Abstract
Due in part to the increasing number of links between autophagy malfunction and human diseases, this field has gained tremendous attention over the past decade. Our increased understanding of the molecular machinery involved in macroautophagy (hereafter autophagy) seems to indicate that the most complex step, or at least the stage of the process where the majority of the autophagy-related (Atg) proteins participate, is in the formation of the double-membrane sequestering vesicle. Thus, it is important to establish reliable approaches to monitor this specific process. One of the most commonly used methods is morphological analysis by electron microscopy of the cytosolic vesicles used in the cytoplasm-to-vacuole targeting (Cvt) pathway and autophagy, or the single-membrane intralumenal products, termed Cvt or autophagic bodies, that are formed after the fusion of these vesicles with the yeast vacuole. This method, however, can be costly and time consuming, and reliable analysis requires expert input. Furthermore, it is extremely difficult to detect an incomplete autophagosome by electron microscopy because of the difficulty of obtaining a section that randomly cuts through the open portion of the phagophore. The primary Cvt pathway cargo, precursor amminopeptidase I (prApe1), is enwrapped within either a Cvt vesicle or autophagosome depending on the nutritional conditions. The proteolytic sensitivity of the prApe1 propeptide can therefore serve as a useful tool to determine the completion status of double-membrane Cvt vesicles/autophagosomes in the presence of exogenously added proteinase. Here, we describe an assay that examines the proteinase protection of prApe1 for determining the completion of Cvt vesicles/autophagosomes.
1. Introduction
Although autophagy is often considered to be nonselective, there are many types of selective autophagy in which specific cargos are enclosed within a phagophore that expands and then seals to become an autophagosome, or a similar sequestering compartment. For example, certain vacuolar hydrolases,Citation1 peroxisomes,Citation2,Citation3 mitochondria,Citation4 or even bacteriaCitation5-Citation12 and virusesCitation13 can be targeted to vacuoles/lysosomes through autophagy. In most cases, the machinery involved in selective and nonselective autophagy overlap. Thus, it is critical to establish reliable methods to monitor both types of autophagy.
In Saccharomyces cerevisiae, one of the best characterized processes involved in selective autophagy is the Cvt pathway. The Cvt pathway is different from nonselective autophagy in many aspects: First, the Cvt pathway is biosynthetic rather than degradative; at least three resident vacuolar hydrolases, amminopeptidase I (Ape1), Ape4 and α-mannosidase (Ams1) are delivered into the vacuole through the Cvt pathway.Citation1,Citation13 Second, these vacuolar hydrolases are sequestered by a double-membrane Cvt vesicle (140–160 nm in diameter), which is smaller than an autophagosome (300–900 nm).Citation14 Third, the sequestering membrane tightly enwraps the Cvt pathway cargos, thus excluding bulk cytoplasm, and suggesting a mechanism for selective recognition and packaging. In line with the latter, the kinetics of transport for individual cargos are much more rapid when delivered to the vacuole through the Cvt pathway as compared with nonselective autophagy. Even with these many differences, both the Cvt and autophagy pathways share most of their molecular components and the two processes have an overall similar morphology. As mentioned above, under nutrient-rich conditions, yeast transport the precursor form of Ape1 to the vacuole through the Cvt pathway, but rely on autophagy under starvation conditions.Citation14 In either case, the process is rapid and efficient due to the use of receptor and adaptor/scaffold proteins.Citation15-Citation17 Once delivered to the vacuole, the propeptide of prApe1 is proteolytically removed to generate the active hydrolase. This processing event can be conveniently monitored as a molecular mass shift by SDS-PAGE, which forms the basis of a proteinase protection assay. Because prApe1 can be transported by these two overlapping mechanisms, it serves as a useful marker to monitor both the Cvt and autophagy pathways.Citation18
2. Materials
2.1. Cells and culture
1. Strains
All strains should contain a pep4Δ deletion (see Note 1). Strains of interest with the pep4Δ deletion should include the experimental strain(s), and strains that will serve as positive and negative controls, such as vam3ts and atg1Δ, respectively (see Note 2).
2. Growth medium
The cells are grown in YPD (Bacto-yeast extract 10 g; Bacto-peptone 20 g; 2% dextrose; double-distilled H2O to 1 L). If the strains of interest carry plasmids, the cells should be grown in SMD selective medium in which the appropriate amino acids and/or nucleic acid bases are omitted. For instance, to grow a strain carrying a plasmid containing the URA3 gene, the cells may be grown in SMD-URA medium [0.67% yeast nitrogen base without amino acids; 2% D-glucose; and appropriate amino acids and nucleic acid bases (except uracil) to satisfy any auxotrophies]. In addition to these minimal requirements, to improve cell growth a complete amino acid mix can be added (0.017% of the following L-amino acids: alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, isoleucine, methionine, phenylalanine, proline, serine, threonine, tyrosine, and valine; 0.3 mM histidine; 1.7 mM leucine; 1 mM lysine; and 0.4 mM tryptophan) along with 0.3 mM adenine; 2.22 mM myoinositol; and 0.00117% p-aminobenzoic acid, omitting any amino acids or nucleic acid bases that are needed for plasmid selection.
3. Culture conditions
If the purpose is to test the function of a strain of interest in the Cvt pathway, cells are grown in YPD or SMD selective medium at 30°C to mid-log phase, harvested (see step 3.4) and then converted to spheroplasts (see step 3.5). To test prApel sensitivity in starvation conditions, cells are grown in YPD or SMD selective medium at 30°C to mid-log phase, then converted to spheroplasts as above. The spheroplasts are then subjected to starvation treatment (see step 3.9) in SD(-N) (0.17% yeast nitrogen base without amino acids and ammonium sulfate, with 2% glucose) containing 1.2 M sorbitol (see Note 3). If the strain of interest is a temperature-sensitive mutant, cells should be grown at a permissive temperature and shifted to a nonpermissive temperature (see step 3.9) for the appropriate period of time before the final harvesting (see Note 4).
2.2. Chemicals
Zymolyase 20T (Seikagaku, NC9934469), and proteinase K (03115879001) are from Roche. The corresponding solutions should be made fresh (see Note 5).
2.3. Solutions
MURB: 50 mM sodium phophate buffer, pH 7.0, 25 mM MES, 1% SDS (w/v), 3 M urea, 1 mM NaN3, 1% β-mercaptoethanol, 0.01% bromophenol blue.
DTT buffer: 10 mM Tris-Sulfate, pH 9.4, 10 mM DTT.
Spheroplasting medium: 1X YNB (0.67% yeast nitrogen base), 2% D-glucose (w/v), appropriate amino acids and nucleic acid bases, 1.2 M sorbitol.
PS200 (lysis buffer): 20 mM K-PIPES, pH 6.8, 200 mM sorbitol, 5 mM MgCl2.
2.4. Antibody
Ape1 antibody.
2.5. Spheroplast lysis
Dounce homogenizer or a 25-mm Swin-Lok holder assembly (Thomas Scientific, 420200) fitted with a 3.0-μm Nucleopore Track-Etch membrane (Whatman, 110612) and a 10 ml syringe.
3. Methods
Overview
The propeptide of prApe1 is sensitive to proteolysis, whereas the mature protein is extremely resistant to further degradation due to its nature as a vacuolar hydrolase. Thus, prApe1 can be processed by an exogenously added proteinase following spheroplast lysis as long as it is not sequestered within a membrane compartment.
In brief, yeast spheroplasts are osmotically lysed in a condition where the integrity of the vacuole and other membrane compartments are maintained. The prApe1-containing membranes are enriched by low-speed centrifugation to generate a P5 pellet fraction (prApe1 remains associated with the membrane fraction in the presence of MgCl2Citation19). The assay uses two different control strains: (A) A wild-type strain carrying a pep4Δ deletion or a vam3ts mutation serves as a positive control (see Note 2). In these strains, prApe1 accumulates within the vacuole (in Cvt or autophagic bodies) or completed Cvt vesicles/autophagosomes in the cytosol, respectively; spheroplast lysis that retains the integrity of the vacuole and the double-membrane sequestering vesicles will result in the accumulation of prApe1 that is protected from exogenous proteinase unless detergent is added. (B) A negative control strain such as atg1Δ pep4Δ (see Note 2) that is defective in the generation of Cvt vesicles/autophagosomes; prApe1 will be sensitive to exogenous proteinase after spheroplast lysis.
The membrane fractions (P5) from each of these strains are split into four aliquots and subjected to the following treatments: (1) No treatment; (2) treatment with proteinase alone; (3) treatment with detergent alone; and (4) treatment with both proteinase and detergent. In a positive control pep4Δ strain prApe1 is enwrapped in completed double-membrane vesicles and is protected from exogenously added proteinase K in the absence of detergent (). Only in the condition where both proteinase K and detergent are present, is the proteinase-sensitive propeptide domain of prApe1 cleaved resulting in a molecular mass shift. In the negative control strain, in which an atg mutation is present along with pep4Δ, vesicle formation is defective, and prApe1 is proteolytically cleaved by exogenously added proteinase K even in the absence of detergent ().
Figure 1. The prApe1 proteinase protection assay. (A) Wild-type (atg8Δ pep4Δ CUP1p-Atg8) cells or (B) atg8Δ pep4Δ cells carrying an empty vector were grown to exponential phase in SMD-URA medium. Cells were first converted to spheroplasts and starved in SD(-N) medium containing 1.2 M sorbitol. Spheroplasts were harvested, resuspended in lysis buffer (PS200), and then disrupted. A preclearing step was carry out to remove unbroken cells by centrifuging cell lysates at 300 × g and to obtain total cell lysates (T). To get prApe1-enriched membrane fractions, the total cell lysates were further separated into 5,000 × g suppernatant (S5) and pellet (P5) fractions. The prApe1-containing P5 fractions were split into four aliquots and subjected to different conditions: No treatment, 0.2% Triton X-100 (TX-100), proteinase K (PK), or proteinase K in the presence of 0.2% Triton X-100. The samples were precipitated using 10% TCA, acetone washed twice and subjected to immunoblot analysis using anti-Ape1 antiserum. For internal controls to verify the complete lysis of spheroplasts and proper membrane separation, samples were analyzed using anti-Pgk1 and anti-Ape1 antisera. The integrity of organelle compartments in the P5 fractions were tested by examining the proteolytic cleavage of the precursor form of Prc1. This figure includes data previously published in reference Citation20 and reproduced by permission of Landes Bioscience.
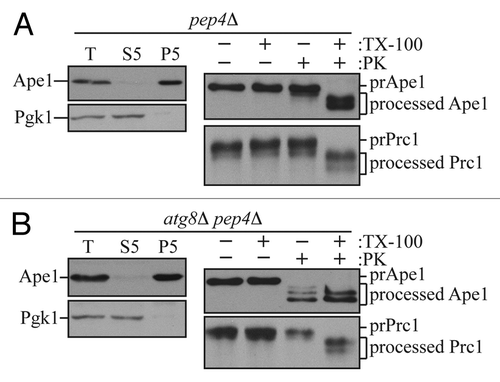
Additional (internal) controls are optimal for reliable interpretation of this experiment. The fractionation profile of a cytosolic marker, such as Pgk1, should be tested in total cell lysate (T), membrane fractions (P5), and supernatant fractions (S5) as an internal control to determine whether there was efficient spheroplast lysis. Pgk1 is predominantly detected in total and supernatant fractions (the presence of a substantial population of Pgk1 in the P5 fraction would indicate the presence of unlysed spheroplasts). Note that prApe1 should be present primarily in the P5 pellet fraction.
Another internal control is needed to verify the integrity of intracellular compartments following spheroplast lysis. In this analysis we assume that the vacuole is the subcellular compartment that is most sensitive to osmotic lysis. It is possible to use various marker proteins to monitor the integrity of the vacuole in a pep4Δ background. One option is to analyze the protease sensitivity of the precursor form of another vacuolar hydrolase, Prc1 (carboxypeptidase Y). Upon delivery to the vacuole, the Prc1 propeptide is proteolytically removed in a Pep4-dependent manner, thus becoming an active hydrolase. In a pep4Δ strain, Prc1 accumulates in the vacuole in a precursor form. In this case, the propeptide is protected from exogenousely added proteinase, but becomes sensitive when the vacuole is solublized by detergent. Thus, the sensitivity of the lumenal propeptide reflects the integrity of membrane compartments including the vacuole. Alternatively, Pho8 can serve as a marker to monitor both spheroplast lysis efficiency and vacuole integrity. Pho8 localizes to the vacuolar membrane, and it contains both a cytosolic tail and a lumenal propeptide domain. The removal of the propeptide is again Pep4 dependent. Thus, the accessibility of the propeptide region indicates the integrity of the vacuole, whereas the sensitivity of the cytosolic tail to exogenouly added proteinase in the absence of detergent reflects the efficiency of spheroplast lysis.Citation21
Detailed protocol
3.1
Yeast strains carrying a pep4Δ deletion are cultured in 5 ml YPD or SMD selective medium at 30°C, shaking at 250 rpm (temperature-sensitive strains should be grown at an appropriate permissive temperature; for the vam3ts strain the permissive temperature is 24°C). Yeast strains containing an integrated version of GFP-Atg8 are grown in YPD (see Note 6). Dilute overnight cultures from mid-log phase to an OD600 = 0.1–0.2 and grow until the cells reach exponential phase (OD600 = 0.8–1.0) in 25–50 ml of medium. Ideally, the cells should be in log phase prior to this dilution, otherwise the dilution may need to be greater.
3.2
Harvest 50 OD600 units of cells by centrifugation at 2,000 rpm for 5 min and wash once with 4 ml of DTT buffer. [If testing temperature-sensitive mutants in nutrient rich conditions, choose an appropriate temperature and incubation time to inactivate the temperature-sensitive protein (see Note 4)].
3.3
Discard the supernatant fraction, resuspend the cells in approximately 25 ml DTT buffer (to 2 OD/ml) and incubate them at 30°C (for testing temperature-sensitive strains for starvation-induced autophagy, cells should be incubated at an appropriate permissive temperature), shaking at 180–220 rpm for 15 min.
3.4
Harvest the cells by centrifugation at 600 × g for 5 min and remove the supernatant fraction thoroughly (residual DTT may affect later reactions).
3.5
Resuspend the cells in 6 ml of spheroplasting medium (preincubated at 30°C) containing 1.2 mg of Zymolyase 20T. To test the spheroplasting efficiency, remove a sample and measure the OD of a 1:10 dilution in water as an initial reading. Incubate cells at 30°C for 30 min while gently shaking at 150–180 rpm (or at an appropriate permissive temperature for temperature-sensitive strains for testing starvation-induced autophagy).
3.6
Invert the tube several times during this incubation to keep the cells in solution.
3.7
Check the spheroplasting percentage by diluting a sample at a 1:10 ratio in water. Continue until there is at least an 80% reduction from the initial reading (see Note 7).
3.8
Harvest the spheroplasts at 2000 × g for 10 min. (If testing the Cvt pathway in nutrient rich conditions, go to step 3.11 directly.)
3.9
Wash the spheroplasts once with SD(-N) containing 1.2 M sorbitol, resuspend them in 50 ml fresh SD(-N) medium containing 1 M sorbitol and incubate the samples for 1 to 4 h at 30°C while shaking (see Note 3). For ts strains, choose an appropriate temperature to inactivate the temperature-sensitive protein (see Note 4; for the vam3ts strain the NPT is 37°C).
3.10
Harvest the spheroplasts at 2000 × g for 10 min, discard the supernatant fraction, and put the samples on ice.
3.11
Osmotically lyse the spheroplasts by resuspending in 2 ml ice-cold PS200 using a cropped tip (maxium spheroplast density in PS200 is 20 OD/ml), and dounce homogenize the spheroplasts with 10 strokes. Alternatively, lyse the spheroplasts by resuspending them in 6 ml PS200 buffer and disrupting them by passage through a a 3.0-μm Nucleopore Track-Etch membrane using a 25-mm Swin-Lok holder assembly and disposable syringe.
3.12
Remove unbroken cells and cell debris by centrifugation at 300–500 × g for 10 min at 4°C. Remove the supernatant fraction to new centrifuge tubes and perform this preclearing step one more time.
3.13
For each strain, split the supernatant fraction into 6 aliquots (one aliquot as total cell lysate, one for analyzing the P5 and S5 fractions, and 4 for the proteinase protection analysis) in microcentrifuge tubes. Add 100% TCA to the total (T) aliquot to a final concentration of 10% and put on ice.
3.14
Spin the resulting 5 aliquots of cell lysates at 5,000 × g for 5 min at 4°C to separate the supernatant (S5) and pellet (P5) fractions. Resuspend the P5 fraction in 300 µl ice-cold PS200. Add TCA to one S5 fraction and one of the resuspended P5 fractions.
3.15
For the remaining 4 aliquots of the P5 fraction resuspend the P5 in 300 µl (3 OD600 units/treatment) of ice-cold PS200 containing the following (see ): (A) PS200 alone (no treatment control), (B) 40–80 µg/ml proteinase K, (C) 0.2% Triton-X 100, (D) 5–10 µg/ml proteinase K with 0.2% Triton X-100. Incubate samples on ice for 30 min (see Note 8).
Table 1. Conditions for sample analysis
3.16
To stop the reactions, add 33 µl of 100% TCA to the samples, vortex to mix thoroughly and incubate on ice for at least 20 min. Centrifuge the reactions 10 min at 13,000 rpm at 4°C.
3.17
Discard the supernatant fractions, and wash the pellet fractions twice with 100% acetone. Dry the pellets, and resuspend in 50 μl MURB buffer by waterbath sonication. Heat the samples for 5 min at 100°C. Centrifuge for 2 min at 10,000 rpm to remove insoluble material.
3.18
Load 10–15 μl of the samples for SDS-PAGE.
3.19
Perform western blotting using anti-Ape1 antibody to detect precursor and processed Ape1, and anti-Pgk1, Pho8 and/or anti-Prc1 antibodies for the appropriate controls. Develop the western blots with appropriate exposure.
Conclusion
The prApe1 protease protection assay is reliable but it has its limits. For instance, some temperature-sensitive strains may show defects in prApe1 import only at the nonpermissive temperature (NPT). In such strains, the complete vesicles have formed and accumulated at the permissive temperature (PT). A short-term incubation at NPT may therefore not be sufficient to see the proteinase-sensitive phenotype (see Note 1). In this case, a radiolabeled prApe1 proteinase protection assay at the NPT has to be performed to examine the specific population of prApe1 that is synthesized at the NPT. These additional steps may make the assay less desirable or difficult in some strains. We also refer readers to the protocol describing the use of the GFP-Atg8 processing assay as an alternative to monitor autophagosome completion.Citation22
4. Notes
1
It is possible to use strains that are wild type for the PEP4 gene. However, the wild-type control strain will have accumulated a large background of mature Ape1 within the vacuole that will make the subsequent analysis more problematic. The same problem can occur with mutants that display only partial defects in prApe1 delivery to the vacuole, or that are defective for autophagy, but not for the Cvt pathway. The pep4Δ strain lacks the major processing hydrolase proteinase A, and will accumulate only the precursor form of Ape1. If the mutant strain being analyzed displays a complete block in prApe1 maturation, it is not necessary to use a pep4Δ background, although this may still be desirable in order to keep the mutant and control strains isogenic (aside from the mutation of interest).
2
Other control strains may be used including a conditional ypt7 mutant to block fusion of completed vesicles with the vacuole, and most atg mutants, which will prevent formation of a completed vesicle. The vam3 or ypt7 mutants are defective in the fusion of completed autophagosomes with the vacuole. Therefore, these strains accumulate prApe1 within cytosolic autophagosomes. Accordingly, the precursor protein should be protected from exogenous proteinase in the absence of detergent, serving as a positive control. Note that it is preferable to use a conditional vam3 or ypt7 mutant rather than a null strain; the complete absence of these proteins results in the generation of a substantial number of membranous compartments, as well as interfering with homotypic vacuole fusion, both of which can make the analysis more difficult. The atg1 mutant, and most other atg mutants, is defective for autophagosome formation. Thus, this strain will accumulate proteinase-sensitive prApe1, and serves as a negative control.
3
The length of starvation needs to be determined empirically, but 1 to 4 h is usually sufficient. It is important to note that we recommend converting cells to spheroplasts prior to initiating starvation, rather than converting starved cells to spheroplasts. Starvation results in a thickening of the yeast cell wall, which renders it much more resistant to lytic degradation. Thus, subsequent conversion of the starved cells to spheroplasts can be problematic.
4
The period of time required for incubation at the nonpermissive temperature, as well as the specific temperature, depend on the particular mutant being examined and must be determined empirically. The minimum length of time within which the block can be clearly detected is desired.
5
Solutions of proteinase K can be aliquoted and frozen, but we do not recommend this due to the potential for self-digestion. Lytic enzyme preparations other than Zymolyase 20T can be used for cell wall digestion, but they need to be relatively free of protease.
6
GFP-Atg8 can also be expressed from a plasmid.
7
The appropriate incubation time for the Zymolyase 20T reaction will vary depending on the strain background. Pilot experiments may be required to optimize this step. Typically, 30–60 min is sufficient to obtain 80–90% spheroplasts.
8
Pilot experiments may be required to determine the concentration and incubation time for the proteinase K treatment.
Acknowledgments
The authors thank Dr. Eduardo Cebollero (University Medical Center Utrecht) for helpful comments. This work was supported by grant GM53396 to D.J.K.
References
- Klionsky DJ, Cueva R, Yaver DS. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J Cell Biol 1992; 119:287 - 99; http://dx.doi.org/10.1083/jcb.119.2.287; PMID: 1400574
- Dunn WA Jr., Cregg JM, Kiel JAKW, van der Klei IJ, Oku M, Sakai Y, et al. Pexophagy: the selective autophagy of peroxisomes. Autophagy 2005; 1:75 - 83; http://dx.doi.org/10.4161/auto.1.2.1737; PMID: 16874024
- Hutchins MU, Veenhuis M, Klionsky DJ. Peroxisome degradation in Saccharomyces cerevisiae is dependent on machinery of macroautophagy and the Cvt pathway. J Cell Sci 1999; 112:4079 - 87; PMID: 10547367
- Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy 2011; 7:297 - 300; http://dx.doi.org/10.4161/auto.7.3.14502; PMID: 21252623
- Yoshimori T. Autophagy vs. Group A Streptococcus. Autophagy 2006; 2:154 - 5; PMID: 16874113
- Webster P. Cytoplasmic bacteria and the autophagic pathway. Autophagy 2006; 2:159 - 61; PMID: 16874112
- Vergne I, Singh S, Roberts E, Kyei G, Master S, Harris J, et al. Autophagy in immune defense against Mycobacterium tuberculosis. Autophagy 2006; 2:175 - 8; PMID: 16874111
- Ogawa M, Sasakawa C. Shigella and autophagy. Autophagy 2006; 2:171 - 4; PMID: 16874102
- Dubuisson JF, Swanson MS. Mouse infection by Legionella, a model to analyze autophagy. Autophagy 2006; 2:179 - 82; PMID: 16874080
- Colombo MI, Gutierrez MG, Romano PS. The two faces of autophagy: Coxiella and Mycobacterium. Autophagy 2006; 2:162 - 4; PMID: 16874070
- Birmingham CL, Brumell JH. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy 2006; 2:156 - 8; PMID: 16874057
- Bélanger M, Rodrigues PH, Dunn WA Jr., Progulske-Fox A. Autophagy: a highway for Porphyromonas gingivalis in endothelial cells. Autophagy 2006; 2:165 - 70; PMID: 16874051
- Sumpter R Jr., Levine B. Selective autophagy and viruses. Autophagy 2011; 7:260 - 5; http://dx.doi.org/10.4161/auto.7.3.14281; PMID: 21150267
- Baba M, Osumi M, Scott SV, Klionsky DJ, Ohsumi Y. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J Cell Biol 1997; 139:1687 - 95; http://dx.doi.org/10.1083/jcb.139.7.1687; PMID: 9412464
- Scott SV, Guan J, Hutchins MU, Kim J, Klionsky DJ. Cvt19 is a receptor for the cytoplasm-to-vacuole targeting pathway. Mol Cell 2001; 7:1131 - 41; http://dx.doi.org/10.1016/S1097-2765(01)00263-5; PMID: 11430817
- Shintani T, Huang W-P, Stromhaug PE, Klionsky DJ. Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev Cell 2002; 3:825 - 37; http://dx.doi.org/10.1016/S1534-5807(02)00373-8; PMID: 12479808
- Yorimitsu T, Klionsky DJ. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol Biol Cell 2005; 16:1593 - 605; http://dx.doi.org/10.1091/mbc.E04-11-1035; PMID: 15659643
- Lynch-Day MA, Klionsky DJ. The Cvt pathway as a model for selective autophagy. FEBS Lett 2010; 584:1359 - 66; http://dx.doi.org/10.1016/j.febslet.2010.02.013; PMID: 20146925
- Kim J, Huang W-P, Stromhaug PE, Klionsky DJ. Convergence of multiple autophagy and cytoplasm to vacuole targeting components to a perivacuolar membrane compartment prior to de novo vesicle formation. J Biol Chem 2002; 277:763 - 73; http://dx.doi.org/10.1074/jbc.M109134200; PMID: 11675395
- Nair U, Yen W-L, Mari M, Cao Y, Xie Z, Baba M, et al. A role for Atg8-PE deconjugation in autophagosome biogenesis. Autophagy 2012; 8:780 - 93; http://dx.doi.org/10.4161/auto.19385; PMID: 22622160
- Klionsky DJ, Emr SD. Membrane protein sorting: biosynthesis, transport and processing of yeast vacuolar alkaline phosphatase. 1989; 8:2241 - 50
- Nair U, Thumm M, Klionsky DJ, Krick R. GFP-Atg8 protease protection as a tool to monitor autophagosome biogenesis. Autophagy 2011; 7:1546 - 50; http://dx.doi.org/10.4161/auto.7.12.18424; PMID: 22108003