Abstract
Reactive oxygen species (ROS) have been implicated as a signal for general autophagy. Both mitochondrial-produced and exogenous ROS induce autophagosome formation. However, it is unclear whether ROS are required for the selective autophagic degradation of mitochondria, a process called mitophagy. Recent work using carbonyl cyanide m-chlorophenylhydrazone (CCCP), a mitochondrial-uncoupling reagent, has been shown to induce mitophagy. However, CCCP treatment may not be biologically relevant since it causes the depolarization of the entire mitochondrial network. Since mitochondria are the main ROS production sites in mammalian cells, we propose that short bursts of ROS produced within mitochondria may be involved in the signaling for mitophagy. To test this hypothesis, we induced an acute burst of ROS within mitochondria using a mitochondrial-targeted photosensitizer, mitochondrial KillerRed (mtKR). Using mtKR, we increased ROS levels in the mitochondrial matrix, which resulted in the loss of membrane potential and the subsequent activation of PARK2-dependent mitophagy. Importantly, we showed that overexpression of the mitochondrial antioxidant protein, superoxide dismutase-2, can squelch mtKR-induced mitophagy, demonstrating that mitochondrial ROS are responsible for mitophagy activation. Using this assay, we examined the impact of mitochondrial morphology on mitophagy. It was shown recently that elongated mitochondria are more resistant to mitophagy through unknown mechanisms. Here, we show that elongated mitochondria are more resistant to ROS-induced damage and mitophagy compared with fragmented mitochondria, suggesting that mitochondrial morphology has an important role in regulating ROS and mitophagy. Together, our results suggest that ROS-induced mitochondrial damage may be an important upstream activator of mitophagy.
Introduction
Mitochondria are essential organelles required for energy production, cell survival, cell death and cell signaling.Citation1 They are also one of the main production sites of reactive oxygen species (ROS) in the cell. The mitochondrion and its contents are prone to oxidative damage resulting from ROS produced in the matrix. The accumulation of damaged mitochondria in the cell can cause cellular oxidative stress that will eventually lead to cell death.Citation2 Damaged and dysfunctional mitochondria need to be selectively removed to protect the cells from excessive oxidative stress and cell death.Citation3 The selective degradation of mitochondria by autophagy (mitophagy) is a process whereby damaged mitochondria are sequestered into double-membrane vesicles called autophagosomes and are transported to lysosomes for degradation.Citation4 It is believed that dysfunction in the mitophagy pathway can lead to several neurodegenerative disorders including Parkinson and Alzheimer disease.Citation4,Citation5 Hence, understanding the mechanism of mitophagy is essential to the development of effective clinical strategies for many human diseases.
The current knowledge of the mitophagy pathway is mainly based on experiments using carbonyl cyanide m-chlorophenylhydrazone (CCCP) as a mitochondrial depolarizing agent.Citation6,Citation7 CCCP acts as an ionophore, quickly and universally dissipating the proton gradient of the mitochondrial inner membrane.Citation8 Results from CCCP experiments suggest that the loss of mitochondrial potential can activate mitophagy.Citation6-Citation9 One of the mechanisms that induces mitophagy in mammalian cells is through the accumulation of PTEN-putative kinase 1 (PINK1) on the depolarized mitochondria. PINK1 subsequently recruits PARK2/PARKIN, an E3 ubiquitin ligase, to the mitochondrial outer membrane (MOM).Citation9 PARK2 ubiquitinates several MOM proteins and the ubiquitin (UB) chains serve as a signal for both proteasome degradation and mitophagy. The removal of MOM proteins such as MFN1 and MFN2 results in mitochondrial fragmentation.Citation10 Fragmented and depolarized mitochondria are sequestered to autophagosomes by autophagy adaptor proteins and are degraded upon fusion of the autophagosomes with lysosomes.Citation4
The in vivo signal that initiates mitophagy in cells is currently unclear but one possible candidate is mitochondrial ROS. Both exogenously added hydrogen peroxide (H2O2) and superoxide (O2●-) produced in the mitochondria have been previously shown to upregulate the formation of autophagosomes.Citation11,Citation12 Furthermore, a recent study demonstrated that photo-irradiation of mitochondria using 488 nm light can cause mitophagy.Citation13 However, it is unclear whether mitophagy occurs due to inactivation of the electron transport chain by photo-irradiation or the subsequent production of ROS by damaged mitochondria. In fact, a recent study by Narendra, et al. group reported that treating cells with an antioxidant such as N-acetyl cysteine (NAC) did not inhibit CCCP-induced mitophagy, suggesting that ROS may not be involved in the mitophagy pathway induced by the uncoupling of the oxidative phosphorylation pathway.Citation6 Therefore, a direct examination of mitochondrial ROS and mitophagy is required.
Several groups have reported short bursts of superoxide production in mitochondria, called superoxide flashes, may cause mitochondrial damage.Citation14,Citation15 Here, we have examined whether mitophagy can be activated by ROS-induced mitochondrial damage. To mimic the acute elevation in ROS seen in vivo, we employed a genetically encoded photosensitizer, KillerRed, which is targeted to mitochondria (mtKR) to examine the effect of a short burst of ROS in the mitochondria. KR is a homolog of green fluorescent protein (GFP) that produces 1000 times more ROS than GFP upon photo-activation (photo-bleaching) with 561 nm light.Citation16 KR can be readily expressed in cultured cells and can be targeted to different subcellular compartments. Attaching two mitochondrial-targeting sequences to the N-terminus of KR (mtKR) promotes its mitochondrial localization.Citation17 Yang and Yang recently reported a new mitophagy-induction technique using mtKR as the damaging-agent. They confirmed that photo-activating mtKR led to PARK2-mediated mitophagy.Citation18
Here we used mtKR to directly investigate the role of mitochondrial ROS in the initiation steps of mitophagy. Using live-cell imaging, we demonstrated that photo-activation of mtKR produces ROS to directly cause mitochondrial damage, which leads to PARK2-dependent mitophagy. Moreover, we showed that overexpression of the mitochondrial antioxidant protein, superoxide dismutase 2 (SOD2), squelched mitophagy induced by ROS. Our results also shed light on an important connection between mitochondrial dynamics and mitophagy regulation.
Results
ROS produced by mtKR activation can be modulated within the mitochondrial matrix
We postulated that an acute elevation of ROS within the matrix of mitochondria could cause damage leading to loss of mitochondrial potential and the activation of mitophagy. To simulate an acute burst of ROS in the mitochondrial matrix we used mtKR, which has been shown to produce ROS both in vitro and in vivo.Citation16-Citation18 To directly demonstrate that the excitation of mtKR produces ROS in mitochondria we measured ROS levels in mitochondria. To monitor changes in mitochondrial ROS, we incubated cells with 10 µM OxyBURST® Green H2DCFDA prior to imaging. Images of cells expressing mtKR before photo-bleaching (pre-pb) and after photo-bleaching (post-pb) were acquired with 561 nm imaging laser power at 1% output (). Before photo-bleaching mtKR, OxyBURST staining was mainly observed in mitochondria (, pre-pb), indicating that these are the major ROS production sites in the cell. After photo-bleaching, the mtKR signal was reduced by 85% (, compare pre-pb to post-pb; Fig. S1A). To test if photo-bleaching mtKR results in a burst of ROS within mitochondria, we compared the OxyBURST level in the pre-pb image to the post-pb image. Upon photo-bleaching mtKR with 561 nm laser light at 100% output for 30 iterations, there was a significant increase in the OxyBURST signal indicating an increase in ROS levels (, post-pb). In contrast, photo-bleaching mitochondrial-targeted red fluorescent protein (mtRFP) under the same conditions did not cause a change in the OxyBURST signal (, compare pre-pb to post-pb).
Figure 1. Photo-bleaching mtKR produces ROS in the mitochondria in a dose-dependent manner. Representative images of HeLa cells transfected with mtKR (A) or mtRFP (B) and incubated with 10 µM OxyBURST® Green H2DCFDA for 15 min prior to irradiating the cells with 561 nm laser light for 30 iterations at 100% output. Images were acquired before (pre-pb) and after photo-bleaching (post-pb) the cells. Scale bar: 10 µm. (C) The mtKR- and mtRFP-expressing cells were incubated with 10 µM OxyBURST and were irradiated with 561 nm laser light for 5, 10, 15 or 30 iterations. Pre-pb and post-pb images were acquired using an open pinhole. The OxyBURST fluorescent signals in the pre-pb and post-pb images were measured with the image-processing software Volocity® and the average percentage increase in OxyBURST fluorescence signal was quantified from three independent experiments (n = 10). (D) HeLa cells expressing mtKR were incubated with 10 µM OxyBURST and irradiated with 561 nm laser light for 5, 10, 15 or 30 iterations (n = 9). The increase in total OxyBURST signal after the photo-bleach was plotted against the corresponding total mtKR fluorescence signal of the cell. Linear regression lines calculated with a zero-intercept are shown with the data points.
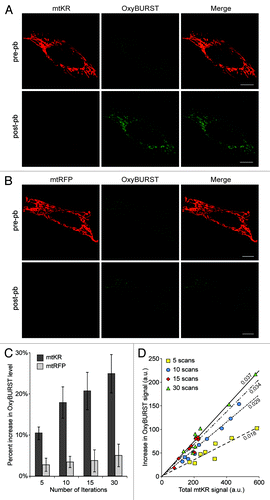
Consistent with the hypothesis that activation of mtKR causes an increase in mitochondrial ROS levels, we also observed that the amount of ROS increase depended on both the number of iterations during photo-bleaching and the expression levels of mtKR. To quantify this observation, HeLa cells expressing mtKR were exposed to 561 nm laser light at 100% for 5, 10, 15, and 30 iterations in the presence of 10µM OxyBURST. We quantified the percentage increase in OxyBURST fluorescence signal and found a direct correlation between the increase in OxyBURST levels and the number of photo-bleaching iterations in the mtKR-expressing HeLa cells, but not in the mtRFP-expressing cells (). Photo-bleaching mtKR for 30 iterations resulted in a 25% ± 5% increase in the OxyBURST fluorescent signal while mtRFP only resulted in a 6% ± 3% increase (). Furthermore, when we plotted the increase in total OxyBURST signal as a function of the total fluorescence intensity of mtKR before photo-bleach, we found a strong linear relationship between the total mtKR signal and the increase in OxyBURST signal over the assayed range (). Using this data, we determined that the optimal number of iteration of photo-bleach required in order to maximize ROS production was 30 iterations (Fig. S1B). Therefore, in all following experiments, mtKR was photo-activated with 30 iterations of 561 nm laser at maximum output. Taken together, these experiments show that the amount of ROS produced within the mitochondrial matrix is determined by the level of mtKR expression as well as the amount of photo-activation. This demonstrates the utility and control of using mtKR to fine-tune the ROS burst levels within the mitochondrial matrix.
Only ROS originating from mitochondria can induce PARK2 recruitment
To test whether the ROS produced by mtKR activation lead to mitophagy, we examined whether PARK2 is recruited to the mitochondria, which is a well-established indicator of mitophagy activation in HeLa cells. PARK2 is an E3 ligase that is recruited to damaged mitochondria during the early stages of CCCP-induced mitophagy.Citation4 To determine whether an acute increase in mitochondrial ROS can activate mitophagy, we co-expressed mtKR and Cerulean-PARK2 (Cer-PARK2) in HeLa cells and photo-bleached mtKR with 30 iterations of 561 nm wavelength laser light at 100% output (, pre-pb and post-pb). The image taken 60 min after photo-bleaching clearly showed both mitochondrial fragmentation and PARK2 recruitment to the mitochondria outer membrane in the photo-bleached cell (, 60 min and zoom; Movie S1).
Figure 2. ROS produced only within the mitochondria leads to PARK2 recruitment to the mitochondria. Representative images of HeLa cells that were transfected with mtKR (A) or mtRFP (B) and Cer-PARK2 16 to 24 h prior to the experiments. Cells were irradiated with 561 nm laser for 30 iterations at 100% output. Images were acquired immediately before and after the photo-bleaching. Cells were then imaged live for 60 min, and the 561 nm laser power was increased from 1% to 2% after the photo-bleaching. The magnification of the square box in 60 min is shown in the panels labeled zoom. (C and D) Representative images of HeLa cells co-transfected with cytosol KR (cyto-KR) (C) or peroxisomal KR (KR-SKL) (D), Venus-OMP25-TM and Cer-PARK2 for 16 to 24 h. The white outline shown in the merge panel indicates the region in which the KR was photo-bleached with the 561 nm laser for 30 iterations at 100% output. A pre-pb image and a post-pb image were acquired and the cells were monitored for 60 min after photo-bleaching. The magnification of the square box in 60 min is shown in the panels label Zoom. Scale bar: 10 µm.
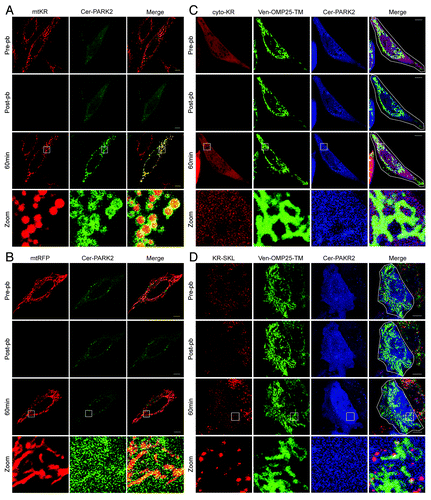
It has been previously demonstrated that photo-irradiation with 488 nm light can induce mitochondrial damage resulting in PARK2 recruitment.Citation13 To ensure that the mitochondrial recruitment of PARK2 was not due to nonspecific effects of exposure to high-powered 561 nm laser light, we irradiated HeLa cells expressing mitochondrial targeted RFP (mtRFP) and Cer-PARK2 with 561 nm laser light at 100% output for 30 iterations. Neither mitochondrial fragmentation nor PARK2 recruitment to mitochondria were observed in our system (). These data demonstrate that exposure to 561 nm laser light does not by itself induce the mitochondrial fragmentation or PARK2 recruitment observed in the cells that express mtKR.
We also tested whether KillerRed needed to be localized to mitochondria to activate PARK2 recruitment. It has been suggested that an increase in cellular ROS, either mitochondrial ROS or exogenously added ROS, can upregulate general autophagy.Citation11,Citation12 Therefore, it is possible that PARK2 recruitment to mitochondria after mtKR-activation is a result of general autophagy induction. To test whether a general increase in cellular ROS can activate PARK2 recruitment to the mitochondria, we examined whether KR expressed in different subcellular locations could promote PARK2 recruitment to mitochondria. We co-expressed Cer-PARK2 with KR in the cytosol (cytoKR), or in the peroxisomes (KR-SKL). Photo-bleaching either cytoKR or KR-SKL did not lead to PARK2 recruitment to mitochondria 60 min post-pb (), thus demonstrating that only ROS produced in the mitochondrial matrix could recruit PARK2 to the mitochondria.
Quantification of PARK2 Recruitment
Previously, mitophagy has been quantified by scoring cells as positive or negative for PARK2 recruitment or by measuring the loss of an endogenous mitochondrial protein.Citation6,Citation8 We believe these methods suffer from subjectivity and may potentially mask important data. Therefore, we developed a method that we call the PARK2 Recruitment Quantification (PARe-Q) to quantify and represent PARK2 recruitment in a more accurate and objective manner. Taking advantage of the fact that PARK2 is freely diffused in the cytosol prior to mitophagy, we used the image-analysis software Volocity® to measure the area of PARK2 recruited to the mitochondria in relation to total mitochondrial area. The percentage of PARK2 positive mitochondria represents the percentage of mitochondria that recruited PARK2 90 min post-pb. We also obtained the mtKR expression level in each cell by measuring the total pixel intensity of the mtKR fluorescence signal (see Materials and Methods for a detailed description).
In order to determine the basal level of PARK2 recruitment to mitochondria, we expressed mtRFP and Cer-PARK2 in HeLa cells and subjected them to 30 iterations of photo-bleaching. Since we found that there was no visual colocalization between mitochondria and PARK2 in mtRFP cells 60 min post-pb () we assumed that the Cer-PARK2 punctate structures observed in the mtRFP expressing cells likely represent a basal level of PARK2 aggregation. The images were analyzed as described above, and the percentage of PARK2-positive mitochondrial area out of the total mitochondrial area was plotted against total mtRFP or mtKR signal for each treatment (). We defined 3% of PARK2-positive mitochondria as the threshold level since the majority of the mtRFP-expressing cells had less than 3% of mitochondria colocalized with PARK2 (). Using the same technique, we also quantified the percentage of PARK2-positive mitochondria in cells expressing mtKR. mtKR-expressing HeLa cells were irradiated with 5, 10, 15 and 30 iterations of 561 nm laser light at 100% output. We found that the percentage of cells with greater than 3% PARK2-positive mitochondria increased with increasing number of photo-bleaching iterations in the mtKR expressing cells (). These data correlated well with the increase in ROS production in mitochondria (), suggesting that increase in ROS production lead to increased PARK2 recruitment.
Figure 3. A quantitative method for measuring PARK2 Recruitment (PARe-Q). (A) HeLa cells expressing mtRFP and Cer-PARK2 were irradiated with 561 nm laser for 30 iterations at 100% output. The PARK2-positive mitochondrial area over the total mitochondria area was quantified in cells 90 min post-pb using Volocity®. 3% PARK2-positive mitochondria was defined to be the basal level of PARK2 recruitment to mitochondria (n = 16). The divide at 3% is shown with a dotted line. (B–E) HeLa cells expressing mtKR and Cer-PARK2 were irradiated with 561 nm laser for 5 (B), 10 (C), 15 (D) or 30 (E) iterations at 100% output (n = 46 for each set). The percentage of PARK2-positive mitochondria was calculated and plotted against the total mtKR fluorescent signal. Shown on the right of the graph is the percentage of cells with PARK2-positive mitochondria above 3% (data points in the red regions) and those below 3% (data points in green region). (F) Lysates from HeLa cells treated with either nontargeting siRNA (siCtrl) or siRNA against PINK1 (siPINK1) for 72 h were separated on a SDS-PAGE and was immunoblotted with antibody against PINK1. The arrows indicate PINK1-specific bands while the asterisks indicate a cross reacting band. (G and H) HeLa cells that received siCtrl (G), or siPINK1 (H) and expressing mtKR and Cer-PARK2 were irradiated with 561 nm laser light for 30 iterations as in (E). These cells were imaged again 90 min after photo-bleaching and the percent of PARK2 recruitment was determined (n = 29). The difference between the siPINK1 and the siCtrl population is significant (p = 5.8 × 10−6). (I) Representative image of a HeLa cell that received siPINK1, and was co-transfected with mtKR and Cer-PARK2. The cell was photo-bleached by the 561 nm laser light. 200 nM MTRED was added after photo-bleaching. An image was acquired before, after and 90 min after photo-bleaching. Scale bars: 10 µm.
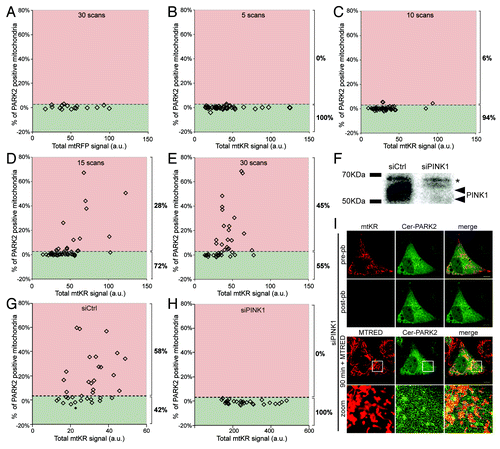
To further validate our PARe-Q, we examined whether the observed recruitment of exogenously expressed PARK2 to the mitochondria was dependent on PINK1. PINK1 has been shown to accumulate on the MOM of depolarized mitochondria and to allow the recruitment of PARK2 in order to activate mitophagy.Citation20 We quantified the percentage of PARK2-positive mitochondria in cells where endogenous PINK1 expression was depleted using siRNA specific for PINK1. In siPINK1 cells, we found that PARK2 recruitment was severely inhibited compared with cells treated with nontargeting siRNA (siCtrl) (). Representative images of a siPINK1 cell expressing mtKR and Cer-PARK2 showed that photo-bleaching mtKR did not lead to the colocalization of PARK2 and the mitochondria ()
In order to determine the statistical significance of the observed activation under experimental and control conditions, we assumed individual cells to constitute independent observations and we quantified the response to activation at a given level of photo-bleaching as the ratio of PARK2 recruitment to mtKR expression. Cells were ranked according to this response and the nonparametric Mann-Whitney rank sum test was applied. To control for variation between experiments, the population of cells that had undergone 30 iterations of photo-bleaching was designated the “standard” protocol () and the population distributions for each different treatment were compared with the standard, as well as the control; we would therefore expect no significant difference between standard and control protocols and significant differences for protocols with an effect on PARK2 recruitment. The siPINK1 population’s response was significantly attenuated relative to both standard and siCtrl populations (p = 1.9 × 10−6 resp. 5.8 × 10−6), whereas the difference between standard and siCtrl was not significant (p = 0.073) (Table S1). Therefore, our data show that ROS-induced PARK2 recruitment depends on the presence of PINK1.
mtKR-induced PARK2 recruitment to mitochondria leads to mitophagy
Based on studies using mitochondrial decouplers such as CCCP, the current view of the molecular events of mitophagy is that once PARK2 is localized to mitochondria, it ubiquitinates several MOM proteins including MFN1, MFN2, VDAC, and TOM20.Citation10 These ubiquitinated mitochondria are then engulfed by autophagosomes (marked by LC3-II) and degraded in lysosomes (marked by LAMP-1).Citation4 To demonstrate that mtKR-mediated PARK2 recruitment to mitochondria can lead to mitophagy, we examined the localization of UB, LC3, and LAMP-1 following the acute burst of mitochondrial ROS. We cotransfected HeLa cells with mtKR, Cer-PARK2, and a plasmid containing GFP-UB, GFP-LC3 or GFP-LAMP-1. As shown in , all three markers of mitophagy colocalized with PARK2-decorated mitochondria 60 min, 90 min, and 4 h after photo-bleaching mtKR, demonstrating that mtKR-induced ROS activates mitophagy (). Importantly, photo-bleaching mtRFP did not lead to colocalization between PARK2-positive mitochondria and GFP-UB or GFP-LC3 (Fig. S2A and S2B). Since HeLa cells lack endogenous PARK2, they did not show GFP-UB or GFP-LC3 recruitment to mitochondria after mtKR was photo-bleached in the absence of Cer-PARK2 co-expression, even after 19 h (Fig. S3A and S3B), thus strongly suggesting that mtKR-mediated mitophagy is PARK2-dependent.Citation21 Interestingly, photo-bleaching mtKR resulted in mitochondrial fragmentation in the absence of PARK2 (Fig. S3A and S3B), suggesting that ROS may simultaneously cause both mitochondrial depolarization and fragmentation independent of PARK2’s E3 ligase activity.
Figure 4. Photo-bleaching of mtKR leads to the sequential events of the PARK2-dependent mitophagy pathway. HeLa cells expressing mtKR, Cer-PARK2, and one of the following constructs: GFP-UB (A), GFP-LC3 (B), or GFP-LAMP-1 (C) were irradiated with 561 nm laser light for 30 iterations and were imaged by time-lapse at 5 min interval for up to 6 h. The representative images are a GFP-UB expressing cell at 60 min post-pb (A), a GFP-LC3 expressing cell at 90 min post-pb (B) and a GFP-LAMP-1 expressing cells at 4 h post-pb (C). The magnification of the area outlined by the white box is shown in the panels label Zoom. (D) Representative images of SH-SY5Y cells expressing mtKR and GFP-LC3 and irradiated with white light for 30 min at 37°C. Cells were imaged live 16 h after the photo-bleaching. The magnification of the areas outlined by the white boxes is shown in the panels that are labeled zoom 1 and zoom 2. Scale bars: 10 µm. (E) Plot of the time when PARK2, Ub or LC3 first show colocalized structures with mitochondria. The dash indicates the average time of recruitment (n = 14). Cer-PARK2 first appeared on mitochondria at 20.4 min post-pb, 34 min for GFP-UB and 40.1 min for GFP-LC3.
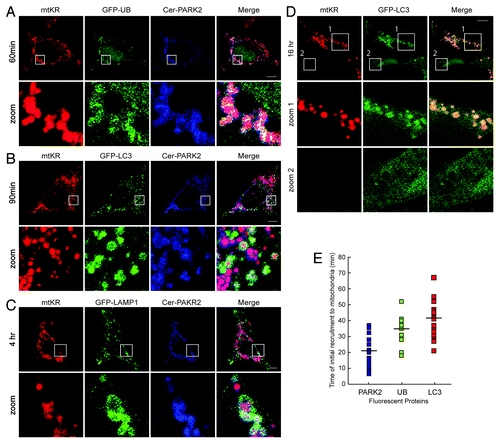
To determine whether mtKR can activate mitophagy in cells with endogenous PARK2 expression, we examined for the colocalization between mitochondria and autophagosomes in SH-SY5Y cell line after photo-bleaching mtKR. SH-SY5Y is a human neuroblastoma cell line that has been shown to express endogenous PARK2 and where mitophagy can be activated by CCCP treatment. Therefore, we used the colocalization between the autophagosomal marker GFP-LC3 and the mitochondria as an indicator of mitophagy activation.Citation22 As seen in , we observed GFP-LC3 to colocalize with mitochondria after the activation of mtKR (, zoom 1) but not in the cells lacking mtKR (, zoom 2).
One advantage of selectively activating mitophagy via mtKR is that it allows for a more reliable temporal measurement of the molecular events that define mitophagy. The timing of Ub, LC3, and LAMP-1 colocalization events following mtKR activation are shown in . After activation of mitophagy with mtKR, PARK2 began to appear on the mitochondria on average 21 min after photo-bleaching (). This was quickly followed by UB, which localized with mitochondria on average at 34 min, and the initial localization of LC3 to mitochondria took on average 41 min (). Mitochondrial localization to the lysosomes (GFP-LAMP-1) began approximately 2 h after photo-bleaching and this colocalization became more prominent after 4 h (). Although these steps of mitophagy have been previously described, we have here determined the specific timing of each of these important molecular events following the ROS burst.Citation8
ROS-induced mitophagy requires the depolarization of mitochondria
Next we examined the mechanism by which mitochondrial ROS induces mitophagy. A previous study has demonstrated that CCCP promotes mitophagy by causing the depolarization of mitochondria and that ROS may not be required for CCCP-induced mitophagy.Citation6 However, given that mitochondria are the main sites for ROS production, we propose that ROS-mediated mitochondrial depolarization may initiate mitophagy. We used the potential-dependent mitochondrial dye, MitoTracker® Red (MTRED), to determine the state of the irradiated mitochondria. MTRED was applied to cells expressing mtKR and mitochondrial-targeted GFP (mtGFP) immediately after photo-bleaching. Although MTRED and mtKR have similar excitation/emission spectra, the mtKR fluorescence signal at 120 min post-pb was negligible due to photo-bleaching and was detected at a very low level at the imaging settings used (Fig. S1C). Therefore, mitochondria that have lost their potential will only emit a GFP fluorescence signal while polarized mitochondria will emit fluorescence from both GFP and MTRED. We selectively photo-bleached a region of the mitochondria indicated by the white outline and followed its fate for 30 min (, pre-pb). We found that photo-bleaching mtKR resulted in mitochondrial depolarization as the majority of the mitochondria within this region only emitted GFP fluorescent signal (, 30 min + MTRED, and zoom in 1). Mitochondria lacking MTRED signal were only seen in the region where mtKR was photo-bleached (green only) while the nonphoto-bleached region emitted signals from both mtGFP and MTRED (yellow) (, 30 min + MTRED). Upon closer examination of the photo-bleached region (, zoom in 1), we observed two populations of mitochondria. One population only emitted the mtGFP signal (solid arrow head) while the other population emitted both the mtGFP and MTRED signals (arrows), indicating that a proportion of mitochondria were somehow recalcitrant to ROS-induced damage. However, in the area that was not photo-bleached, mitochondria did not lose their potential (, zoom in 2). Furthermore, the loss of membrane potential was not due to the exposure of the cells to high energy 561 nm laser light alone since the photo-beaching of mtRFP did not cause mitochondria depolarization (Fig. S1D).
Figure 5. PARK2 is recruited to mitochondria deploarized by mtKR photo-bleaching. (A) Representative images of HeLa cells transfected with mtKR and mtGFP and irradiated with 561 nm laser light in the region indicated by the white outline (pre-pb). 200 nM MitoTracker® Red (MTRED) was added to the cells after photo-bleaching and the cells were imaged live for 30 min. Two areas indicated by the white box in the 30 min + MTRED panel have been magnified. In zoom 1, the solid arrowheads indicate mitochondria with absent MTRED staining and the arrows indicate mitochondria with both mtGFP and MTRED signal. (B) Representative images of HeLa cells expressing mtKR and GFP-PARK2 and irradiated with 561 nm laser in the region indicated by the white outline (pre-pb). 200 nM MTRED was added to the cells following photo-bleaching and the cells were imaged again for 30 min. The white square box area is magnified in the panel labeled as “zoom in.” The arrow path in the “zoom in” panel indicates the pixel intensity of the fluorescent signal from GFP-PARK2 (green) and MTRED (red) shown in (C). Scale bar: 10 µm. (C) The pixel intensity of red and green channels was plotted against the pixel position along line scan.
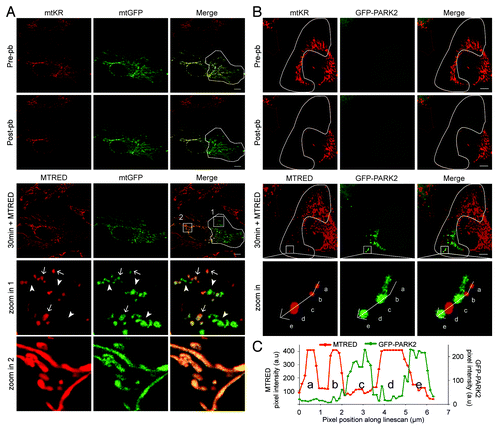
Next we asked whether PARK2 is specifically recruited to these depolarized mitochondria. To test this, we photo-bleached a selected region of the mitochondrial network in a cell expressing mtKR and GFP-PARK2 (, pre-pb, indicated by the white line), added 200 nM MTRED immediately after photo-bleaching, and analyzed the cell by live imaging for 30 min. We found that only the mitochondria in the photo-bleached region were fragmented and colocalized with PARK2 (, 30 min + MTRED). The MTRED signal revealed that PARK2-positive mitochondria were depolarized. In contrast, mitochondria that retained their membrane potential did not recruit PARK2 (, zoom in). This differential location of PARK2 and MTRED can be readily seen in the fluorescent intensity line analysis of the image (), which showed that only mitochondria negative for MTRED signal contained signal from GFP-PARK2.
SOD2 prevents mtKR-induced mitophagy
Computational simulation studies predicted that superoxide (O2●-), a highly reactive and membrane-impermeable ROS, is produced when KR is photo-activated.Citation16 O2●- produced in the mitochondrial matrix is converted to hydrogen peroxide (H2O2) by the mitochondrial-antioxidant protein superoxide dismutase 2 (SOD2). H2O2 is membrane-permeable and is further converted to water and oxygen by catalase either inside or outside the mitochondria.Citation12 To determine which reactive oxygen species is responsible for mtKR-induced mitophagy, we examined the effect of overexpressing SOD2 in mitochondria. We hypothesized that if O2●- is responsible for inducing mitophagy, an overexpression of SOD2 should prevent or diminish PARK2 recruitment to mitochondria following mtKR activation. However, if H2O2 is the ROS responsible for inducing mitophagy, then SOD2 overexpression should increase mtKR-induced mitophagy. HeLa cells expressing mtKR, Cer-PARK2, and SOD2-GFP were photo-bleached and were imaged at 90 min post-pb. As seen in , neither mitochondrial fragmentation nor PARK2 recruitment was observed in SOD2-GFP expressing cells (, 90 min + MTRED) whereas in control cells expressing mtGFP instead of SOD2-GFP, PARK2 recruitment was readily visible (, 90 min + MTRED). Quantification of the PARK2 recruitment area using the PARe-Q confirmed the protective nature of SOD2-GFP since 37% of the mitochondria expressing mtGFP showed PARK2 recruitment compared with 8% of the cells expressing SOD2-GFP (). Therefore, the overexpression of SOD2 significantly impeded ROS-induced mitophagy compared with overexpression of mtGFP (Table S2). In addition, treating cells with the O2●- scavenger mitoTempo, which mimics SOD2 activity, also impeded mtKR-induced mitophagy, further suggesting that it is the reduction of O2●- that is responsible for inhibiting ROS-induced mitophagy (Fig. S4). Interestingly, treating cells with 10 mM NAC, which eliminates H2O2, did not inhibit ROS-induced mitophagy suggesting that H2O2 is not required for mitophagy activation. Thus, these data strongly suggest that O2●-, not H2O2, is the ROS that initiates PARK2 recruitment to mitochondria.
Figure 6. Overexpression of SOD2 impedes ROS-induced mitophagy. (A and B) Representative images of HeLa cells expressing mtKR and Cer-PARK2 with either SOD2-GFP (A), or mtGFP (B) and irradiated with 561 nm laser light for 30 iterations. Pre-pb and post-pb images were acquired. Fifteen min prior to the 90 min post-pb image was taken, 200 nM MTRED was added to the photo-bleached cell. The magnifications of the area outlined with the white box are shown in panels labeled as “zoom.” (C and D) PARe-Q was done on the images of cells treated as in (A and B) (n = 35). 8% of SOD2-GFP expressing cells showed PARK2 recruitment 90 min post-pb (C), while 37% of mtGFP expressing cells showed PARK2 recruitment (D) This difference is significant (p = 0.033). (E–G) The total area of PARK2 recruitment (µm2) was compared with the total area of depolarized mitochondria (µm2) in HeLa cells expressing mtKR and Cer-PARK2 (E), mtKR, Cer-PARK2, and SOD2-GFP (F), and mtKR, Cer-PARK2, and mtGFP (G) (refer to Materials and Methods for details). PARK2 recruitment area was plotted against the corresponding area of depolarized mitochondria (n = 35). (H) HeLa cells expressing mtRFP, Cer-PARK2, and SOD2-GFP were treated with 10 µM CCCP or DMSO for 1 h as indicated. Cells were imaged live 1 h after the treatment. Scale bar: 10 µm.
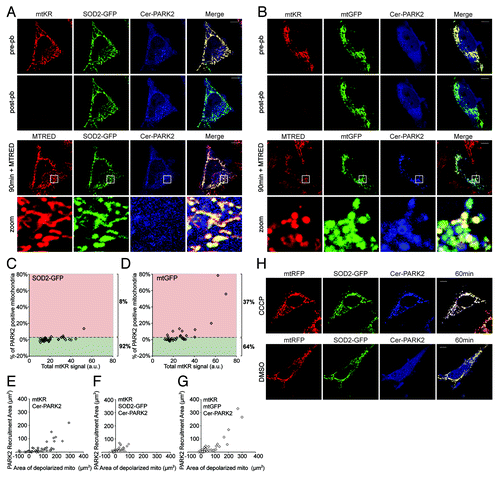
To further understand how SOD2 inhibited PARK2 recruitment, we examined the integrity of the mitochondria in our assay. We postulated that overexpression of SOD2 metabolized more O2●- and prevented mitophagy by protecting the mitochondria from depolarization. To test this hypothesis, we added MTRED 15 min prior to imaging the 90 min post-pb time point to cells overexpressing SOD2-GFP and quantified the area of depolarized mitochondria and compared it to the area of PARK2 recruitment (refer to Materials and Methods for details). As expected, we found a positive correlation between the PARK2 recruitment area and the area of depolarized mitochondria when these two values were plotted on a scatter plot (). Expression of SOD2 reduced both mitochondrial depolarization area and PARK2 recruitment area ( compared with ); whereas cells expressing mtGFP did not have reduced mitochondrial depolarization nor reduced PARK2 recruitment area compared with control ( compared with ). Interestingly, the protective effect of SOD2 was not observed in cells where mitophagy was induced with CCCP () supporting previous observations that ROS are not required for CCCP to induce mitophagy. Taken together, our data strongly suggest that O2●- induces mitochondrial depolarization leading to PARK2 recruitment. The data also suggest that ROS acts upstream of mitochondrial depolarization in the mitophagy pathway, which is an important distinction in understanding the signals that promote mitochondrial quality control.
Mitochondrial morphology directly affects ROS-induced mitophagy
Recently, it has been shown that mitochondrial elongation induced by starvation prevents their degradation by general autophagy.Citation23,Citation24 We postulated that mitochondrial morphology might be an important determinant in the mitophagy pathway. To test this, we determined whether mitochondrial elongation could reduce mitochondrial degradation by ROS-induced mitophagy; and conversely whether mitochondrial fragmentation could promote ROS-induced mitophagy. In HeLa cells expressing mtKR and Cer-PARK2, we induced mitochondrial elongation by reducing endogenous DRP1 and induced mitochondrial fragmentation by reducing endogenous OPA1 using siRNA.Citation25,Citation26 We photo-bleached mtKR in the cells and imaged them for 90 min (). Fifteen min prior to acquiring an image at 90 min post-pb, 200 nM MTRED was added to the photo-bleached cells to visualize mitochondrial potential. 90 min after photo-bleaching mtKR, elongated mitochondria retained their potential and did not appear to colocalize with PARK2 (, 90 min), while the majority of fragmented mitochondria had lost their potential and recruited PARK2 (, 90 min). The PARe-Q further validated the observation that elongated mitochondria are more resistant to PARK2 recruitment than the ones in the control cells during ROS-induced mitophagy (). Conversely, fragmented mitochondria () were more susceptible to ROS-induced mitophagy compared with control cells (). In addition, our statistical analysis demonstrated that both the siDRP1 cells and the siOPA1 cells behaved significantly differently from the siCtrl cells (Table S3). Thus, our data demonstrate that elongated mitochondria possess some property that makes them more resistant to ROS-induced mitophagy.
Figure 7. Mitochondrial morphology plays a role in ROS-induced mitophagy. (A and B) HeLa cells transfected with siRNA against DRP1 (A) or OPA1 (B) for 72 h were also transfected with mtKR and Cer-PARK2 constructs during the last 16 h before imaging. Cells were irradiated with 561 nm laser for 30 iterations. An image was acquired at pre-pb and at post-pb. 200 nM of MTRED was added to the photo-bleached cells 15 min before the 90 min images were acquired (90 min + MTRED). The magnification of the area outlined with the white boxes is shown in the panels labeled as “zoom.” Scale bar: 10 µm. (C–E) PARe-Q on the data collected from DRP1 and OPA1 knockdown experiments (n = 44). siDRP1 treated cells show only 23% PARK2 recruitment (C) while siOPA1 treated cells had 80% of the cells with above basal level of PARK2 recruitment (D). (E) Treating cells with control siRNA lead to 51% PARK2 recruitment 90 min after mtKR was photo-bleached. The differences between the siDRP1 and the siOPA1 respectively the siCtrl population are significant (p = 2.1 × 10−10 resp. 6.9 × 10−5); the difference between the siOPA1 population and the siCtrl population is also significant (p = 6.4 × 10−6). (F and G) HeLa cells were transfected with siRNA against DRP1 or scrambled siRNA for 72 h. During the last 16 h, cells were also transfected with mtRFP and Cer-PARK2. Cells were treated with 10 µM CCCP or DMSO for 1 h prior to live-cell imaging.
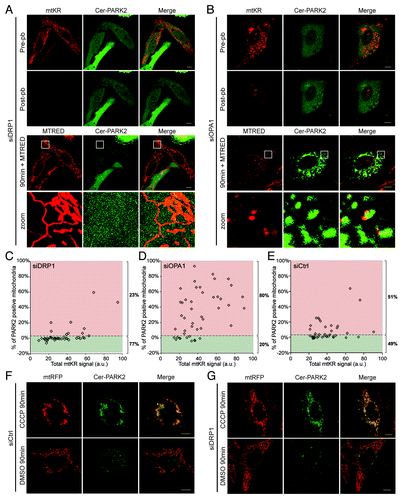
One hypothesis has suggested that elongated mitochondria are resistant to mitophagy since the elongated morphology might not be congruent for PARK2 recruitment.Citation24 To test this possibility, we examined whether elongated mitochondria were resistant to CCCP-induced PARK2 recruitment. When cells treated with DRP1 siRNA and scramble siRNA were incubated with 10 µM CCCP, we found that PARK2 was readily recruited to almost all mitochondria in both DRP1 knockdown and control cells (). These data suggest that the resistance of elongated mitochondria to ROS-induced mitophagy is not due to an inability of PARK2 to be recruited to the elongated mitochondria but likely due to their increased resistance to ROS-induced mitophagy.
Discussion
ROS is a cellular paradox as it acts both as an intracellular signaling molecule and as a lethal toxin; hence, cellular ROS homeostasis is tightly controlled. Autophagy activated by ROS is likely one such mechanism that detects changes in cellular redox homeostasis. Both exogenously added H2O2 and mitochondrial ROS have been shown to activate general autophagy. Autophagy is essential to degrade ROS-producing organelles such as mitochondria and peroxisomes.Citation27 However, beyond activating general autophagy, it is not clear whether ROS is also involved in the activation of selective autophagy such as mitophagy.
Based on our current findings, we propose that superoxide (O2●-) acts as a signal to activate mitophagy by depolarizing the mitochondrial inner membrane. We have demonstrated that an acute burst of ROS specifically in the mitochondrial matrix resulted in mitochondrial damage, depolarization, and the subsequent activation of mitophagy. These depolarized mitochondria became fragmented and recruited PARK2, the mitophagy E3 ubiquitin ligase. Following PARK2 recruitment, the damaged mitochondria were ubiquitinated, engulfed by LC3-positive autophagosomes, and finally were localized to lysosomes. O2●- was determined to be the specific ROS that damages mitochondria since the overexpression of SOD2 or O2●- scavenger inhibited both the mitochondrial fragmentation and the mitophagy induced by mtKR-activation. These experiments suggest that the O2●- produced by mtKR within the mitochondria induces damage on the mitochondria, which leads to mitochondrial depolarization, fragmentation, and the activation of mitophagy. The observation that an elevation in O2●- leads to mitophagy is physiologically relevant given that the electron transport chain produces O2●- in the mitochondria. Therefore we suggest that ROS is the initiating signal for mitophagy and that mitophagy is amplified by the resulting loss of membrane potential.
Our data suggest a possible relationship between mitochondrial morphology and the antioxidant capacity of mitochondria. It was recently reported that mitochondria reconfigure into an elongated and interconnected network during starvation. These elongated mitochondria were shown to be more resistant to starvation-induced autophagy through a mechanism yet to be understood.Citation23,Citation24 Although the mechanism of how elongated mitochondria resist an acute burst of superoxide is not known, our data showed that interconnected mitochondria are less susceptible to mtKR-induced damage and mitophagy. The elongated mitochondria clearly have the ability to resist ROS-induced depolarization, which protects them from mitophagy. Interestingly, we do know that these mitochondria are still susceptible to mitophagy because CCCP treatment resulted in the loss of polarity and the subsequent recruitment of PARK2 (). Thus, elongated mitochondria may have a higher antioxidant capability than fragmented mitochondria. The resistance of elongated mitochondria to higher ROS concentration may be related to the recent finding that elongated mitochondria have increased oxidative phosphorylation activity.Citation23 Since oxidative phosphorylation is likely the main cause of superoxide formation, it is conceivable that elongated mitochondria have mechanisms to cope with an increase in ROS production.
Consistent with our findings, we note that during the preparation of this manuscript, two other groups have reported the method of using mtKR to activate mitophagy.Citation18,Citation28 In particular, Yang and Yang showed that local photo-activation of mtKR leads to PARK2 recruitment followed by LC3 colocalization to the affected mitochondria. In addition to demonstrating the technological advance over CCCP-treatment, we have directly identified the chronology of each molecular stage of mitophagy, and we also characterized the mechanism of mitophagy activation by mtKR. Moreover, we have deduced the role of antioxidants and mitochondrial morphology in the regulation of the cellular mitophagic response.
There are many questions remaining about the mechanism of mitophagy and we believe the mtKR methodology will be useful in the endeavor to address these questions. A major question is the molecular basis for the inherent oxidative capacity of mitochondria with respect to mitochondrial morphology and mitophagy as discussed above. It has been speculated that a mitochondrion’s ability to cope with oxidative stress depends on mitochondrial morphology.Citation29 MtKR may be the ideal tool to study the relationship between oxidative stress and mitophagy since it causes an acute increase in ROS in the mitochondrial matrix. In addition, the ability to induce damage in a specific mitochondrion can help compare morphologically different mitochondria within a single cell. Notably, mitochondrial-decouplers such as CCCP are not ideal tools to study the morphological dependence of mitophagy since they cause a global damage to the mitochondrial network and bypass the ROS signaling step.Citation6 Furthermore, the ability to fine-tune the amount of mtKR damage can be used to determine the resilience of different mitochondrial morphologies to combat different amounts of ROS.
Finally, the ability to spatially and temporally activate mitophagy can be used to determine the relationship between mitochondrial location, cell-cycle, and even possible inter-cellular communication events that could impact the orchestration of mitophagy. For example, it is possible that mitochondrial localization within the cell affects the degree of mitophagy.Citation10 Mitochondrial-enriched autophagosomes tend to preferentially localize to the periphery of cells. The efficiency of autophagic clearance of damaged mitochondria at the cell periphery and perinuclear regions could be easily assayed using the mtKR methodology. Similar hypotheses could be addressed with respect to cell cycle progression and cell-cell contact sites underscoring the utility of this methodology in understanding the biology of mitophagy.
Materials and Methods
Plasmids
The plasmid of mitochondria-targeting KillerRed (mtKR) was purchased from Evrogen (FP964). The Cerulean-PARK2 (Cer-PARK2) plasmid was constructed by replacing YFP in the YFP-PARK2 plasmid with monomerized Cerulean using the common NheI and BsrGI restriction sites. The YFP-PARK2 plasmid was generously provided by Richard Youle (NIH, Bethesda). Mitochondria-targeting GFP (mtGFP) was constructed by replacing YFP in the mtYFP plasmid with a monomerized version of EGFP (Clontech, 6085-1). The mtYFP plasmid and mEGFP-N1 plasmid were digested by BamHI and BsrGI. The mtYFP, GFP-Ubiquitin, GFP-LC3, and GFP-LAMP-1 plasmids were previously described. SOD2-GFP was a generous gift from Andre Melendes (Albany Medical College). siRNA against DRP1 (5′-AACGCAGAGCAGCGGAAAGAGTT-3′) was purchased from Santa Cruz Biotechnology INC. (sc43732). A pool of three siRNA against OPA1 1: (5′-GAACAGCUCUGAAAGCAUUTT-3′), 2: (5′-GCAAUGGGAUGCAGCUAUUTT-3′), and 3: (5′-GCAAUGAUGUGGUCUUGUUTT-3′) was purchased from Santa Cruz Biotechnology. Two siRNA against PINK1 1: (5′-GGAGAUCCAGGCAAUUUUUUTT-3′) and 2: (5′-CCGGACGCUGUUCCUCGUUAUTT-3′) were synthesized by GenePharma Co. Ltd.
Cell culture and transfection
HeLa cells were grown in Dulbecco's modified Eagle's medium (HyClone, SH3008101) supplemented with 2 mM L-glutamine (HyClone, SH30034.01) and 10% FBS (Invitrogen, A12617) at 37°C and 5% CO2. SH-SY5Y cells were grown in Gibco® RPMI Media (Invitrogen, 11875-093) supplemented with 2 mM GlutaMAX™ (Invitrogen, 35050) and 10% FBS. For live-cell imaging, cells were grown in 4-well LabTek chamber slides (Thermo, 155383). Plasmids were transfected with Lipofectamine-2000 (Invitrogen, 11668019) according to the manufacturer’s instructions 16 to 24 h prior to imaging. siRNA was transfected with Lipofectamine-2000 according to the manufacturer’s instruction for 48 to 72 h prior to imaging. Cells were changed into CO2-independent medium (Invitrogen, 18045) prior to live-cell imaging. When imaging mitochondria in lysosomes, cells were treated with 0.5 µM Leupeptin (Bioshop, Leu001.50) and 2 µM E-64 (Alexis Biochemical ALX260-007-m025) to inhibit lysosomal protease. For experiments in which ROS levels were measured, cells were incubated with medium containing 10 µM OxyBURST® Green H2DCFDA (Invitrogen, D2935) at 37°C for 15 min. To measure mitochondrial potential following photo-bleaching, 200 nM MitoTracker® REDCMXRos (Invitrogen, M-7512) was added to the chamber on the stage of the microscope and cells were imaged 5 min later. For antioxidant experiments, 10 nM mtTempo (Enzo Life Sciences, ALX-430-150) or 1 mM N-acetyl-l-cysteine (NAC) (Sigma-Aldrich, A7250) was added to the CO2-independent medium prior to live-cell imaging. For long-term time-lapse experiments, 1× antibiotic cocktail (Wisent, 450-115-EL) was added to the CO2-independent medium to prevent bacterial growth.
Microscopy
All fluorescent imaging was performed on a Zeiss LSM710 equipped with a 63× 1.4 NA oil immersion objective and the appropriate lasers. The images were acquired in 1024 × 1024 pixels at the depth of 12 bits. For visual presentation only the brightness was adjusted. All quantification was done using the imaging software Volocity® 5.0 (Perkin Elmer).
Induction of mitophagy
In our assay, mtKR was used both to visualize the mitochondria and to cause an acute elevation of mitochondrial ROS. We found that mtKR can be readily visualized using the 561 diode laser at low power (1–2%) with minimal photo-bleaching (Fig. S1A). Significant photo-bleaching (greater than 50% reduction in fluorescent signal) was only observed after photo-bleaching with 15 iterations of the 561 nm laser at full power (100%). At 30 iterations of 561 nm laser light at 100%, we typically observed approximately 85% reduction in mtKR fluorescent signal (Fig. S1A). Importantly, similar treatments with the 561 nm laser (15–30 iterations at 100% output) did not photo-bleach other fluorescent proteins such as GFP (Fig. S1A) and Cerulean ().
To photo-bleach mtKR or mtRFP, the target region was drawn on the field view of the monitor and the Definite Focus system on the Zeiss LSM 710 was used to maintain focus on a single focal plane. The 561 nm imaging laser was set to 1% output power prior to the photo-bleaching process, and was maintained at 1% or increased to 2% output after photo-bleaching in order to continue visualizing the mitochondria using the mtKR fluorescence signal. When MTRED was used, the imaging laser power was kept at 1% throughout the experiment. 561 nm laser light at 100% output was used to photo-bleach mtKR and mtRFP, and the number of iterations was indicative of the number of times the laser bleached the defined region. Images were taken immediately before and after photo-bleaching the cells, and at the specified time points. For time-lapse experiments, multiple locations in the chamber were selected and saved with Zeiss ZEN imaging software. Each location was imaged at 5–10 min intervals.
To photo-bleach mtKR in SH-SY5Y cells, cells were incubated in CO2-independent DMEM containing 25 µM Leupeptin and 1µM E64 and were irradiated with strong white light for 30 min at 37°C. Cells were imaged live 16 h after photo-bleaching.
For the CCCP treatment, cells were kept at 37°C in CO2-independent media. Time-lapse imaging of multiple locations was set up using Zeiss ZEN as above. After the initial image was acquired (0 min), CCCP was added to the chamber containing the cells to achieve a final concentration of 10 µM. Cells were imaged at 5 min intervals for 1–2 h.
To confirm that after photo-bleaching the mtKR signal did not interfere with the signal of MTRED, HeLa cells transfected with mtKR and mtGFP were photo-bleached by the 561nm laser for 30 iterations. The cells were imaged live at 5 min intervals for 105 min. 200 nM MTRED was added at 105 min post-pb, and the imaging process continued for another 15 min. The total pixel intensity of the green and the red channels was measured using the image analysis software Volocity®, and results were plotted against time (Fig. S1B). To visualize the mitochondrial potential of the cells during live-cell imaging, cells were photo-bleached using the protocol described above. The imaging laser power before photo-bleaching (pre-pb) and after photo-bleaching (post-pb) was kept constant at 1% laser output, while photo-bleaching laser power was at 100% laser output. MTRED was added into the LabTek chamber containing the photo-bleached cells to achieve a final concentration of 200 nM. Cells were treated with MTRED 15 min prior to the final post-pb image (indicated in the Figures).
ROS detection
To measure the increase in ROS levels after photo-bleaching, cells were incubated in 10 µM OxyBURST® Green H2DCFDA at 37°C for 15 min. To prepare for the live-cell imaging, cells were washed with 37°C PBS once and were replenished with CO2-independent medium. To determine the total fluorescent intensity of the OxyBURST signal within the cell an open pinhole (9.1 µm) was used when acquiring the fluorescent signal from OxyBURST. Photo-bleaching was conducted as described above and images were acquired. The area of the cell was determined by drawing a region of interest (ROI) around individual cells, in which the total pixel intensity of the green signal was measured using the image analysis software Volocity® 5.0 (PerkinElmer) in arbitrary units. The total green signal was measured in the pre-pb images and post-pb images. For each cell, the percentage increase was calculated as the increase in total OxyBURST signal post-pb over the total OxyBURST signal in pre-pb image.
The PARK2 requirement quantification (PARe-Q)
To quantify PARK2 recruitment to mitochondria, we used Volocity® to determine the area of PARK2 positive mitochondria. A ROI was used to identify and select Cer-PARK2 positive structures above a predetermined threshold level in each cell in the 90 min post-pb image. Due to the different expression levels of Cer-PARK2 in each cell, the minimal threshold for the Cer-PARK2 signal was adjusted accordingly to ensure that all PARK2 aggregates were recognized. To measure the background area due to auto-fluorescence, the same threshold was used to measure the area of auto-fluorescence in the pre-pb image. The auto-fluorescence area was then subtracted from the PARK2 recruitment area to obtain the “calculated PARK2 recruitment area.” To measure the area of mitochondria, we measured the area of mtKR that was above a given threshold in the pre-pb image. The total signal of mtKR was obtained by measuring the total pixel intensity of the red channel in the pre-pb image. The percentage of PARK2-positive mitochondria was calculated by the ratio of “calculated PARK2 recruitment area” over “total area of mitochondria.” The percentage of PARK2-positive mitochondria was plotted against its corresponding total mtKR signal in a scatter plot.
Statistical analysis of the PARe-Q
We considered each experimental protocol to generate a population of cells, and each cell within the population to constitute an independent observation. For all PARK2 recruitment quantifications, the ratio of “the percentage PARK2 recruitment area” over “total mtKR signal” of each treated cell was ranked and compared with the corresponding ratios of “standard” protocol respectively the control protocols. Calculations were performed with the R statistical workbench using the Wilcox test function and a normal approximation for the calculation of p-values.Citation30 The usual p = 0.05 threshold was applied to label differences as “significant.”
Additional material
Download Zip (6.8 MB)Acknowledgment
We thank Dr. John Brumell for critical and constructive comments on the manuscript. We thank Drs. Richard Youle and Andre Melendes for reagents. This work was supported in part by operating research grants (to P.K.K. and G.A.M.) from the Canada Institute of Health Research Grant and Discovery Grants (to P.K.K. and G.A.M.) from the Natural Sciences and Engineering Research Council of Canada.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/21211
References
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol 2006; 16:R551 - 60; http://dx.doi.org/10.1016/j.cub.2006.06.054; PMID: 16860735
- Mammucari C, Rizzuto R. Signaling pathways in mitochondrial dysfunction and aging. Mech Ageing Dev 2010; 131:536 - 43; http://dx.doi.org/10.1016/j.mad.2010.07.003; PMID: 20655326
- Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J 2012; 441:523 - 40; http://dx.doi.org/10.1042/BJ20111451; PMID: 22187934
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12:9 - 14; http://dx.doi.org/10.1038/nrm3028; PMID: 21179058
- Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI, et al. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J Alzheimers Dis 2010; 20:Suppl 2 S401 - 12; PMID: 20463393
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795 - 803; http://dx.doi.org/10.1083/jcb.200809125; PMID: 19029340
- Shi G, Lee JR, Grimes DA, Racacho L, Ye D, Yang H, et al. Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson’s disease. Hum Mol Genet 2011; 20:1966 - 74; http://dx.doi.org/10.1093/hmg/ddr077; PMID: 21355049
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RLA, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 2010; 107:378 - 83; http://dx.doi.org/10.1073/pnas.0911187107; PMID: 19966284
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8:e1000298; http://dx.doi.org/10.1371/journal.pbio.1000298; PMID: 20126261
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RLJ, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 2011; 20:1726 - 37; http://dx.doi.org/10.1093/hmg/ddr048; PMID: 21296869
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749 - 60; http://dx.doi.org/10.1038/sj.emboj.7601623; PMID: 17347651
- Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 2009; 16:1040 - 52; http://dx.doi.org/10.1038/cdd.2009.49; PMID: 19407826
- Kim I, Lemasters JJ. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal 2011; 14:1919 - 28; http://dx.doi.org/10.1089/ars.2010.3768; PMID: 21126216
- Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, et al. Superoxide flashes in single mitochondria. Cell 2008; 134:279 - 90; http://dx.doi.org/10.1016/j.cell.2008.06.017; PMID: 18662543
- Ma Q, Fang H, Shang W, Liu L, Xu Z, Ye T, et al. Superoxide flashes: early mitochondrial signals for oxidative stress-induced apoptosis. J Biol Chem 2011; 286:27573 - 81; http://dx.doi.org/10.1074/jbc.M111.241794; PMID: 21659534
- Roy A, Carpentier P, Bourgeois D, Field M. Photofunctional proteins: from understanding to engineering Diffusion pathways of oxygen species in the phototoxic fluorescent protein KillerRed. Photoch Photobio Sci 2010; 9:1342 - 50; http://dx.doi.org/10.1039/C0PP00167H; PMID: 20820672
- Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV, et al. A genetically encoded photosensitizer. Nat Biotechnol 2006; 24:95 - 9; http://dx.doi.org/10.1038/nbt1175; PMID: 16369538
- Yang JY, Yang WY. Spatiotemporally controlled initiation of Parkin-mediated mitophagy within single cells. Autophagy 2011; 7:1230 - 8; http://dx.doi.org/10.4161/auto.7.10.16626; PMID: 22011618
- Ivashchenko O, Van Veldhoven PP, Brees C, Ho YS, Terlecky SR, Fransen M. Intraperoxisomal redox balance in mammalian cells: oxidative stress and interorganellar crosstalk. Mol Biol Cell 2011; 22:1440 - 51; http://dx.doi.org/10.1091/mbc.E10-11-0919; PMID: 21118999
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 2012; 22:320 - 33; http://dx.doi.org/10.1016/j.devcel.2011.12.014; PMID: 22280891
- Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, Berman SB. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet 2011; 20:927 - 40; http://dx.doi.org/10.1093/hmg/ddq531; PMID: 21147754
- Zhu J, Dagda RK, Chu CT. Monitoring mitophagy in neuronal cell cultures. Methods Mol Biol 2011; 793:325 - 39; http://dx.doi.org/10.1007/978-1-61779-328-8_21; PMID: 21913110
- Gomes LC, Benedetto GD, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011; 13:589 - 98; http://dx.doi.org/10.1038/ncb2220; PMID: 21173798
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A 2011; 108:10190 - 5; http://dx.doi.org/10.1073/pnas.1107402108; PMID: 21646527
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 2007; 282:11521 - 9; http://dx.doi.org/10.1074/jbc.M607279200; PMID: 17301055
- Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem 2004; 279:18792 - 8; http://dx.doi.org/10.1074/jbc.M400920200; PMID: 14970223
- Liu Z, Lenardo MJ. Reactive oxygen species regulate autophagy through redox-sensitive proteases. Dev Cell 2007; 12:484 - 5; http://dx.doi.org/10.1016/j.devcel.2007.03.016; PMID: 17419989
- Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, et al. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 2011; 286:10814 - 24; http://dx.doi.org/10.1074/jbc.M110.132514; PMID: 21252228
- Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal 2011; 14:1939 - 51; http://dx.doi.org/10.1089/ars.2010.3779; PMID: 21128700
- R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, 2011. doi: http://www.R-project.org/.