Abstract
We previously reported that autophagy is upregulated in Prnp-deficient (Prnp0/0) hippocampal neuronal cells in comparison to cellular prion protein (PrPC)-expressing (Prnp+/+) control cells under conditions of serum deprivation. In this study, we determined whether a protective mechanism of PrPC is associated with autophagy using Prnp0/0 hippocampal neuronal cells under hydrogen peroxide (H2O2)-induced oxidative stress. We found that Prnp0/0 cells were more susceptible to oxidative stress than Prnp+/+ cells in a dose- and time-dependent manner. In addition, we observed enhanced autophagy by immunoblotting, which detected the conversion of microtubule-associated protein 1 light chain 3 β (LC3B)-I to LC3B-II, and we observed increased punctate LC3B immunostaining in H2O2-treated Prnp0/0 cells compared with H2O2-treated control cells. Interestingly, this enhanced autophagy was due to impaired autophagic flux in the H2O2-treated Prnp0/0 cells, while the H2O2-treated Prnp+/+ cells showed enhanced autophagic flux. Furthermore, caspase-dependent and independent apoptosis was observed when both cell lines were exposed to H2O2. Moreover, the inhibition of autophagosome formation by Atg7 siRNA revealed that increased autophagic flux in Prnp+/+ cells contributes to the prosurvival effect of autophagy against H2O2 cytotoxicity. Taken together, our results provide the first experimental evidence that the deficiency of PrPC may impair autophagic flux via H2O2-induced oxidative stress.
Introduction
Prion diseases or transmissible spongiform encephalopathies (TSEs) are a group of fatal neurodegenerative disorders in animals and humans. These disorders are characterized by the accumulation of the pathological scrapie isoform (PrPSc) protein, which is post-translationally generated from PrPC.Citation1 PrPC is a glycosylphosphatidylinositol (GPI)-anchored, membrane-bound protein encoded by Prnp and is highly conserved in mammals.Citation2 The expression of PrPC is most abundantly detected in neuronal cells, but it has also been observed in non-neuronal cells.Citation3,Citation4 Therefore, PrPC may play an important role in the central nervous system, although its exact physiological function remains unknown. Prnp0/0 mice are comparatively healthy and do not develop a prion-like disease.Citation5 These mice have also been reported to show abnormalities in synaptic physiology,Citation6 circadian rhythms, and sleep.Citation7 In the last several years, various biological functions for PrPC have been suggested, including neurotransmitter metabolism,Citation8 signal transduction,Citation9 copper metabolism,Citation10 cell adhesion,Citation11 neuritogenesis,Citation12 antioxidant activity,Citation13 and immune cell activation.Citation14 Moreover, many studies have suggested that PrPC plays a neuroprotective role in response to serum deprivation and oxidative stress in neuronal and non-neuronal cell lines.Citation15-Citation17 In response to oxidative stress, primary neurons cultured from the brains of Prnp0/0 mice are more susceptible to cell death than those cultured from the brains of Prnp+/+ mice.Citation18,Citation19 However, the molecular mechanism related to the protective role of PrPC against oxidative stress is unclear.
Autophagy is a major lysosomal degradation pathway for the elimination of damaged or long-lived proteins and organelles and plays important roles in physiology and pathophysiology in all cell types. Autophagy was originally described as a response to nutrient deprivation, thus aiding in cell survival through the turnover and recycling of cytoplasmic contents.Citation20 Autophagy is activated during the differentiation and development of multicellular organisms, such as flies, worms and slime molds,Citation21 and is also involved in cancer, neurodegenerative diseases, pathogen infections, liver injury and myopathies.Citation22,Citation23 Despite its role in cell survival, autophagy is also associated with cell death. Indeed, cell death occurs through three mechanisms: apoptosis (Type I), autophagic cell death (Type II) and necrosis (Type III).Citation24 Although autophagic cell death is caspase-independent and is clearly distinct from apoptotic cell death, these mechanisms can occur in mixed forms with autophagic and caspase-related apoptotic features.Citation25-Citation28
Ultrastructural analyses of prion-infected cultured cellsCitation29 and neurons from the brain tissues of TSE-affected patients and rodentsCitation30 show the presence of autophagic vacuoles and multivesicular bodies. In addition, stimulator of chondrogenesis 1/scrapie responsive gene 1 (SCRG1) expression is increased in the autophagic vacuoles of neurons from scrapie-infected mice.Citation31 Recently, it is reported that autophagy induction achieved by pharmacological autophagy inducers, such as trehalose or lithium, can reduce PrPSc expression in persistently prion-infected neuronal cells.Citation32,Citation33 Interestingly, the ectopic overexpression of PrP protein 2 (dublet)/PrP-like protein doppel (PRND) in PrPC-deficient central neurons of Ngsk prion protein-deficient (NP0/0) mice showed that the expression levels of SCRG1 and the autophagic markers LC3B-II and sequestosome 1 (SQSTM1)/p62 were increased both before and during Purkinje cell death, whereas the mRNA expression levels of Lc3b and Sqstm1/p62 remained stable. These results may reflect an impairment of autophagic flux in the Purkinje cells of NP0/0 mice.Citation34 A recent report revealed that downregulation of PrPC expression using antisense oligonucleotides targeting the PRNP transcript led to autophagy-dependent cell death, with the absence of markers of apoptotic cell death in human malignant glioma cell lines and nonglial tumor cells.Citation35 This report suggested that PrPC could directly modulate the autophagy-dependent cell death pathway.
In a previous report, we showed that autophagy is increased in Prnp0/0 hippocampal neuronal cells compared with wild-type control cells under serum deprivation conditions. This increased autophagy is suppressed in Prnp0/0 cells transfected with the Prnp gene.Citation36 However, it is unclear whether increased autophagy contributes to the activation of cell survival or cell death pathways. In transformed and cancer cell lines, oxidative stress-induced autophagic cell death persisted upon treatment with reactive oxygen species (ROS)-generating agents, such as hydrogen peroxide (H2O2) and 2-methoxyestradiol.Citation37 Low levels of such ROS as superoxide (O2•–), hydroxyl radical (HO•) and H2O2 can serve as signaling molecules in various cellular pathways that lead to growth and survival. However, high levels of ROS can promote cellular damage and cell death through the oxidation of lipids, proteins and DNA.Citation38,Citation39
To investigate further a mechanism for the protective role of PrPC in oxidative stress, we examined the alteration and differentiation of autophagy under H2O2-induced oxidative stress in hippocampal neuronal cell lines expressing PrPC (Prnp+/+) and a control vector (Prnp0/0).Citation36,Citation40 In addition, we explored whether the inhibition or induction of autophagy could promote cell survival or cell death. In these experiments, we show that the expression of the autophagy marker LC3B-II was more increased in Prnp0/0 neuronal cells than in Prnp+/+ cells under oxidative stress conditions. Furthermore, we demonstrated that the enhanced expression of LC3B-II in Prnp0/0 cells is due to impaired autophagic flux. Moreover, we show that increased autophagy may be associated with cell survival and cell death in Prnp+/+ and Prnp0/0 cells, respectively, under oxidative stress. Our findings suggest that PrPC not only plays a novel protective role against oxidative stress-induced cell death but may also regulate autophagic flux during oxidative stress in neuronal cells.
Results
Prnp0/0 neuronal cells exhibit increased susceptibility to H2O2-induced oxidative stress
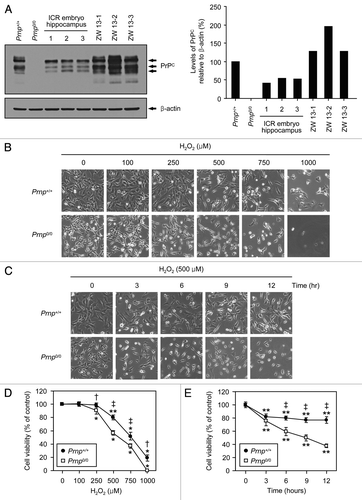
In a previous report, it was shown that cultured primary neurons of Zürich I Prnp0/0 mice are more susceptible to H2O2-induced oxidative stress than are Prnp+/+ neurons.Citation18 Recently, we established Zürich I Prnp0/0 hippocampal neuronal cell lines, which are stably transfected with a vector encoding murine Prnp (Prnp+/+ cells) or an empty vector (Prnp0/0 cells).Citation36 The cells transfected with the empty vector were shown to be Prnp0/0, while the cells transfected with Prnp were used as PrPC controls. To confirm the expression levels of PrPC in Prnp+/+ and Prnp0/0 cells, we performed western blot analysis using embryonic hippocampus lysates from ICR mice and previously established ICR hippocampal neuronal cell lines (ZW 13-1, ZW 13-2 and ZW 13-3). The results showed that PrPC expression was slightly lower in the Prnp+/+ cells than in the ICR hippocampal neuronal cell lines, whereas the expression level of PrPC in the Prnp+/+ cells was higher than in the ICR embryonic hippocampus (). Using Prnp+/+ and Prnp0/0 cell lines, we investigated the neuroprotective role of PrPC under H2O2-induced oxidative stress using light microscopy and a 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate (WST-1)-based assay. As shown in , the morphological features of Prnp+/+ and Prnp0/0 cells exposed to H2O2 revealed irregular shrinkage and cell rounding compared with the untreated control cells. Although the number of cells attached to the surfaces of culture dishes gradually decreased in both cell lines, there were fewer attached Prnp0/0 cells than Prnp+/+ cells in a dose- and time-dependent manner following H2O2 treatment. Subsequently, we examined whether the cell lines differed in susceptibility to H2O2 toxicity using a WST-1-based assay. Although treatment with H2O2 significantly decreased cell viability in both Prnp+/+ (doses of 500–1,000 µM) and Prnp0/0 (doses of 250–1,000 µM) cells, the Prnp0/0 cells were significantly more susceptible to the H2O2 toxicity (). Moreover, the viability of Prnp0/0 cells was significantly lower than that of Prnp+/+ cells in a time-dependent manner (3–12 h) after treatment with 500 µM H2O2 (). These findings clearly demonstrate that Zürich I Prnp0/0 neuronal cells are more susceptible to H2O2-induced oxidative stress than Prnp+/+ control cells, indicating that PrPC plays a protective role against H2O2 cytotoxicity.
Figure 1. The protective effect of PrPC against H2O2-induced cell death. (A) The expression of PrPC in distinct stable cell lines and the embryo hippocampus of ICR mice. Western blot analysis was conducted using anti-PrP (3F10) in lysates from Zürich I Prnp0/0 hippocampal neuronal cell lines transfected with murine Prnp (Prnp+/+) or with an empty vector (Prnp0/0), several hippocampi from 14-d-old embryos of ICR mice and 3 different WT hippocampal neuronal cell lines of ICR mice (ZW 13–2, ZW 13–2 and ZW 13–3). The expression of PrPC was quantified using densitometry analysis (right panel). (B and C) Morphological appearance of Prnp+/+ and Prnp0/0 cells as observed under an inverted microscope. In (B and D), Prnp+/+ and Prnp0/0 cells were incubated with the indicated concentrations of H2O2 for 6 h. In (C and E), cells were treated with 500 μM H2O2 for the indicated time periods. (D and E) Cell viability was measured using the WST-1-based assay. The data are presented as the means ± standard deviation (S.D.) of 3 independent experiments performed in triplicate. *p < 0.02 and **p < 0.001, significant differences between treated and untreated cells; †p < 0.01 and ‡p < 0.001, significant differences between Prnp+/+ and Prnp0/0 cells.
Prnp0/0 neuronal cells are more susceptible to autophagic induction during H2O2-induced oxidative stress
To determine whether H2O2 cytotoxicity is associated with autophagy, we measured the expression of endogenous LC3B-II using western blot analysis. During the induction of autophagy, the cytoplasmic form of LC3B (LC3B-I, 18 kDa) is converted to the autophagosomal membrane-bound form (LC3B-II, 16 kDa). This conversion is a reliable marker for autophagy activation.Citation41 As shown in , no differences in the basal expression levels of LC3B-II were detected between the untreated Prnp+/+ and Prnp0/0 cells. However, increased expression levels of LC3B-II were observed at doses of 250–750 µM H2O2 in both cell lines (). In addition, enhanced expression of LC3B-II was detected at 3–24 h after treatment with 500 µM H2O2 in both cell lines (). Interestingly, the expression of LC3B-II in the Prnp+/+ cells was lower than that in the Prnp0/0 cells after H2O2 treatment in a dose- and time-dependent manner ().
Figure 2. The induction of autophagy in Prnp+/+ and Prnp0/0 cells exposed to H2O2. (A) Cells were incubated with the indicated concentrations of H2O2 for 6 h. (B) Cells were treated with 500 μM H2O2 for the indicated time periods. Cell lysates were subjected to western blot analysis using anti-LC3B or anti-β-actin antibodies (A and B), and the expression of LC3B-II was quantified using densitometry analysis (right panels). At least 3 independent experiments were used to assess the percentage of LC3B-II expression relative to that of β-actin. (C) Immunofluorescence microscopy analysis using anti-LC3B antibody and DAPI counterstaining in both Prnp+/+ and Prnp0/0 cells treated with 500 μM H2O2 for 3 h. The average number of LC3B puncta per cell is shown in the lower panel. At least 60 cells in several fields were counted in each experiment. The data represent the means ± SD of 3 independent experiments. *p < 0.05 and **p < 0.01, significant differences between H2O2-treated and untreated cells; †p < 0.05 and ‡p < 0.01, significant differences between Prnp+/+ and Prnp0/0 cells. Scale bar: 20 µm.
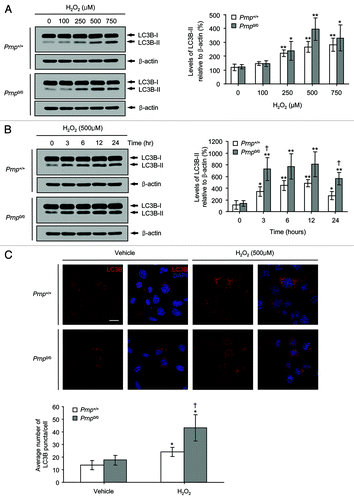
When autophagy was induced, LC3B was redistributed from a cytosolic diffuse pattern to a punctate staining pattern in the vacuolar membranes, which indicates the presence of autophagosomes.Citation41 Therefore, we counted the number of endogenous LC3B-positive puncta per cell in both Prnp+/+ and Prnp0/0 cells exposed to 500 µM H2O2 for 3 h using immunofluorescence microscopy and an anti-LC3B antibody. Upon exposure to H2O2, the average number of LC3B puncta per cell was higher in the Prnp0/0 cells than in the Prnp+/+ cells (). These results suggest that H2O2-induced cell death is associated with enhanced levels of autophagy in both neuronal cell lines.
H2O2-induced oxidative stress is impaired during the early stages of autophagic flux in Prnp0/0 neuronal cells
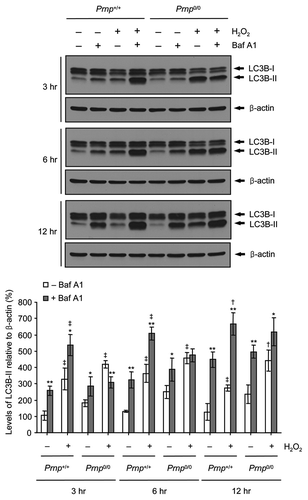
To confirm further whether the H2O2-induced enhancement of autophagy arose from autophagy stimulation or impaired autophagic flux, we used a vacuolar H+-ATPase inhibitor, bafilomycin A1 (Baf A1), which prevents lysosome acidification and autophagosome-lysosome fusion. Prnp+/+ and Prnp0/0 cells were treated in the absence or presence of Baf A1 combined with H2O2 for various time periods (3, 6 and 12 h). Treatment with Baf A1 in combination with H2O2 increased the expression of LC3B-II at each time point (3–12 h) in the Prnp+/+ cells compared with H2O2 treatment alone, indicating that H2O2 did not inhibit lysosomal activity or autophagic flux in the Prnp+/+ cells (). In contrast, the expression of LC3B-II at early time points (3 and 6 h) in Prnp0/0 cells treated with Baf A1 and H2O2 was similar to that of Prnp0/0 cells treated with H2O2 alone, suggesting that H2O2 treatment impairs autophagic flux in Prnp0/0 cells. Interestingly, this impaired autophagic flux is rescued at a later time point (12 h) in Prnp0/0 cells. To confirm whether PrPC is associated with autophagic flux, we examined basal autophagic flux in the absence or presence of Baf A1 without H2O2 in both cell lines. Interestingly, the expression of LC3B-II was higher in both cell lines treated with Baf A1 for 3–12 h compared with untreated cells. This result indicates that the PrPC deficiency did not affect the basal autophagy flux under normal culture conditions. Taken together, these data suggested that PrPC is indirectly associated with autophagic flux under H2O2-induced oxidative stress.
Figure 3. Impaired autophagic flux in Prnp0/0 cells exposed to H2O2. Cells were incubated with or without 500 μM H2O2 in the presence or absence of 20 nM bafilomycin A1 (Baf A1) for the indicated time periods. Cell lysates were subjected to western blot analysis using anti-LC3B or anti-β-actin antibody (upper panel), and the expression of LC3B-II was quantified using densitometry analysis (lower panel). At least 3 independent experiments were used to assess the percentage of LC3B-II expression relative to that of β-actin. The data represent the means ± SD of 3 independent experiments. *p < 0.05 and **p < 0.01, significant differences between Baf A1-treated and untreated cells; †p < 0.05 and ‡p < 0.01, significant differences between H2O2-treated and untreated cells.
H2O2-induced oxidative stress activates caspase-dependent and caspase-independent apoptosis at different time points in both Prnp+/+ and Prnp0/0 cells
Our previous study showed that Prnp0/0 neuronal cells are more sensitive to autophagy and apoptosis than Prnp+/+ neuronal cells, although expression levels of LC3-II and cleaved caspase 3 (CASP3) were elevated in both cell types under conditions of serum deprivation.Citation36 Therefore, we examined the generation of cleaved CASP3, a marker for apoptosis,Citation42 in both cell types under exposure to H2O2. Interestingly, cleaved CASP3 was initially detected at a dose of 750 µM H2O2 or at 12 h after treatment with 500 µM H2O2 in both Prnp+/+ and Prnp0/0 cells (). Unexpectedly, the expression of cleaved CASP3 in Prnp+/+ cells was higher than that of Prnp0/0 cells in a dose- and time-dependent manner. This result suggests that Prnp+/+ cells are more susceptible to apoptotic cell death than Prnp0/0 cells in the later stages of H2O2-induced oxidative stress.
Figure 4. Apoptosis activation in Prnp+/+ and Prnp0/0 cells exposed to H2O2. (A) Cells were incubated with the indicated concentrations of H2O2 for 6 h. (B) Cells were treated with 500 μM H2O2 for the indicated time periods. Cell lysates were subjected to western blot analysis with anti- CASP3 or anti-β-actin antibodies (A and B), and the expression of cleaved CASP3 was quantified using densitometry analysis (right panels). At least 3 independent experiments were used to assess the percentages of cleaved CASP3 expression relative to that of β-actin. †p < 0.05, significant differences between Prnp+/+ and Prnp0/0 cells. (C) Cells were preincubated with the indicated concentrations of zVAD-fmk for 3 h and treated with or without 500 μM H2O2 for 6 h. Cell viability was determined using the WST-1 reagent assay. The data are represented as the percent viability normalized to mock-treated control groups (100%). The data are presented as the means ± SD of 3 independent experiments performed in triplicate. (D) Cells were treated with 500 μM H2O2 for the indicated time periods and fractionated into cytoplasmic and nuclear fractions. Each fraction was analyzed by western blotting with anti-AIFM1, anti-ENDOG, anti-ENO1 (cytosolic fraction markers) and anti-HDAC2 antibody (nuclear fraction marker).
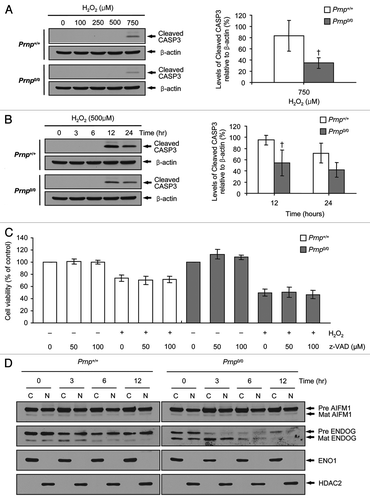
To confirm further the absence of CASP3-dependent apoptosis in H2O2 treatment at early time points (3–6 h), we investigated whether z-VAD-fmk, a pan-caspase inhibitor, could prevent H2O2-induced cell death. As expected, z-VAD-fmk treatment did not suppress H2O2-induced cell death in either cell type (). These results are in agreement with the fact that cleaved CASP3 was not detected in either Prnp+/+ or Prnp0/0 cells at 3–6 h after treatment with 500 µM H2O2, which suggests that CASP3-dependent apoptosis does not contribute to H2O2-induced cell death at early stages.
Next, we investigated whether H2O2-induced cell death is associated with caspase-independent apoptosis using apoptosis-inducing factor, mitochondrion-associated, 1 (AIFM1) and endonuclease G (ENDOG). AIFM1 and ENDOG can directly activate nuclear DNA fragmentation when translocated from the mitochondrial intermembrane space to the nucleus through regulation of the pro-apoptotic BCL2 family.Citation43-Citation46 Both AIFM1 and ENDOG are expressed as precursor proteins that are converted to mature forms upon mitochondrial import and cleavage of the N-terminal targeting sequence.Citation47,Citation48 The cells were treated with H2O2 and fractionated into cytoplasmic and nuclear fractions. As shown in , the expression of mature AIFM1 was unchanged from 0 h to 12 h in both Prnp+/+ and Prnp0/0 cells, whereas mature ENDOG expression was markedly increased at 3 h. Enolase 1 (ENO1) and histone deacetylase 2 (HDAC2) were recovered exclusively in the cytoplasmic and nuclear fractions, respectively, confirming the adequate separation of cytoplasmic and nuclear fractions. This result indicates that H2O2-induced cell death is associated with ENDOG expression at an early stage (3 h) in both Prnp+/+ and Prnp0/0 cells. Taken together, these results suggest that H2O2 cytotoxicity is associated with caspase-independent apoptosis at an early stage (at 3 h after treatment with 500 µM H2O2) and caspase-dependent apoptosis at a later stage (at 12 h after treatment with 500 µM H2O2) in both cell types.
The administration of 3-MA reduced H2O2-induced autophagy, whereas treatment with rapamycin or trehalose promoted H2O2-induced autophagy in Prnp0/0 cells
To evaluate the role of H2O2-induced autophagy, we investigated the effect of 3-methyladenine (3-MA), an inhibitor of phosphatidylinositol 3-kinase (PtdIns3K), which is a component of the autophagy pathway,Citation49 on H2O2-induced autophagy in both Prnp+/+ and Prnp0/0 cells. In this study, increased autophagy activation and reduced CASP3 cleavage were observed in both Prnp+/+ and Prnp0/0 cells at 6 h after treatment with 500 µM H2O2. We therefore examined the effect of 3-MA at 6 h after treatment with 500 µM H2O2 using western blot analysis and cell viability assays. Treatment with 3-MA slightly reduced the expression of LC3B-II in Prnp0/0 cells treated with or without H2O2 and had no effect on LC3B-II expression in Prnp+/+ cells (). The WST-1 cell viability assay revealed that 3-MA significantly decreased H2O2-induced cell death in Prnp0/0 cells but had no significant effects in Prnp+/+ cells (). Importantly, 3-MA is a widely used autophagy inhibitor that suppresses class III PtdIns3K activity.Citation49 However, it was recently reported that 3-MA can also activate autophagy under nutrient-rich conditions, such as normal culture conditions, via persistent inhibition of class I PtdIns3K, disruption of mechanistic target of rapamycin (MTOR) complex I activity and transient suppression of class III PtdIns3K.Citation50 Hence, we further explored the expression of phospho-AKT1, a major downstream target of class I PtdIns3K, to confirm the inhibition of class I PtdIns3K. As shown in , the treatment of 3-MA did not reduce the expression of phospho-AKT1, suggesting that the effect of 3-MA in Prnp+/+ cells may occur through a class I PtdIns3K-independent pathway.
Figure 5. Effects of the autophagy inhibitor 3-MA and the autophagy inducers rapamycin (Rapa) and trehalose (Treha) on H2O2-induced cell death. (A–F) Prnp+/+ and Prnp0/0 cells were pretreated with or without 5 mM 3-MA, 10 nM Rapa or 100 mM Treha for 1 h (Rapa) or 3 h (3-MA and Treha) and with or without 500 μM H2O2 for 6 h. (A, C and E) Cell lysates were subjected to western blot analysis using an anti-LC3B, antibody in PathScan® Multiplex Western Cocktail I to detect phosphorylated AKT1 or RPS6 or anti-β-actin antibody. (B, D and F) Cell viability was determined using the WST-1 reagent assay. The data are represented as the percent viability normalized to mock-treated control groups (100%). The data are presented as the means ± SD of 3 independent experiments performed in triplicate.
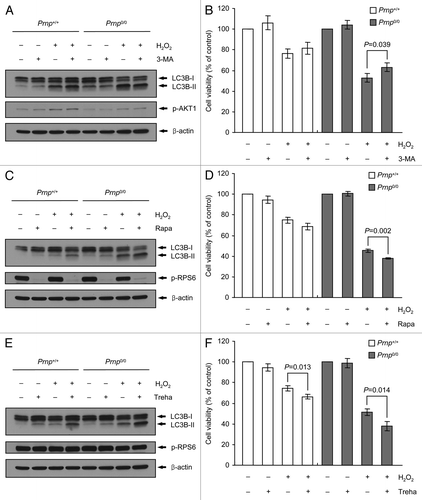
To study further the effect of H2O2-induced autophagy, we used two autophagy inducers, namely, rapamycin (MTOR-dependent)Citation51 and trehalose (MTOR-independent).Citation52 The MTOR-dependent and independent effects of these inducers were confirmed through the expression of phospho-ribosomal protein S6 (RPS6), a surrogate marker for MTOR activity, using western blot analysis (). These two autophagy inducers markedly increased the expression levels of LC3B-II upon treatment with or without H2O2 in both Prnp+/+ and Prnp0/0 cells (). Furthermore, these two autophagy inducers significantly enhanced H2O2-induced cell death in Prnp0/0 cells. Likewise, trehalose significantly increased H2O2-induced cell death in Prnp+/+ cells, although the effects of rapamycin in these cells were not significant (). Taken together, these experiments suggest that the role of autophagy in H2O2 cytotoxicity might be associated with cell death in Prnp0/0 cells. However, these results remain controversial, and further investigations are required to establish the effects of pharmacological inhibition or activation of autophagy in Prnp+/+ and Prnp0/0 cells.
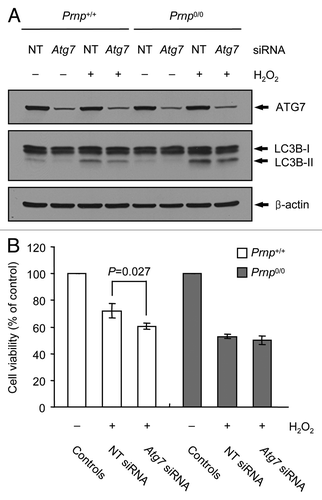
The knockdown of Atg7 inhibits autophagy and promotes H2O2 cytotoxicity in Prnp+/+ cells, but not Prnp0/0 cells
The chemical inhibition of autophagy may be a consequence of the side effects from the drug used;Citation53 therefore, we assessed the role of autophagy in H2O2 cytotoxicity using an siRNA knockdown of Atg7, which is essential for the formation of autophagosomes. The expression of ATG7 protein was markedly decreased in Prnp+/+ and Prnp0/0 cells transfected with Atg7 siRNA. The knockdown of Atg7 also led to decreased expression levels of LC3B-II in both Prnp+/+ and Prnp0/0 cells treated with or without H2O2 (). Interestingly, the knockdown of Atg7 resulted in enhanced H2O2-induced cell death in Prnp+/+ cells but did not affect cell viability in the H2O2-treated Prnp0/0 cells (). Taken together, these results suggest that autophagy may be involved in cell survival in H2O2-treated Prnp+/+ cells and that impaired autophagic flux in H2O2-treated Prnp0/0 cells may contribute to cell death. Furthermore, the inhibition of autophagy may not affect cell death in H2O2-treated Prnp0/0 cells because the autophagic flux is already impaired in Prnp0/0 cells.
Figure 6. Augmentation of H2O2-induced cell death using Atg7 knockdown in Prnp+/+ cells. (A and B) Prnp+/+ and Prnp0/0 cells were transfected with nontargeting (NT) or Atg7 siRNA, respectively. The siRNA-transfected cells were subsequently treated with or without 500 μM H2O2 for 6 h. (A) Cell lysates were subjected to western blot analysis using anti-ATG7, anti-LC3B or anti-β-actin antibody. (B) Cell viability was determined using the WST-1 reagent assay. The data are represented as the percent viability normalized to H2O2-untreated control groups (100%). The data are presented as the means ± SD of 3 independent experiments performed in triplicate.
Discussion
Building upon previous results,Citation18 we showed that Prnp0/0 cells are more susceptible to H2O2-induced oxidative stress than Prnp+/+ cells. These results indicate that PrPC has anti-oxidant activity. Indeed, many reports have shown that PrPC can act as a superoxide dismutase 1 (SOD1)Citation54 and can modulate the activity of Cu/Zn SOD1 by binding to 5 Cu2+ ions on octapeptide repeat motifs in the N-terminal region.Citation10,Citation55 Nevertheless, the molecular mechanism associated with the neuroprotective role of PrPC against oxidative stress remains unclear. Therefore, we investigated whether neuronal autophagy and apoptosis are activated during H2O2-induced oxidative stress in Prnp+/+ and Prnp0/0 cells. In this study, we demonstrate that the induction of autophagy following H2O2 treatment was due to enhanced and impaired autophagic flux in Prnp+/+ and Prnp0/0 cells, respectively. Moreover, autophagy inhibition achieved by Atg7 siRNA promotes H2O2 cytotoxicity in Prnp+/+ cells but does not affect Prnp0/0 cells, indicating that the increased autophagic flux in Prnp+/+ cells (at 3–12 h) and the impaired autophagic flux in Prnp0/0 cells (at 3–6 h) contribute to neuronal cell protection and the acceleration of neuronal cell death, respectively, during H2O2-induced oxidative stress. Interestingly, the impaired autophagic flux is rescued at a later time (12 h), suggesting that reactivated or restored autophagic flux may play a protective role against H2O2-induced oxidative stress ().
Figure 7. Schematic representation of time-dependent H2O2-induced cell death in Prnp+/+ and Prnp0/0 cells. In 2 types of cells, caspase-independent and caspase-dependent apoptosis were observed through ENDOG and cleaved CASP3, respectively. Activated autophagic flux protects against oxidative stress in Prnp+/+ cells, while impaired autophagic flux plays a role in neuronal cell death in Prnp0/0 cells. This impaired autophagic flux is rescued at a later time point (12 h), suggesting that the reactivated or restored autophagic flux may play an additional protective role.
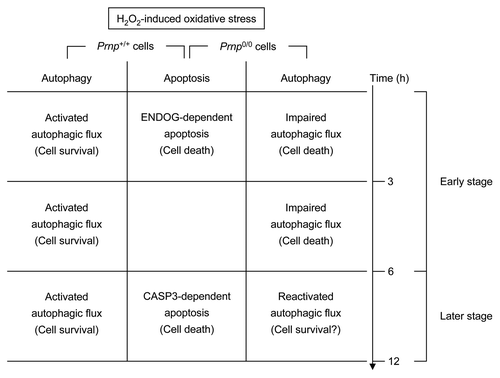
In addition, caspase-independent (at an early time point) and caspase-dependent apoptosis (at later time points) was observed through ENDOG and cleaved CASP3 in both Prnp+/+ and Prnp0/0 cells exposed to H2O2 (). This result is somewhat consistent with the previous finding that H2O2 treatment leads to ENDOG-dependent death but not CASP3 activation at an early time point in primary cortical neurons.Citation56 Unexpectedly, we found that Prnp0/0 cells were less sensitive to apoptosis at later time points after treatment with H2O2 than Prnp+/+ cells. This result suggests that the presence of PrPC may promote CASP3-dependent apoptosis during H2O2-induced oxidative stress. However, this result remains controversial, and further investigation is required to establish the role of PrPC in H2O2-induced CASP3-dependent apoptosis.
Although H2O2-induced oxidative stress increased autophagy in both cell types, it was found that autophagy induction resulted from enhanced and impaired autophagic flux in Prnp+/+ and Prnp0/0 cells, respectively. It was recently reported that H2O2 induces autophagic cell death with elevated autophagic activity in primary cortical neurons.Citation57 However, it is reasonable to propose that enhanced autophagy could eliminate oxidized or damaged intracellular components as a cellular defense mechanism against oxidative stress.
A number of studies have shown that autophagy dysfunction is directly associated with several neurodegenerative diseases, such as Alzheimer disease, Huntington disease, Parkinson disease, frontotemporal dementia and acute injuries.Citation58,Citation59 This impairment of autophagy could result from alterations in the autophagy process, which is composed of several steps, resulting in neurodegeneration. However, concerning prion diseases, the evidence of impaired autophagic flux remains unclear. Oxidative stress is induced through ROS or free radicals and may play an important role in the pathogenesis of several neurodegenerative diseases.Citation60 Notably, increased markers of oxidative damage and decreased antioxidants are observed in the brains of prion diseases.Citation61,Citation62 These changes imply that the conversion of PrPC to the nonfunctional form PrPSc or the absence of PrPC expression causes oxidative stress in prion diseases. Hence, the appearance of impaired autophagic flux through oxidative damage in prion diseases could accelerate PrPSc accumulation.
We observed inconsistent results in the viability assay using either pharmacological autophagy inhibition with 3-MA or induction using rapamycin and trehalose with H2O2 treatment, although the treatment of these drugs affected the Prnp+/+ and Prnp0/0 cells differently. However, the observed chemical inhibition and activation of autophagy may be a consequence of the side effects of the drug used. Hence, this result remains controversial, and further investigations are required to establish the effects of pharmacological autophagy inhibition and activation under H2O2-induced oxidative stress in neuronal cells. Interestingly, in H2O2-treated Prnp+/+ cells, the viability was significantly decreased upon transfection with Atg7 siRNA compared with non-targeting (NT) siRNA. These data suggest that enhanced autophagy in H2O2-treated Prnp+/+ cells is associated with a protective role. In contrast, in H2O2-treated Prnp0/0 cells, Atg7 siRNA transfection did not affect cell viability. In fact, it is possible that reduced formation of autophagosomes through Atg7 siRNA could not rescue impaired autophagic flux because autophagic flux is already disrupted in H2O2-treated Prnp0/0 cells. Hence, it is also possible that H2O2-induced oxidative stress may impair autophagosome/lysosome fusion or lysosomal degradation after successful formation of autophagosomes against H2O2 toxicity in Prnp0/0 cells, as shown in the H2O2-treated Prnp+/+ cells.
In addition, PrPC may not be directly involved in autophagic flux because natural autophagic flux was shown in both Prnp+/+ and Prnp0/0 cells under normal culture conditions, suggesting that the deficiency of PrPC may contribute to the impairment of autophagic flux under stress-induced conditions, such as H2O2 treatment or PRND overexpression. As previously described, it was shown in NP0/0 mice that the impairment of autophagic flux resulted in the loss of Purkinje cells through the autophagic cell death. Interestingly, it has been reported that PRND alone is toxic to neurons, and this toxicity is associated with upregulated heme oxygenase 1 (HMOX1), an enzyme that mediates the formation of antioxidants, and nitric oxide synthases (NOSs), such as neuronal NOS (nNOS) and inducible NOS (iNOS), in cell culture models and mice.Citation63,Citation64 Together, this evidence suggests that impaired autophagic flux in NP0/0 mice could result from PRND toxicity-induced oxidative stress and the lack of PrPC expression. However, we did not observe any relationship between PrPC and autophagic flux in this study; therefore, further investigations are required to determine the relationship between PrPC and autophagic flux.
Taken together, these results demonstrate that the deficiency of PrPC contributes to H2O2-induced autophagic cell death in hippocampal neuronal cells via impaired autophagic flux. Furthermore, our present results provide insight into the protective mechanism of PrPC in neuronal cells and the pathogenesis of prion diseases associated with autophagy.
Materials and Methods
Reagents
Reagents were obtained from commercial sources. The cell proliferation reagent WST-1 (11644807001) was purchased from Roche. H2O2 (1.07210) and methanol (1.13351) were obtained from Merck. Acetone (179124), 3-MA (M9281), trehalose dihydrate (T5251), rapamycin (R8781), bafilomycin A1 (B1793) and dimethyl sulfoxide (DMSO; D2650) were purchased from Sigma-Aldrich. z-VAD-fmk (4800-520) was obtained from MBL. Dulbecco’s modified Eagle’s medium (DMEM; SH30243.01), fetal bovine serum (FBS, SH30396.03), HaltTM protease inhibitor cocktail (87785) and HaltTM phosphatase inhibitor cocktail (78420) were obtained from Thermo Scientific. Penicillin-streptomycin (15140) and puromycin (A11138-03) were purchased from Invitrogen. Hanks’ balanced salt solution (HBSS, LB203-04) was obtained from WelGENE. A bicinchoninic acid (BCA) protein assay kit (23225) and the Supersignal West Pico Chemiluminescent Substrate (34080) were purchased from Pierce.
Antibodies
Anti-PrP (3F10) antibody was employed as described in ref. Citation65. Anti-CASP3 (9662), anti-ATG7 (SAB1407006), anti-AIFM1 (4642S), anti-ENDOG (4969S) and PathScan Multiplex Western Cocktail I (5301), including anti-phospho-AKT1 and anti-phospho-RPS6, were purchased from Cell Signaling. Anti-LC3B (L7543) and anti-β-actin (A5441) antibodies were purchased from Sigma-Aldrich. Anti-LC3B antibody (L10382) for immunocytochemistry was purchased from Invitrogen. Anti-ENO1 antibody (LF-MA10098) was purchased from Ab Frontier. Anti-HDAC2 antibody (sc-9959) was purchased from Santa Cruz Biotechnology.
Cell culture
Prnp+/+ and Prnp0/0 cell lines were previously established as described in ref. Citation36. All cell lines were cultivated in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin in a 5% CO2 atmosphere at 37°C.
To analyze the effects of 3-MA, z-VAD-fmk, rapamycin and trehalose under H2O2 treatment, 8 × 103 cells or 1 × 106 cells were plated in 0.1 ml or 10 ml of culture media per well in a 96-well plate or 100-mm Petri dish, respectively. The next day, the various compounds were preincubated with cells for 1 h or 3 h before H2O2 treatment.
Cell viability assay
Cell viability was determined using a Cell Proliferation Reagent WST-1 (4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate)-based assay. Briefly, 8 × 103 cells per well were plated in 96-well flat-bottomed plates with 0.1 ml of culture medium. The next day, the cells were incubated in fresh culture media without (control) or with designated concentrations of H2O2 for various durations. The Cell Proliferation Reagent WST-1 assay was performed according to the manufacturer’s instructions. The absorbance at 440 nm (A440) was quantified using a plate reader. The A440 value in the control cells was regarded as 100% viability.
Western blot analysis
To obtain lysates, cells (1 × 106 cells per 100-mm Petri dish) were harvested through scraping in HBSS containing a protease inhibitor cocktail (Roche, 11697498001). The cell pellet was obtained after centrifugation at 2500 × g for 5 min at 4°C. The pellet was resuspended using a 1-ml syringe (26-gauge) in lysis buffer containing 10 mM TRIS-HCl (pH 7.4), 1 mM EDTA, 150 mM NaCl, 1% Nonidet P-40, 1% deoxycholic acid, 1 mM phenylmethyl sulfonyl fluoride, HaltTM phosphatase inhibitor cocktail and protease inhibitor cocktail and was subsequently solubilized on ice for 30 min. To remove the insoluble materials, the cells were centrifuged at 5000 × g for 10 min at 4°C. The protein concentration was determined using a BCA protein assay, and equal amounts of protein were separated on 10% (ATG7, AIFM1, ENDOG, ENO1 and HDAC2), 12% (PathScan Multiplex Western Cocktail I) or 15% (PrP, LC3B, CASP3 and β-actin) SDS-PA gels and transferred to 0.45-μm nitrocellulose membranes. The blots were preincubated with 3% nonfat dried milk in Tris-buffered saline (TBS) containing 0.05% Tween 20 (TBS-T) for 1 h at room temperature and subsequently incubated with primary antibodies against ATG7 (1:1000), AIFM1 (1:1000), ENDOG (1:1000), ENO1 (1:5000), HDAC2 (1:200), PrP (1:3000), LC3B (1:1000), CASP3 (1:1000), PathScan Multiplex Western Cocktail I (1:200) or β-actin (1:10,000) for 16 h at 4°C or 1.5 h at room temperature (RT). The membranes were washed in TBS-T and subsequently incubated with species-specific secondary antibodies conjugated to horseradish peroxidase (goat anti-mouse IgG and goat anti-rabbit IgG; Pierce; dilution 1:3000) for 1 h at RT. The reactive protein bands were visualized using the Supersignal West Pico Chemiluminescent Substrate and quantified by densitometric analysis with ImageJ software.
Immunofluorescence microscopy
The cells were washed quickly with ice-cold HBSS and fixed using 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at RT. After 3 washes with PBS, the cells were permeabilized using ice-cold acetone:methanol (1:1) for 5 min at -20°C and then washed 3 more times with PBS. For endogenous LC3B staining, the cells were incubated with rabbit primary antibody against LC3B (dilution 1:1000 in 1% BSA blocking solution) for 2 h at RT. After washing 6 times with PBS, the cells were incubated with Alexa Fluor 568 goat anti-rabbit IgG antibody (Molecular Probes, A11011) for 2 h. After washing 6 times with PBS, the cells were subsequently mounted using Vectashield with DAPI (Vector Laboratories, H-1200). Imaging was performed using a Zeiss LSM-700 confocal laser-scanning microscope imaging system. The number of LC3B-positive puncta was determined using ImageJ software and expressed as the number of LC3B-positive puncta per cell.
siRNA knockdown
Nontargeting siRNA (sc-37007) and Atg7 siRNA (sc-41448) were purchased from Santa Cruz Biotechnology. Prnp+/+ and Prnp0/0 cells were seeded at 1.2 × 106 cells per 100-mm Petri dish and cultured for 6 h in culture medium without antibiotics. Subsequently, the cells were transfected with 50 nM nontargeting siRNA or 50 nM Atg7 siRNA using Lipofectamine RNAiMAX (Invitrogen, 13778–150) for 18 h according to the manufacturer’s protocol. After transfection, the cells were seeded onto 96-well plates or a 100-mm Petri dish at a concentration of 8 × 103 cells or 1 × 106 cells, respectively. After 12 h, the cells were treated with H2O2 for 6 h.
Cell fractionation
Cell fractionation was performed using NE-PER® Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, 78833) according to the manufacturer’s instructions. ENO1 and HDAC2 served as cytosolic and nuclear markers, respectively.
Statistical analysis
All experiments were repeated at least 3 times. The data are expressed as the means ± standard deviation (S.D) All analyses were performed using the SIGMA STAT 3.5 software (Systat Software, Inc.). The statistical significance of the data was determined using one-way ANOVA followed by Tukey’s test.
| Abbreviations: | ||
| PrPC | = | cellular prion protein |
| Prnp 0/0 | = | Prnp-deficient |
| H2O2 | = | hydrogen peroxide |
| LC3B | = | microtubule-associated protein 1 light chain 3 beta |
| TSE | = | transmissible spongiform encephalopathy |
| PrPSc | = | pathological scrapie isoform |
| GPI | = | glycosylphosphatidylinositol |
| 3-MA | = | 3-methyladenine |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| MTOR | = | mechanistic target of rapamycin |
| SOD1 | = | superoxide dismutase 1 |
| Baf A1 | = | bafilomycin A1 |
| AIFM1 | = | apoptosis-inducing factor, mitochondrion-associated, 1 |
| ENDOG | = | endonuclease G |
| NOS | = | nitric oxide synthase |
| RPS6 | = | ribosomal protein S6 |
| SQSTM1 | = | sequestosome 1 |
| HDAC2 | = | histone deacetylase 2 |
| CASP3 | = | caspase 3 |
| ENO1 | = | enolase 1 |
| SCRG1 | = | stimulator of chondrogenesis 1/scrapie responsive gene 1 |
| PRND | = | PrP protein 2 (dublet)/PrP-like protein doppel |
| HMOX1 | = | heme oxygenase 1 |
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012-0000308) and by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2011-619-E0001).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Basler K, Oesch B, Scott M, Westaway D, Wälchli M, Groth DF, et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 1986; 46:417 - 28; http://dx.doi.org/10.1016/0092-8674(86)90662-8; PMID: 2873895
- Kretzschmar HA, Prusiner SB, Stowring LE, DeArmond SJ. Scrapie prion proteins are synthesized in neurons. Am J Pathol 1986; 122:1 - 5; PMID: 3079955
- Manson J, West JD, Thomson V, McBride P, Kaufman MH, Hope J. The prion protein gene: a role in mouse embryogenesis?. Development 1992; 115:117 - 22; PMID: 1353438
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 47; http://dx.doi.org/10.1016/0092-8674(93)90360-3; PMID: 8100741
- Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, et al. Prion protein is necessary for normal synaptic function. Nature 1994; 370:295 - 7; http://dx.doi.org/10.1038/370295a0; PMID: 8035877
- Tobler I, Gaus SE, Deboer T, Achermann P, Fischer M, Rülicke T, et al. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 1996; 380:639 - 42; http://dx.doi.org/10.1038/380639a0; PMID: 8602267
- Lee HG, Park SJ, Choi EK, Carp RI, Kim YS. Increased expression of prion protein is associated with changes in dopamine metabolism and MAO activity in PC12 cells. J Mol Neurosci 1999; 13:121 - 6; http://dx.doi.org/10.1385/JMN:13:1-2:121; PMID: 10691299
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, et al. Signal transduction through prion protein. Science 2000; 289:1925 - 8; http://dx.doi.org/10.1126/science.289.5486.1925; PMID: 10988071
- Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, et al. The cellular prion protein binds copper in vivo. Nature 1997; 390:684 - 7; http://dx.doi.org/10.1038/37733; PMID: 9414160
- Mangé A, Milhavet O, Umlauf D, Harris D, Lehmann S. PrP-dependent cell adhesion in N2a neuroblastoma cells. FEBS Lett 2002; 514:159 - 62; http://dx.doi.org/10.1016/S0014-5793(02)02338-4; PMID: 11943143
- Graner E, Mercadante AF, Zanata SM, Forlenza OV, Cabral AL, Veiga SS, et al. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res Mol Brain Res 2000; 76:85 - 92; http://dx.doi.org/10.1016/S0169-328X(99)00334-4; PMID: 10719218
- Brown DR, Schulz-Schaeffer WJ, Schmidt B, Kretzschmar HA. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp Neurol 1997; 146:104 - 12; http://dx.doi.org/10.1006/exnr.1997.6505; PMID: 9225743
- Cashman NR, Loertscher R, Nalbantoglu J, Shaw I, Kascsak RJ, Bolton DC, et al. Cellular isoform of the scrapie agent protein participates in lymphocyte activation. Cell 1990; 61:185 - 92; http://dx.doi.org/10.1016/0092-8674(90)90225-4; PMID: 1969332
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, et al. Prions prevent neuronal cell-line death. Nature 1999; 400:225 - 6; http://dx.doi.org/10.1038/22241; PMID: 10421360
- Kim BH, Lee HG, Choi JK, Kim JI, Choi EK, Carp RI, et al. The cellular prion protein (PrPC) prevents apoptotic neuronal cell death and mitochondrial dysfunction induced by serum deprivation. Brain Res Mol Brain Res 2004; 124:40 - 50; http://dx.doi.org/10.1016/j.molbrainres.2004.02.005; PMID: 15093684
- Senator A, Rachidi W, Lehmann S, Favier A, Benboubetra M. Prion protein protects against DNA damage induced by paraquat in cultured cells. Free Radic Biol Med 2004; 37:1224 - 30; http://dx.doi.org/10.1016/j.freeradbiomed.2004.07.006; PMID: 15451062
- White AR, Collins SJ, Maher F, Jobling MF, Stewart LR, Thyer JM, et al. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am J Pathol 1999; 155:1723 - 30; http://dx.doi.org/10.1016/S0002-9440(10)65487-9; PMID: 10550328
- Brown DR, Nicholas RS, Canevari L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res 2002; 67:211 - 24; http://dx.doi.org/10.1002/jnr.10118; PMID: 11782965
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717 - 21; http://dx.doi.org/10.1126/science.290.5497.1717; PMID: 11099404
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 77; http://dx.doi.org/10.1016/S1534-5807(04)00099-1; PMID: 15068787
- Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol 2004; 14:70 - 7; http://dx.doi.org/10.1016/j.tcb.2003.12.002; PMID: 15102438
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990 - 5; http://dx.doi.org/10.1126/science.1099993; PMID: 15528435
- Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990; 181:195 - 213; http://dx.doi.org/10.1007/BF00174615; PMID: 2186664
- Bursch W, Ellinger A, Kienzl H, Török L, Pandey S, Sikorska M, et al. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis 1996; 17:1595 - 607; http://dx.doi.org/10.1093/carcin/17.8.1595; PMID: 8761415
- Ohsawa Y, Isahara K, Kanamori S, Shibata M, Kametaka S, Gotow T, et al. An ultrastructural and immunohistochemical study of PC12 cells during apoptosis induced by serum deprivation with special reference to autophagy and lysosomal cathepsins. Arch Histol Cytol 1998; 61:395 - 403; http://dx.doi.org/10.1679/aohc.61.395; PMID: 9990423
- Martin DN, Baehrecke EH. Caspases function in autophagic programmed cell death in Drosophila. Development 2004; 131:275 - 84; http://dx.doi.org/10.1242/dev.00933; PMID: 14668412
- Guillon-Munos A, van Bemmelen MX, Clarke PG. Role of phosphoinositide 3-kinase in the autophagic death of serum-deprived PC12 cells. Apoptosis 2005; 10:1031 - 41; http://dx.doi.org/10.1007/s10495-005-0741-6; PMID: 16151638
- Schätzl HM, Laszlo L, Holtzman DM, Tatzelt J, DeArmond SJ, Weiner RI, et al. A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J Virol 1997; 71:8821 - 31; PMID: 9343242
- Liberski PP, Sikorska B, Bratosiewicz-Wasik J, Gajdusek DC, Brown P. Neuronal cell death in transmissible spongiform encephalopathies (prion diseases) revisited: from apoptosis to autophagy. Int J Biochem Cell Biol 2004; 36:2473 - 90; http://dx.doi.org/10.1016/j.biocel.2004.04.016; PMID: 15325586
- Dron M, Bailly Y, Beringue V, Haeberlé AM, Griffond B, Risold PY, et al. Scrg1 is induced in TSE and brain injuries, and associated with autophagy. Eur J Neurosci 2005; 22:133 - 46; http://dx.doi.org/10.1111/j.1460-9568.2005.04172.x; PMID: 16029203
- Aguib Y, Heiseke A, Gilch S, Riemer C, Baier M, Schätzl HM, et al. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009; 5:361 - 9; http://dx.doi.org/10.4161/auto.5.3.7662; PMID: 19182537
- Heiseke A, Aguib Y, Riemer C, Baier M, Schätzl HM. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem 2009; 109:25 - 34; http://dx.doi.org/10.1111/j.1471-4159.2009.05906.x; PMID: 19183256
- Heitz S, Grant NJ, Leschiera R, Haeberlé AM, Demais V, Bombarde G, et al. Autophagy and cell death of Purkinje cells overexpressing Doppel in Ngsk Prnp-deficient mice. Brain Pathol 2010; 20:119 - 32; http://dx.doi.org/10.1111/j.1750-3639.2008.00245.x; PMID: 19055638
- Barbieri G, Palumbo S, Gabrusiewicz K, Azzalin A, Marchesi N, Spedito A, et al. Silencing of cellular prion protein (PrPC) expression by DNA-antisense oligonucleotides induces autophagy-dependent cell death in glioma cells. Autophagy 2011; 7:840 - 53; http://dx.doi.org/10.4161/auto.7.8.15615; PMID: 21478678
- Oh JM, Shin HY, Park SJ, Kim BH, Choi JK, Choi EK, et al. The involvement of cellular prion protein in the autophagy pathway in neuronal cells. Mol Cell Neurosci 2008; 39:238 - 47; http://dx.doi.org/10.1016/j.mcn.2008.07.003; PMID: 18674620
- Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 2008; 15:171 - 82; http://dx.doi.org/10.1038/sj.cdd.4402233; PMID: 17917680
- Jabs T. Reactive oxygen intermediates as mediators of programmed cell death in plants and animals. Biochem Pharmacol 1999; 57:231 - 45; http://dx.doi.org/10.1016/S0006-2952(98)00227-5; PMID: 9890550
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749 - 60; http://dx.doi.org/10.1038/sj.emboj.7601623; PMID: 17347651
- Kim BH, Kim JI, Choi EK, Carp RI, Kim YS. A neuronal cell line that does not express either prion or doppel proteins. Neuroreport 2005; 16:425 - 9; http://dx.doi.org/10.1097/00001756-200504040-00002; PMID: 15770145
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Green D, Kroemer G. The central executioners of apoptosis: caspases or mitochondria?. Trends Cell Biol 1998; 8:267 - 71; http://dx.doi.org/10.1016/S0962-8924(98)01273-2; PMID: 9714597
- Wang H, Yu SW, Koh DW, Lew J, Coombs C, Bowers W, et al. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci 2004; 24:10963 - 73; http://dx.doi.org/10.1523/JNEUROSCI.3461-04.2004; PMID: 15574746
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol 2002; 158:507 - 17; http://dx.doi.org/10.1083/jcb.200202130; PMID: 12147675
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001; 412:95 - 9; http://dx.doi.org/10.1038/35083620; PMID: 11452314
- van Loo G, Schotte P, van Gurp M, Demol H, Hoorelbeke B, Gevaert K, et al. Endonuclease G: a mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ 2001; 8:1136 - 42; http://dx.doi.org/10.1038/sj.cdd.4400944; PMID: 11753562
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999; 397:441 - 6; http://dx.doi.org/10.1038/17135; PMID: 9989411
- Côté J, Ruiz-Carrillo A. Primers for mitochondrial DNA replication generated by endonuclease G. Science 1993; 261:765 - 9; http://dx.doi.org/10.1126/science.7688144; PMID: 7688144
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000; 275:992 - 8; http://dx.doi.org/10.1074/jbc.275.2.992; PMID: 10625637
- Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010; 285:10850 - 61; http://dx.doi.org/10.1074/jbc.M109.080796; PMID: 20123989
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 1998; 273:3963 - 6; http://dx.doi.org/10.1074/jbc.273.7.3963; PMID: 9461583
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 2007; 282:5641 - 52; http://dx.doi.org/10.1074/jbc.M609532200; PMID: 17182613
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Brown DR, Wong BS, Hafiz F, Clive C, Haswell SJ, Jones IM. Normal prion protein has an activity like that of superoxide dismutase. Biochem J 1999; 344:1 - 5; http://dx.doi.org/10.1042/0264-6021:3440001; PMID: 10548526
- Brown DR, Besinger A. Prion protein expression and superoxide dismutase activity. Biochem J 1998; 334:423 - 9; PMID: 9716501
- Higgins GC, Beart PM, Nagley P. Oxidative stress triggers neuronal caspase-independent death: endonuclease G involvement in programmed cell death-type III. Cell Mol Life Sci 2009; 66:2773 - 87; http://dx.doi.org/10.1007/s00018-009-0079-2; PMID: 19582370
- Higgins GC, Devenish RJ, Beart PM, Nagley P. Autophagic activity in cortical neurons under acute oxidative stress directly contributes to cell death. Cell Mol Life Sci 2011; 68:3725 - 40; http://dx.doi.org/10.1007/s00018-011-0667-9; PMID: 21437645
- Lee JA. Autophagy in neurodegeneration: two sides of the same coin. BMB Rep 2009; 42:324 - 30; http://dx.doi.org/10.5483/BMBRep.2009.42.6.324; PMID: 19558789
- Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 2010; 13:805 - 11; http://dx.doi.org/10.1038/nn.2575; PMID: 20581817
- Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol 1995; 38:357 - 66; http://dx.doi.org/10.1002/ana.410380304; PMID: 7668820
- Kim JI, Choi SI, Kim NH, Jin JK, Choi EK, Carp RI, et al. Oxidative stress and neurodegeneration in prion diseases. Ann N Y Acad Sci 2001; 928:182 - 6; http://dx.doi.org/10.1111/j.1749-6632.2001.tb05648.x; PMID: 11795509
- Brown DR. Neurodegeneration and oxidative stress: prion disease results from loss of antioxidant defence. Folia Neuropathol 2005; 43:229 - 43; PMID: 16416388
- Wong BS, Liu T, Paisley D, Li R, Pan T, Chen SG, et al. Induction of HO-1 and NOS in doppel-expressing mice devoid of PrP: implications for doppel function. Mol Cell Neurosci 2001; 17:768 - 75; http://dx.doi.org/10.1006/mcne.2001.0963; PMID: 11312611
- Cui T, Holme A, Sassoon J, Brown DR. Analysis of doppel protein toxicity. Mol Cell Neurosci 2003; 23:144 - 55; http://dx.doi.org/10.1016/S1044-7431(03)00017-4; PMID: 12799144
- Choi JK, Park SJ, Jun YC, Oh JM, Jeong BH, Lee HP, et al. Generation of monoclonal antibody recognized by the GXXXG motif (glycine zipper) of prion protein. Hybridoma (Larchmt) 2006; 25:271 - 7; http://dx.doi.org/10.1089/hyb.2006.25.271; PMID: 17044782