Abstract
Low vitamin D levels in human immunodeficiency virus type-1 (HIV) infected persons are associated with more rapid disease progression and increased risk for Mycobacterium tuberculosis infection. We report that physiological concentrations of 1α,25-dihydroxycholecalciferol (1,25D3), the active form of vitamin D, inhibits M. tuberculosis and HIV replication in co-infected macrophages through human cathelicidin microbial peptide-dependent autophagy that requires phagosomal maturation. These findings provide a biological explanation for the importance of vitamin D sufficiency in HIV and M. tuberculosis-infected persons, and provide new insights into novel approaches to prevent and treat HIV infection and related opportunistic infections.
One-third of HIV-infected individuals are estimated to be co-infected with Mycobacterium tuberculosis (Mtb), a leading cause of death among people living with HIV. Despite the high prevalence and mortality of HIV-Mtb co-infection, little is understood about the pathogenesis of dual infection, and treatment options remain limited. Knowledge of the innate immune defense mechanisms targeting Mtb and HIV infection can provide insights into the development of safe, simple and cost-effective adjunctive strategies to prevent and treat both pathogens. Mtb and HIV have developed elaborate methods to subvert the immune system to establish persistent and latent infection. One such mechanism is the ability to inhibit macroautophagy (hereafter referred to as autophagy) by blocking autophagosome-lysosome biogenesis that enables their intracellular survival. In single infection studies, inducers of autophagy including amino acid starvation, rapamycin, and 1,25D3, the active form of vitamin D, overcome the phagosome maturation block leading to enhanced mycobacterial and HIV destruction.
Although no studies had examined the effect of autophagy inducers on HIV or Mtb in a co-infection model, recent research from our laboratory and that of others had identified a potential role for vitamin D in the control of both pathogens. In addition to the documented in vitro antimicrobial effects of vitamin D, several clinical studies had also identified an association of low levels of the inactive form of vitamin D, 25-hydroxycholecalciferol, and/or 1,25D3 with increased risk or severity of infection with HIV and Mtb.
In the article discussed here, we describe the effect of physiologically relevant concentrations of 1,25D3 on autophagy-mediated killing of Mtb and inhibition of HIV in productively co-infected macrophages. 1,25D3 induces dose-dependent inhibition of HIV and mycobacterial growth both alone and in the presence of co-infection that becomes statistically significant at physiological concentrations above 50 pmol/L. The observed effect on mycobacterial viability is dependent upon macrophage infection by Mtb, as 1,25D3 has no effect in a cell-free environment.
Several groups, including our own, have demonstrated that autophagy is a major, previously unrecognized mechanism for elimination of intracellular Mtb and the inhibition of HIV. However, no study has investigated the effect of 1,25D3 on autophagy during co-infection. 1,25D3 induces the significant conversion of LC3B-I to LC3B-II, the turnover of which is inhibited using lysosomal protease inhibitors regardless of infection status. Polyubiquitin-binding protein SQSTM1/p62 (sequestosome 1) degradation in HIV and/or Mtb infected macrophages is also increased post-1,25D3 treatment in the absence of cytotoxicity. Collectively, these data suggest that 1,25D3 induces autophagic flux in HIV- and/or Mtb-infected macrophages.
The relationship between infection status and the 1,25D3-mediated induction of autophagy was also investigated. In HIV-infected macrophages, 1,25D3 reduces the number of cells with detectable HIV infection while reducing the overall level of HIV replication that is greatest in cells undergoing induced autophagy. Untreated Mtb-infected macrophages display an increase in saponin-resistant LC3B-II, while 1,25D3 further increases the number of saponin-resistant LC3B-II-positive cells, but decreases the number of Mtb-infected cells. Interestingly, HIV co-infection inhibits the saponin-resistant LC3B-II that appears in Mtb-only infected macrophages. 1,25D3 is able to overcome this inhibition and induce equivalent levels of LC3B-II saponin resistance while reducing HIV and Mtb infection.
The contribution of autophagy to the 1,25D3-mediated inhibition of HIV and mycobacterial growth was investigated by inhibiting sequential steps of the autophagy pathway. RNA interference for BECN1 and the gene encoding the autophagy-related 5 homolog, combined with chemical inhibitors of autophagic flux, bafilomycin A1, an inhibitor of lysosome acidification and subsequent autophagosome-lysosome fusion, and SID 26681509 an inhibitor of lysosomal hydrolases, demonstrated that the 1,25D3-mediated inhibition of HIV replication and mycobacterial growth both during single infection and co-infection is dependent upon the induction of autophagy through phagosomal maturation.
As previous studies have demonstrated that the human cathelicidin antimicrobial peptide (CAMP) is required for both 1,25D3-mediated antimycobacterial activity and 1,25D3-mediated autophagy in human macrophages, the role of CAMP in 1,25D3-induced antimicrobial activity was investigated. CAMP silencing inhibits 1,25D3-induced autophagy. Also concomitant with our findings that autophagy is required for the 1,25D3 restriction of HIV replication and anti-mycobacterial activities, CAMP silencing also reduces the 1,25D3-mediated inhibition of HIV and Mtb alone and in co-infected cells.
Collectively, our data demonstrate that physiological concentrations of 1,25D3 can act as a potent stimulator of innate antimicrobial responses that can induce autophagy and overcome the HIV-Mtb-imposed autophagosome maturation block through a CAMP-dependent mechanism that inhibits HIV replication and causes mycobacterial destruction (). Well-controlled clinical trials are needed to determine if vitamin D supplementation or other inducers of autophagy are of value as adjunctive treatment in HIV-Mtb co-infected persons. To date, all treatment strategies of persons co-infected with HIV and Mtb have targeted the individual pathogens. Although there has been considerable success with current approaches, the emergence of multiple drug resistant (MDR) and extremely drug resistant (XDR) Mtb strains combined with undesirable interactions between anti-Mtb medications and antiretrovirals highlight the critical need for new approaches to combat HIV-Mtb co-infection. The induction and modulation of autophagy through immunological and pharmacological means to enhance HIV-Mtb treatment is attractive because autophagy works at the host cellular level to improve intracellular killing of both replicating and nonreplicating HIV and Mtb, including MDR- and XDR-TB while resistance by either pathogen is unlikely to develop. Dissecting the molecular mechanisms by which HIV and Mtb utilize autophagy has the potential to lead to the identification of novel pharmacological candidates and immunological approaches that can be rapidly moved into clinical trials as adjunctive treatment for persons with Mtb-HIV co-infection.
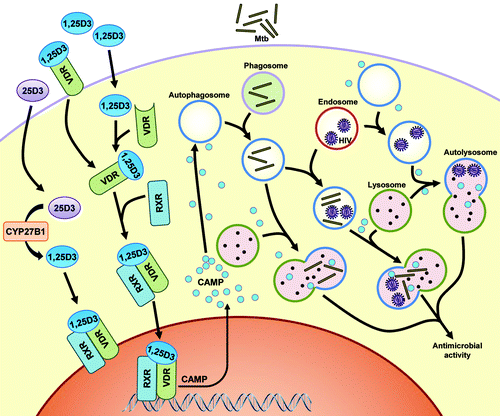
Figure 1. The role of autophagy and CAMP in 1,25D3-mediated inhibition of Mtb and HIV. HIV budding occurs into the multivesicular endosomes of macrophages. Mtb enters through phagocytosis. Cytochrome P450, family 27, subfamily B, polypeptide 1 (CYP27B1) 1α-hydroxylates the inactive 25D3 into the active 1,25D3. 1,25D3 induces the expression of CAMP presumably through binding to the vitamin D (1,25D3) receptor (VDR), which heterodimerizes with the retinoid X receptor (RXR) and directly regulates transcription by binding to the vitamin D response element (VDRE) consensus sequence located upstream of the Camp gene. The expression of CAMP is required both for autophagosome and phagolysosome biogenesis, which leads to killing of the microbial pathogens through autophagy. Adapted from Jo EK. Innate immunity to mycobacteria: vitamin D and autophagy. Cell Microbiol 2010; 12:1026–35; 10.1111/j.1462-5822.2010.01491.x.
Acknowledgements
This work was supported by the NIAID, NIH (grant AI084573) and the International Maternal Perinatal Adolescent AIDS Clinical Trials (IMPAACT) Network. Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials Group (IMPAACT) was provided by the National Institute of Allergy and Infectious Diseases (NIAID) (U01 AI068632), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), and the National Institute of Mental Health (NIMH) (AI068632). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work was supported by the Statistical and Data Analysis Center at Harvard School of Public Health, under the National Institute of Allergy and Infectious Diseases cooperative agreement #5 U01 AI41110 with the Pediatric AIDS Clinical Trials Group (PACTG) and U01 AI068616 with the IMPAACT Group. Support of the sites was provided by the National Institute of Allergy and Infectious Diseases (NIAID) and the NICHD International and Domestic Pediatric and Maternal HIV Clinical Trials Network funded by NICHD (contract number N01-DK-9-001/HHSN267200800001C).