Abstract
Cyclosporine A (CsA), which revolutionized transplantology due to its ability to block the activation of lymphocytes and other immune system cells, triggers autophagy in malignant glioma cell lines via stimulation of endoplasmic reticulum (ER) stress. We also found that autophagy serves as a protective mechanism against CsA toxicity.
Cyclosporine A is a cyclic undecapeptide, which has revolutionized human organ transplantation due to its ability to block the activation of lymphocytes and other immune system cells. Despite its beneficial effects in the prevention of graft rejection and promising results in animal models of chronic inflammatory disorders, the clinical use of CsA has been hampered due to cytotoxic side effects involving neuro-, liver- and nephrotoxicity. Furthermore, CsA at higher doses (> 10–20 µM) exerts antitumor activity toward several types of tumor cells, including malignant gliomas resistant to many traditional chemotherapeutics. Molecular mechanisms underlying CsA-induced cytotoxicity are not well elucidated. CsA binds to specific intracellular proteins, cyclophilins, which are peptidyl-propyl cis-trans isomerases implicated in the maturation and folding of native proteins. CsA inhibits cyclophilins, but blocking of the propyl isomerase activity of these proteins is not essential for CsA immunosuppressive effects. The CsA-cyclophilin complex binds to calcineurin (calcium- and calmodulin-dependent phosphatase) and inhibits its activity. CsA treatment blocks calcineurin-mediated dephosphorylation of NFAT, thus preventing transcriptional induction of several cytokines and their receptor genes crucial for the immune response.
We demonstrated that rat glioma cells express NFAT proteins, and CsA inhibits calcineurin-NFAT signaling; however, blocking this pathway is not sufficient to induce death of glioma cells. We have previously found that CsA induces cell death with apoptotic features in rat C6 glioma cells that is mediated by activation of TP53/p53 and its transcriptional target gene Bax resulting in stimulation of the intrinsic mitochondrial death pathway. Molecular analysis revealed that CsA, through an as-yet unknown mechanism, downregulates AKT1 signaling and facilitates the activation of a forkhead family member (FOXO1) resulting in transcriptional activation of Faslg/FasL in C6 glioma cells. Interestingly, in human malignant glioma cells, defective in TP53 and/or PTEN tumor suppressors, CsA induces either senescence-like growth arrest (U87 cells) or nonapoptotic programmed cell death (U373, T89G cells). Those events involve a prolonged activation of MAPK1/2 (ERK) signaling and accumulation of the cell cycle inhibitor CDKN1A/p21CIP1/WAF1, even in the absence of functional TP53 (). CsA-induced death is accompanied by the appearance of numerous cytoplasmic vacuoles.
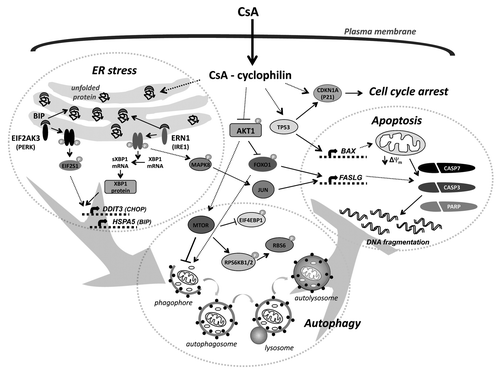
Figure 1. Molecular mechanism of CsA-induced death of malignant glioma cells (see text for details). CsA can induce ER stress, autophagy, cell cycle arrest and cell death. CsA, likely due to inhibition of cyclophilins, induces accumulation of unfolded proteins in the endoplasmic reticulum (ER) that triggers an unfolded protein response. Activation of EIF2AK3-EIF2S1 and the ERN1-XBP1-MAPK8 pathways upregulates the expression of DDIT3 and HSPA5. Induction of ER stress and/or decreased activity of the AKT1-MTOR pathway contribute to initiation of autophagy. Inhibition of the prosurvival pathway (AKT1) results in the activation of FOXO1, that cooperates with the JUN transcription factor in the transcriptional upregulation of FASLG. Autophagy inhibits apoptotic cell death induced by CsA. In cells with a functional TP53, CsA induces caspase-dependent apoptotic cell death via the induction of the pro-apoptotic protein BAX, which reduces the mitochondrial transmembrane potential. In TP53-deficient cells CsA induces MAPK1/3-dependent activation of CDKN1A that results in cell cycle arrest.
Recently, autophagy emerged as a promising therapeutic approach for chemoresistant cells. Autophagy is an intracellular degradation process accompanied by formation of double-membrane autophagosomes, which fuse with acidic compartments, endosomes and/or lysosomes, to degrade the materials inside the autophagosomes. Most chemotherapeutics induce cellular damage that triggers autophagy, but the impact of autophagy on tumor cell death or survival is unclear. On the one hand, autophagy mediates the cytotoxic or cytostatic effect of anticancer agents, and blockade of autophagy abolishes the therapeutic actions. On the other hand, autophagy could be a prosurvival response to withstand a toxicity of the therapy, and inhibition of autophagy enhances drug-induced cytotoxicity. Thus, a final outcome of autophagy activation would be highly dependent on the cellular context (e.g., the genetic background of the cancer cells, and the influence of other signaling pathways), and the strength and duration of the stress-inducing signals. Therefore, estimation of the role of autophagy in particular cells under specific conditions is necessary.
The endoplasmic reticulum stress response represents another adaptive mechanism that basically supports survival and chemoresistance of tumor cells, but after crossing some thresholds of severe conditions, this process will trigger cell death. Several reports have shown that ER stress can activate autophagy, although reciprocal interaction is not well explored, it is likely that accumulation of misfolded proteins and protein aggregates may represent an important trigger interdependently affecting both processes. We investigated whether CsA-induced cell death of malignant glioma cells is associated with autophagy and ER stress and how blocking of these processes would affect glioma cell death. We demonstrate activation of autophagy and ER stress responses in malignant glioma cells exposed to toxic doses of CsA. We observed formation of large cytoplasmic vacuoles in CsA-treated glioma cells that were immunostained for ER markers. The induction of ER stress in glioma cells by CsA was evidenced by accumulation of ubiquitinated (unfolded) proteins and activation of the unfolded protein response (UPR). We found that CsA treatment triggers accumulation of ERN1/IRE1α and phosphorylation of EIF2AK3/PERK in human glioma cells (two UPR sensors). The functional activation of ERN1 and EIF2AK3 was confirmed by the appearance of Xbp1 mRNA splice variants and the increase of EIF2S1/eIF2α phosphorylation upon CsA-treatment, respectively. Active ERN1 may regulate stress response by MAPK8/JNK activation. We have previously shown a prolonged increase of MAPK8 activity, JUN phosphorylation and complex formation in CsA-treated cells, that implicates an involvement of MAPK8 signaling in the induction of ER stress. HSPA5/BIP is a critical player in reestablishing ER homeostasis, in contrast to DDIT3/CHOP, which triggers ER stress-induced cell death despite the presence of a high level of HSPA5. Accumulation of HSPA5 and DDIT3 follows the activation of ERN1 and EIF2AK3 kinases in CsA-treated glioma cells ().
We found that CsA stimulates autophagy in glioma cells. We demonstrate the appearance of several autophagy features after CsA treatment such as: formation of acidic vesicular organelles, increase of autophagic vacuoles, GFP-LC3 puncta formation and LC3-II accumulation upon CsA treatment. The increase of LC3 staining in intracranial gliomas in CsA-treated animals demonstrates that autophagy also occurs in vivo. CsA-induced autophagy is associated with inhibition of the MTOR pathway, which acts as a negative regulator of autophagy. Decrease of phosphorylation of EIF4EBP1/4E-BP1, and RPS6KB1/2/p70S6K (the two direct downstream effectors of MTORC1) is observed in human glioma cells. MTORC1 is regulated by the prosurvival kinase AKT1, inhibition of which leads to MTORC1 inactivation. CsA inhibits AKT1 signaling and activates the FOXO1 transcription factor. Several studies showed that FOXO family members can induce the expression of multiple autophagy genes ().
We also studied interdependence between ER stress, autophagy and cell death. We found that an ER stress response develops prior to autophagy, as its pharmacological inhibition reduces autophagy. Salubrinal (inhibitor of EIF2S1 dephosporylation) or silencing of EIF2AK3 or/and ERN1 partially blocks CsA-induced accumulation of LC3-II. Consistently, salubrinal alleviates the CsA-induced decline of phospho-RPS6KB1/2 and EIF4EBP1 levels. As discussed above, autophagy can play a dual role in cancer cells. In our study silencing of autophagy effectors ULK1, ATG5 or ATG7 increase cell death of treated cells as evidenced by the increased efficacy of CsA in the reduction of colony formation, increased level of active CASP3, CASP7 and PARP degradation, as well as enhancement of CASP3 activity in CsA-treated cells (). Interestingly, the strongest effect was observed in Atg5 siRNA-transfected cells. Our findings show that genetic inhibition of autophagy at different levels enhances the CsA-induced cell death, suggesting a mainly protective role of autophagy in those cells. These observations lead us to conclude that CsA induces both apoptosis and autophagy in malignant glioma cells via induction of ER stress and inhibition of the MTOR-RPS6KB1/2 pathway; however, autophagy is cytoprotective in this context.
| Abbreviations: | ||
| Atg | = | autophagy related |
| CDKN1A | = | cyclin-dependent kinase inhibitor 1A (p21) |
| CsA | = | cyclosporine A |
| DDIT3 | = | DNA-damage-inducible transcript 3 (C/EBP homologous protein, CHOP) |
| EIF2AK3 | = | eukaryotic translation initiation factor 2-alpha kinase 3 (protein kinase-like endoplasmic reticulum kinase, PERK) |
| EIF2S1 | = | α subunit of the eukaryotic initiation factor 2 (eIF2α) |
| EIF4EBP1 | = | eukaryotic translation initiation factor 4E binding protein 1 (4EBP1) |
| ERN1 | = | inositol requiring enzyme 1 (IRE1α) |
| FOXO1 | = | Forkhead box protein O1 |
| HSPA5 | = | heat shock 70kDa protein 5 (binding immunoglobulin protein, BIP) |
| JUN | = | jun proto-oncogene (AP-1) |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| MAPK1/3 | = | mitogen-activated protein kinase 1/3 (extracellular signal-regulated kinases, ERK1/2) |
| MAPK8 | = | mitogen-activated protein kinase 8 (c-Jun-N-terminal kinase, JNK) |
| MTOR | = | mechanistic target of rapamycin |
| NFAT | = | nuclear factor of activated T cells |
| PARP | = | poly (ADP-ribose) polymerase |
| PTEN | = | phosphatase and tensin homolog |
| RPS6KB1/2 | = | ribosomal protein S6 kinase (p70S6K) |
| ULK1 | = | unc-51-like kinase 1 |
| UPR | = | unfolded protein response |
| XBP1 | = | X-box binding protein 1 |
Acknowledgments
This work was supported by a grant N N301 092036 from The Polish Ministry of Science and Higher Education (I.A.C.).