Abstract
Autophagy is a conserved and highly regulated catabolic pathway, transferring cytoplasmic components in autophagosomes to lysosomes for degradation and providing amino acids during starvation. In multicellular organisms autophagy plays an important role for tissue homeostasis, and deregulation of autophagy has been implicated in a broad range of diseases, including cancer and neurodegenerative disorders. In mammals, many aspects of autophagy still need to be fully elucidated: what is the exact hierarchy and relationship between ATG proteins and other factors that lead to the formation and expansion of phagophores? Where does the membrane source for autophagosome formation originate? Which signaling events trigger amino acid starvation-induced autophagy? How are the activities of ULK1/2 and the class III PtdIns3K regulated and linked to each other? To develop therapeutic strategies to manipulate autophagy in human disease, a comprehensive understanding of the molecular protein machinery mediating and regulating autophagy is required.
In order to uncover new proteins involved in mammalian autophagy we performed a genome-wide siRNA screen. HEK293A cells stably expressing the autophagosome marker GFP-LC3 were induced to form GFP-LC3 puncta by amino acid starvation. We identified many genes whose knockdown increased or decreased autophagosomes and performed multiple rounds of validation in order to generate a list of 51 high-confidence hits. These were enriched with cytoplasmic and Golgi apparatus proteins but not plasma membrane proteins. We chose to characterize two hits, SCOC and WAC, because of the strength of their three validated individual siRNAs, their connection to the autophagy interaction network (AIN), and their strong inhibition of LC3 lipidation in HeLa cells.
SCOC Interacts with FEZ1 to Regulate Autophagy
SCOC (short coiled-coil protein) is a Golgi protein implicated in Golgi transport through its interaction with ARL1. SCOC colocalizes with the trans-Golgi network protein TGOLN2/TGN46 and the autophagy protein ATG9A. In starved cells the partial colocalization of SCOC with LC3 suggests that SCOC functions at the site of autophagosome formation. SCOC is a positive regulator of autophagy, as depletion of SCOC decreases LC3 lipidation and inhibits starvation-induced SQSTM1/p62 degradation. In addition, overexpression of SCOC increases LC3 lipidation.
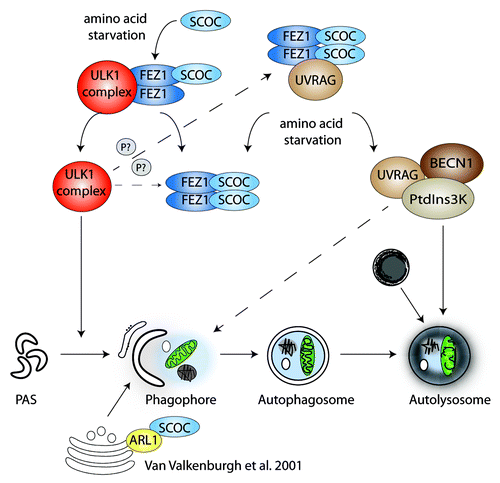
In C. elegans the SCOC homolog UNC-69 interacts with the FEZ1 (fasciculation and elongation protein zeta 1) homolog UNC-76, and both proteins are required for axonal outgrowth. Interestingly, Drosophila Atg1 (the ULK1 homolog) interacts with and regulates UNC-76 by phosphorylation. We confirmed the interaction between FEZ1 and SCOC in HEK293A cells, which is mediated via their highly conserved coiled-coil domains. Moreover, we observed that SCOC and FEZ1 form a trimeric complex with ULK1. While SCOC does not directly bind ULK1, increasing amounts of SCOC reduce ULK1 binding to FEZ1. Furthermore, SCOC knockdown affects the formation of FEZ1-ULK1 high molecular weight complexes, indicating that SCOC may regulate ULK1 binding to FEZ1.
In the autophagy interaction network (AIN) described by the Harper laboratory, SCOC is a low-confidence interactor with the tumor suppressor UVRAG (UV irradiation resistance gene). UVRAG associates with BECN1, which is a stoichiometric component of the class III PtdIns3K complex essential for autophagosome formation. Co-immunoprecipitation experiments confirmed the interaction between SCOC and UVRAG, and this interaction is starvation sensitive. Interestingly, we found that FEZ1 stabilizes the interaction between SCOC and UVRAG.
It might be surprising that the small protein SCOC, consisting mainly of a coiled-coil region, was a high-ranking hit in our screen. Our results show that SCOC and FEZ1 act on key signaling complexes in amino acid starvation-induced autophagy, and might be important membrane proximal scaffold proteins that link ULK1 complex activation with BECN1-PtdIns3K complex activity. In contrast to SCOC, FEZ1 negatively regulates autophagy and at the moment we do not fully understand the opposing effects of FEZ1 and SCOC on autophagy induction. We speculate that SCOC promotes autophagy by binding to FEZ1, thereby releasing and allowing ULK1 complex activation (). FEZ1 may inhibit autophagy by sequestering both the ULK1 and UVRAG-BECN1-PtdIns3K complex, thus preventing their activation. The dynamics of the association between SCOC and FEZ1 remains to be explored, however SCOC and FEZ1 function might be regulated through phosphorylation, e.g., by ULK1. Our current studies are aimed at the analysis of the hierarchical and spatial regulation of the SCOC-FEZ1 interaction with the ULK1 and the UVRAG-BECN1-PtdIns3K complexes. This will provide a better understanding of the role of SCOC and FEZ1 in the autophagic pathway.
Figure 1. SCOC and FEZ1 regulate amino acid starvation-induced autophagy through interactions with ULK1 and UVRAG. We hypothesize that SCOC and FEZ1 coordinate the activities of both the ULK1 and UVRAG-BECN1-PtdIns3K complexes. Amino acid starvation may favor SCOC binding to FEZ1, thereby releasing the ULK1 complex, which becomes activated and induces phagophore formation. Amino acid starvation may also trigger dissociation of UVRAG from FEZ1-SCOC allowing formation of an active UVRAG-BECN1-PtdIns3K complex required for autophagosome formation and maturation. The interaction of FEZ1 and SCOC as well as the SCOC-FEZ1-UVRAG complex may be modulated by ULK1 phosphorylation.
WAC is Required for Autophagy and may Negatively Regulate Proteasomal Degradation
WW domain containing adaptor with coiled-coil (WAC) is a protein located mostly in the nucleus. The WW and coiled-coil domains are well conserved between WAC homologs, which can be found in vertebrates but also insects, schistosoma and sea anemones. WAC interacts with a deubiquitinase, VCPIP1/VCIP135, to mediate Golgi biogenesis at mitosis (). In addition, WAC regulates gene transcription and histone ubiquitination via interaction with the E3 ligase RNF20-RNF40. This regulation is important in cell-cycle checkpoint activation in response to DNA damage. WAC also interacts with UBQLN4/Ubiquilin 4 a UBQ-UBA-containing protein that binds a regulatory subunit of the 26S proteasome.
Figure 2. WAC regulates autophagy, proteasomal degradation and ubiquitination. (A) WAC is required for autophagosome formation and we speculate this may depend on its interaction with BECN1. We have also shown WAC negatively regulates the degradation of proteasomal substrates. (B) WAC binds to, and activates, the deubiquitinase VCPIP1. This function of WAC is important in reforming the Golgi apparatus at mitosis. VCPIP1 binds VCP/p97 and NSFL1C/p47, a complex important in the ubiquitin-proteasome system and autophagosome maturation in mammals. (C) In the cell nucleus, WAC binds to the phosphorylated C-terminal domain of RNA polymerase II via its WW domain and to the E3 ligase RNF20-RNF40 via its coiled-coil domain. This binding activates RNF20-RNF40 to ubiquitinate histone 2B and results in transcriptional upregulation of select genes.
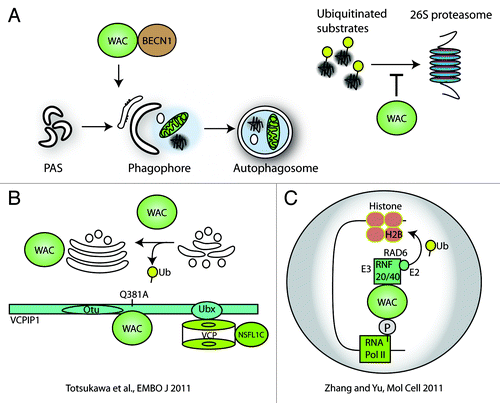
Our results demonstrate that WAC is required for autophagosome formation. Knockdown of WAC reduces endogenous LC3 lipidation in HEK293A and HeLa cells, as well as GFP-LC3 spot number, intensity and area. Additionally, we saw an inhibition of starvation-induced SQSTM1 degradation by WAC siRNA. WAC knockdown also increases basal levels of SQSTM1. In addition to WAC’s positive role in autophagy, it may negatively regulate proteasomal degradation. This finding is in line with the known functions of WAC, which have the common theme of regulating ubiquitin modulation. We used UbG76V-YFP as a direct reporter of the ubiquitin-proteasome system and aggregating huntingtin (HTT)Q103-CFP and nonaggregating HTTQ25-CFP, to investigate WAC’s role in nonautophagic protein clearance. Following knockdown of WAC, we observed a considerable decrease of UbG76V-YFP, and the levels of HTTQ25-CFP and the soluble fraction of HTTQ103-CFP are reduced. Thus, our data suggest that in addition to the requirement of WAC in starvation-induced autophagy, this protein also mediates the negative regulation of the proteasome toward substrates such as HTTQ25-CFP and UbG76V-YFP. In support of WAC’s role in autophagy, we confirmed the interaction of WAC and BECN1 by co-immunoprecipitation, as they were identified as low-confidence interactors in the AIN.
It will be important to elucidate the molecular mechanism of WAC’s control of autophagy. Considering its nuclear localization and function, it may control the transcription of autophagy-related genes. With respect to WAC’s role in the cytoplasm, its control of proteasomal and autophagic turnover may not be mutually exclusive; WAC may regulate the proteasomal degradation of the ATG proteins themselves, for example BECN1.
Future Outlook
Our siGenome screen provides a number of candidates for future research into amino acid starvation-induced autophagy. Two other recent siGenome screens for candidates required for basal autophagy and selective autophagy from the Yuan and Levine labs, respectively, provide two additional and largely nonoverlapping lists of candidates. The complexity of the autophagic response may well encompass all the new top candidates recently identified. Our candidate list and our data on two novel autophagy regulators, SCOC and WAC, increase our repertoire of proteins to further understand starvation-induced autophagy.
Acknowledgments
This work was supported by Cancer Research UK. We thank Dr. Harold B. Jefferies for his comments and suggestions.