Photodynamic therapy for localized infections-State of the art. Photodiagn Photodyn Ther 2009; 6:170 - 88; http://dx.doi.org/10.1016/j.pdpdt.2009.10.008
- Choudhary S, Nouri K, Elsaie ML. Photodynamic therapy in dermatology: a review. Lasers Med Sci 2009; 24:971 - 80; http://dx.doi.org/10.1007/s10103-009-0716-x; PMID: 19653060
- Riddle CC, Terrell SN, Menser MB, Aires DJ, Schweiger ES. A review of photodynamic therapy (PDT) for the treatment of acne vulgaris. J Drugs Dermatol 2009; 8:1010 - 9; PMID: 19894368
- Babilas P, Schreml S, Landthaler M, Szeimies RM. Photodynamic therapy in dermatology: state-of-the-art. Photodermatol Photoimmunol Photomed 2010; 26:118 - 32; http://dx.doi.org/10.1111/j.1600-0781.2010.00507.x; PMID: 20584250
- Ouédraogo GD, Redmond RW. Secondary reactive oxygen species extend the range of photosensitization effects in cells: DNA damage produced via initial membrane photosensitization. Photochem Photobiol 2003; 77:192 - 203; http://dx.doi.org/10.1562/0031-8655(2003)0770192SROSET2.0.CO2; PMID: 12785059
- Rubio N, Fleury SP, Redmond RW. Spatial and temporal dynamics of in vitro photodynamic cell killing: extracellular hydrogen peroxide mediates neighbouring cell death. Photochem Photobiol Sci 2009; 8:457 - 64; http://dx.doi.org/10.1039/b815343d; PMID: 19337658
- Rubio N, Rajadurai A, Held KD, Prise KM, Liber HL, Redmond RW. Real-time imaging of novel spatial and temporal responses to photodynamic stress. Free Radic Biol Med 2009; 47:283 - 90; http://dx.doi.org/10.1016/j.freeradbiomed.2009.04.024; PMID: 19409981
- Chakraborty A, Held KD, Prise KM, Liber HL, Redmond RW. Bystander effects induced by diffusing mediators after photodynamic stress. Radiat Res 2009; 172:74 - 81; http://dx.doi.org/10.1667/RR1669.1; PMID: 19580509
- Buytaert E, Callewaert G, Hendrickx N, Scorrano L, Hartmann D, Missiaen L, et al. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J 2006; 20:756 - 8; PMID: 16455754
- Dewaele M, Martinet W, Rubio N, Verfaillie T, de Witte PA, Piette J, et al. Autophagy pathways activated in response to PDT contribute to cell resistance against ROS damage. J Cell Mol Med 2011; 15:1402 - 14; http://dx.doi.org/10.1111/j.1582-4934.2010.01118.x; PMID: 20626525
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A 1982; 79:1889 - 92; http://dx.doi.org/10.1073/pnas.79.6.1889; PMID: 6952238
- Fedorko M. Effect of chloroquine on morphology of cytoplasmic granules in maturing human leukocytes--an ultrastructural study. J Clin Invest 1967; 46:1932 - 42; http://dx.doi.org/10.1172/JCI105683; PMID: 6073998
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008; 27:433 - 46; http://dx.doi.org/10.1038/sj.emboj.7601963; PMID: 18200046
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011; 13:589 - 98; http://dx.doi.org/10.1038/ncb2220; PMID: 21478857
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A 2011; 108:10190 - 5; http://dx.doi.org/10.1073/pnas.1107402108; PMID: 21646527
- Heirman I, Ginneberge D, Brigelius-Flohé R, Hendrickx N, Agostinis P, Brouckaert P, et al. Blocking tumor cell eicosanoid synthesis by GP x 4 impedes tumor growth and malignancy. Free Radic Biol Med 2006; 40:285 - 94; http://dx.doi.org/10.1016/j.freeradbiomed.2005.08.033; PMID: 16413410
- Thomas JP, Maiorino M, Ursini F, Girotti AW. Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. In situ reduction of phospholipid and cholesterol hydroperoxides. J Biol Chem 1990; 265:454 - 61; PMID: 2294113
- Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med 2003; 34:145 - 69; http://dx.doi.org/10.1016/S0891-5849(02)01197-8; PMID: 12521597
- Nakagawa Y. Role of mitochondrial phospholipid hydroperoxide glutathione peroxidase (PHGPx) as an antiapoptotic factor. Biol Pharm Bull 2004; 27:956 - 60; http://dx.doi.org/10.1248/bpb.27.956; PMID: 15256721
- Sneddon AA, Wu HC, Farquharson A, Grant I, Arthur JR, Rotondo D, et al. Regulation of selenoprotein GPx4 expression and activity in human endothelial cells by fatty acids, cytokines and antioxidants. Atherosclerosis 2003; 171:57 - 65; http://dx.doi.org/10.1016/j.atherosclerosis.2003.08.008; PMID: 14642406
- Kagan VE, Tyurin VA, Jiang JF, Tyurina YY, Ritov VB, Amoscato AA, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 2005; 1:223 - 32; http://dx.doi.org/10.1038/nchembio727; PMID: 16408039
- Belikova NA, Vladimirov YA, Osipov AN, Kapralov AA, Tyurin VA, Potapovich MV, et al. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry 2006; 45:4998 - 5009; http://dx.doi.org/10.1021/bi0525573; PMID: 16605268
- Gonzalvez F, Gottlieb E. Cardiolipin: setting the beat of apoptosis. Apoptosis 2007; 12:877 - 85; http://dx.doi.org/10.1007/s10495-007-0718-8; PMID: 17294083
- Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, et al. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic Biol Med 2009; 46:1439 - 53; http://dx.doi.org/10.1016/j.freeradbiomed.2009.03.004; PMID: 19285551
- Buytaert E, Callewaert G, Vandenheede JR, Agostinis P. Deficiency in apoptotic effectors Bax and Bak reveals an autophagic cell death pathway initiated by photodamage to the endoplasmic reticulum. Autophagy 2006; 2:238 - 40; PMID: 16874066
- Kessel D, Vicente MG, Reiners JJ Jr.. Initiation of apoptosis and autophagy by photodynamic therapy. Lasers Surg Med 2006; 38:482 - 8; http://dx.doi.org/10.1002/lsm.20334; PMID: 16615135
- Kessel D, Vicente MG, Reiners JJ Jr.. Initiation of apoptosis and autophagy by photodynamic therapy. Autophagy 2006; 2:289 - 90; PMID: 16921269
- Reiners JJ Jr., Agostinis P, Berg K, Oleinick NL, Kessel D. Assessing autophagy in the context of photodynamic therapy. Autophagy 2010; 6:7 - 18; http://dx.doi.org/10.4161/auto.6.1.10220; PMID: 19855190
- Kessel D, Castelli M. Evidence that bcl-2 is the target of three photosensitizers that induce a rapid apoptotic response. Photochem Photobiol 2001; 74:318 - 22; http://dx.doi.org/10.1562/0031-8655(2001)074<0318:ETBITT>2.0.CO;2; PMID: 11547571
- Xue LY, Chiu SM, Oleinick NL. Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene 2001; 20:3420 - 7; http://dx.doi.org/10.1038/sj.onc.1204441; PMID: 11423992
- Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part one- photosensitizers, photochemistry and celular localization. Photodiagn Photodyn Ther 2004; 1:279 - 93; http://dx.doi.org/10.1016/S1572-1000(05)00007-4
- Hill BG, Haberzettl P, Ahmed Y, Srivastava S, Bhatnagar A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochem J 2008; 410:525 - 34; http://dx.doi.org/10.1042/BJ20071063; PMID: 18052926
- Lee JY, Jung GY, Heo HJ, Yun MR, Park JY, Bae SS, et al. 4-Hydroxynonenal induces vascular smooth muscle cell apoptosis through mitochondrial generation of reactive oxygen species. Toxicol Lett 2006; 166:212 - 21; http://dx.doi.org/10.1016/j.toxlet.2006.07.305; PMID: 16919899
- Ji C, Amarnath V, Pietenpol JA, Marnett LJ. 4-hydroxynonenal induces apoptosis via caspase-3 activation and cytochrome c release. Chem Res Toxicol 2001; 14:1090 - 6; http://dx.doi.org/10.1021/tx000186f; PMID: 11511183
- Vila A, Korytowski W, Girotti AW. Spontaneous transfer of phospholipid and cholesterol hydroperoxides between cell membranes and low-density lipoprotein: assessment of reaction kinetics and prooxidant effects. Biochemistry 2002; 41:13705 - 16; http://dx.doi.org/10.1021/bi026467z; PMID: 12427033
- Vila A, Korytowski W, Girotti AW. Spontaneous intermembrane transfer of various cholesterol-derived hydroperoxide species: kinetic studies with model membranes and cells. Biochemistry 2001; 40:14715 - 26; http://dx.doi.org/10.1021/bi011408r; PMID: 11724586
- Vila A, Korytowski W, Girotti AW. Dissemination of peroxidative stress via intermembrane transfer of lipid hydroperoxides: model studies with cholesterol hydroperoxides. Arch Biochem Biophys 2000; 380:208 - 18; http://dx.doi.org/10.1006/abbi.2000.1928; PMID: 10900151
- Girotti AW. Translocation as a means of disseminating lipid hydroperoxide-induced oxidative damage and effector action. Free Radic Biol Med 2008; 44:956 - 68; http://dx.doi.org/10.1016/j.freeradbiomed.2007.12.004; PMID: 18206663
- Gallegos AM, Atshaves BP, Storey SM, Starodub O, Petrescu AD, Huang H, et al. Gene structure, intracellular localization, and functional roles of sterol carrier protein-2. Prog Lipid Res 2001; 40:498 - 563; http://dx.doi.org/10.1016/S0163-7827(01)00015-7; PMID: 11591437
- Vila A, Levchenko VV, Korytowski W, Girotti AW. Sterol carrier protein-2-facilitated intermembrane transfer of cholesterol- and phospholipid-derived hydroperoxides. Biochemistry 2004; 43:12592 - 605; http://dx.doi.org/10.1021/bi0491200; PMID: 15449949
- Giorgi C, De Stefani D, Bononi A, Rizzuto R, Pinton P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int J Biochem Cell Biol 2009; 41:1817 - 27; http://dx.doi.org/10.1016/j.biocel.2009.04.010; PMID: 19389485
- Belikova NA, Tyurina YY, Borisenko G, Tyurin V, Samhan Arias AK, Yanamala N, et al. Heterolytic reduction of fatty acid hydroperoxides by cytochrome c/cardiolipin complexes: antioxidant function in mitochondria. J Am Chem Soc 2009; 131:11288 - 9; http://dx.doi.org/10.1021/ja904343c; PMID: 19627079
- Yang JY, Yang WY. Spatiotemporally controlled initiation of Parkin-mediated mitophagy within single cells. Autophagy 2011; 7:1230 - 8; http://dx.doi.org/10.4161/auto.7.10.16626; PMID: 22011618
- Mai S, Muster B, Bereiter-Hahn J, Jendrach M. Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence lifespan. Autophagy 2012; 8:47 - 62; http://dx.doi.org/10.4161/auto.8.1.18174; PMID: 22170153
- Kissová I, Deffieu M, Samokhvalov V, Velours G, Bessoule JJ, Manon S, et al. Lipid oxidation and autophagy in yeast. Free Radic Biol Med 2006; 41:1655 - 61; http://dx.doi.org/10.1016/j.freeradbiomed.2006.08.012; PMID: 17145553
- Nakagawa Y. Initiation of apoptotic signal by the peroxidation of cardiolipin of mitochondria. Ann N Y Acad Sci 2004; 1011:177 - 84; http://dx.doi.org/10.1196/annals.1293.018; PMID: 15126295
- Kriska T, Korytowski W, Girotti AW. Role of mitochondrial cardiolipin peroxidation in apoptotic photokilling of 5-aminolevulinate-treated tumor cells. Arch Biochem Biophys 2005; 433:435 - 46; http://dx.doi.org/10.1016/j.abb.2004.09.025; PMID: 15581600
- Wang HP, Qian SY, Schafer FQ, Domann FE, Oberley LW, Buettner GR. Phospholipid hydroperoxide glutathione peroxidase protects against singlet oxygen-induced cell damage of photodynamic therapy. Free Radic Biol Med 2001; 30:825 - 35; http://dx.doi.org/10.1016/S0891-5849(01)00469-5; PMID: 11295525
- Liu XS, Kim CN, Yang J, Jemmerson R, Wang XD. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996; 86:147 - 57; http://dx.doi.org/10.1016/S0092-8674(00)80085-9; PMID: 8689682
- Korytowski W, Basova LV, Pilat A, Kernstock RM, Girotti AW. Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. J Biol Chem 2011; 286:26334 - 43; http://dx.doi.org/10.1074/jbc.M110.188516; PMID: 21642428
- Chen B, Roskams T, Xu Y, Agostinis P, de Witte PAM. Photodynamic therapy with hypericin induces vascular damage and apoptosis in the RIF-1 mouse tumor model. Int J Cancer 2002; 98:284 - 90; http://dx.doi.org/10.1002/ijc.10175; PMID: 11857421
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19:797 - 806; http://dx.doi.org/10.1091/mbc.E07-10-1092; PMID: 18094039
- Rasband WS. ImageJ. 1997. Bethesda, MD, National Institute of Health.
- Vantieghem A, Assefa Z, Vandenabeele P, Declercq W, Courtois S, Vandenheede JR, et al. Hypericin-induced photosensitization of HeLa cells leads to apoptosis or necrosis. Involvement of cytochrome c and procaspase-3 activation in the mechanism of apoptosis. FEBS Lett 1998; 440:19 - 24; http://dx.doi.org/10.1016/S0014-5793(98)01416-1; PMID: 9862416
- Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem 1971; 44:276 - 87; http://dx.doi.org/10.1016/0003-2697(71)90370-8; PMID: 4943714
- Brigelius-Flohé R, Lötzer K, Maurer S, Schultz M, Leist M. Utilization of selenium from different chemical entities for selenoprotein biosynthesis by mammalian cell lines. Biofactors 1995-1996; 5:125 - 31; PMID: 8922268
- Ursini F, Maiorino M, Gregolin C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim Biophys Acta 1985; 839:62 - 70; http://dx.doi.org/10.1016/0304-4165(85)90182-5; PMID: 3978121
- Hawkins CL, Morgan PE, Davies MJ. Quantification of protein modification by oxidants. Free Radic Biol Med 2009; 46:965 - 88; http://dx.doi.org/10.1016/j.freeradbiomed.2009.01.007; PMID: 19439229