Abstract
Proline dehydrogenase (oxidase, PRODH/POX), the first enzyme in the pathway of proline catabolism, has been identified as a mitochondrial, metabolic tumor suppressor, which is downregulated in a variety of human tumors. However, our recent findings show that PRODH/POX is upregulated by hypoxia in vitro and in vivo. The combination of low glucose and hypoxia produces additive effects on PRODH/POX expression. Both hypoxia and glucose depletion enhance PRODH/POX expression through AMP-activated protein kinase (AMPK) activation to promote tumor cell survival. Nevertheless, the mechanisms underlying PRODH/POX prosurvival functions are different for hypoxia and low-glucose conditions. Glucose depletion with or without hypoxia elevates PRODH/POX and proline utilization to supply ATP for cellular energy needs. Interestingly, under hypoxia PRODH/POX induces protective autophagy by generating reactive oxygen species (ROS). AMPK is the main initiator of stress-triggered autophagy. Thus, PRODH/POX acts as a downstream effector of AMPK in the activation of autophagy under hypoxia. This regulation was confirmed to be independent of the mechanistic target of rapamycin (MTOR) pathway, a major downstream target of AMPK signaling.
Proline is an abundant component of collagen, the major protein in the extracellular matrix (ECM). The upregulation of metalloproteinases (MMPs) degrading ECM in tumors makes proline available in the tumor microenvironment. PRODH/POX is the first enzyme degrading proline in the cells. It tightly binds to mitochondrial inner membranes and contains a flavin adenine dinucleotide (FAD) at its active site. During the conversion of proline to Δ1-pyrroline-5-carboxylate (P5C), PRODH/POX donates electrons through FAD into the electron transport chain (ETC) to generate ROS. By ROS signaling, PRODH/POX initiates apoptosis, inhibits tumor growth, and blocks the cell cycle in vitro, and suppresses tumor formation in a mouse xenograft model. Thus, PRODH/POX was identified as a mitochondrial tumor suppressor protein. Tumor cells can downregulate PRODH/POX expression to eliminate its tumor suppressor role through onco-miRNA MIR23B* and oncogenic transcription factor c-MYC.
However, our recent findings show that PRODH/POX is upregulated in a hypoxic microenvironment in various tumor cells in vitro and in a mouse xenograft model of human breast tumor in vivo. The combination of hypoxia and low glucose has additive effects on PRODH/POX expression. As we know, tumor cells primarily utilize glucose and glutamine for the bioenergetic and biosynthetic demands of proliferation and growth. With rapid growth of tumors, many tumor cells have inadequate blood supply and are depleted of oxygen and nutrients. Since tumor cells with an activated aerobic glycolysis or anaerobic glycolysis use much more glucose than oxidative cells to provide ATP, they tend to deplete glucose more easily. Thus, even with pathological neovascularization, tumor cells will be under different hostile microenvironments, including hypoxia, low glucose or hypoxia combined with low glucose. Under those conditions, alternative energy sources are necessary for tumor cell growth or survival. Proline catalyzed by PRODH/POX not only contributes to ROS generation, but also to ATP. With the high availability of proline in the tumor microenvironment, proline becomes a potential stress substrate.
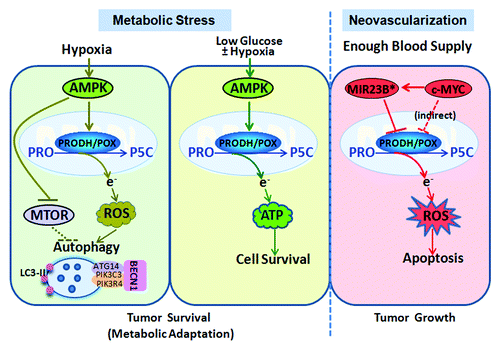
We showed that both hypoxia and glucose deprivation induce PRODH/POX expression to promote tumor cell survival. Nevertheless, upregulated PRODH/POX is used for ATP production only with glucose depletion with or without hypoxia. When only hypoxia is present, enhanced PRODH/POX generates ROS, which is not associated with apoptosis, its recognized tumor-suppressor function, but to yield protective autophagy for cell survival. Knocking down PRODH/POX or using the ROS scavenger N-acetylcysteine dramatically decreases the conversion of LC3-I to LC3-II and the production of autophagosomes as monitored by western blot and punctate distribution of GFP-LC3-II, respectively.
Autophagy is a survival strategy of cancer cells under metabolic stress. It is a cellular self-digestive process whereby macromolecules and cellular organelles are degraded in lysosomes for amino acid recovery and energy recycling. AMPK signaling is the main pathway mediating autophagy, which is activated by a high AMP/ATP ratio following metabolic stress. In addition to suppression of MTOR, the major downstream pathway of AMPK signaling, other autophagy targets of AMPK include CDKN1B/p27, and eukaryotic elongation factor-2 kinase (EEF2K), etc. Our findings indicate that PRODH/POX is also an important target of AMPK activating autophagy under metabolic stress.
The autophagic regulatory mechanism of PRODH/POX is subject to ROS-dependent regulation. Although previous work has shown that MTOR inhibition by rapamycin upregulates PRODH/POX, and the MTOR pathway can induce autophagy through ROS, the AMPK-PRODH/POX-ROS-mediated autophagic response is not dependent on the MTOR pathway. ROS have been long recognized as critical signaling molecules in metabolic stress-induced autophagy. ROS initiate autophagy through several distinct mechanisms involving ATG4, catalase, and the mitochondrial ETC. BECN1 plays a central role in autophagy. Our previous study shows that PRODH/POX-induced ROS can increase the expression of BECN1 by an as-yet unknown mechanism. BECN1 interacts with multiple cofactors and promotes formation of a PIK3C3/VPS34-PIK3R4/VPS15-BECN1-ATG14 core complex, to induce autophagy. Overexpression of BECN1 is associated with tumor hypoxia and poor clinical prognosis in cancer patients, but the underlying mechanism remains unclear. Our findings indicate that hypoxia-induced AMPK-PRODH/POX-ROS is an important regulator of BECN1 expression.
In summary, our results demonstrate that upregulated PRODH/POX through AMPK activation by hypoxia and glucose depletion promotes tumor cell survival through distinct mechanisms (). Glucose depletion with or without hypoxia induces PRODH/POX to provide ATP, whereas hypoxia with adequate glucose activates PRODH/POX-mediated ROS production for protective autophagy. PRODH/POX is a critical factor in AMPK-mediated autophagy. Metabolic stress in the context of the tumor microenvironment switches PRODH/POX functionally from tumor suppressor to tumor survival factor. Upregulation of PRODH/POX producing ATP for cell energy or ROS for autophagy are manifestations of that switch responding to specific metabolic stress. Further exploration of the mechanism underlying this functional switch will help to understand tumor metabolism within different hostile microenvironments and may lead to new therapeutic targets.
Figure 1. PRODH/POX and autophagy under the hypoxia microenvironment. Overexpression of PRODH/POX degrades proline to generate ROS, initiating apoptosis and inhibiting tumor growth. With enough blood supply, PRODH/POX is inhibited by onco-miRNA MIR23B* and oncogenic transcription factor c-MYC, acting as a mitochondrial tumor suppressor, whereas, under metabolic stress, PRODH/POX is induced to function as a tumor survival factor. Hypoxia upregulates PRODH/POX to induce protective autophagy through the AMPK-PRODH/POX-ROS pathway. With low glucose, with or without concurrent hypoxia, PRODH/POX is channeled to produce ATP for cell survival.