Abstract
The link between the deregulation of autophagy and cell death processes can be essential in the development of several neurodegenerative diseases, such as Parkinson disease (PD). However, the molecular mechanism of deregulation of this degradative process in PD patients is unknown. The leucine-rich repeat kinase 2 (LRRK2) gene is related to PD and its implication in autophagy regulation has been described. Our recent work shows that the presence of the G2019S LRRK2 mutation, one of the most prevalent in LRRK2, is accompanied by a deregulation of autophagy basal levels dependent on the MAPK1/3 (ERK2/1) pathway.
Keywords: :
Parkinson disease (PD) is a neurodegenerative disorder caused by specific loss of dopaminergic neurons of the substantia nigra of the midbrain, which project into the striatum via the nigrostriatal pathway. This disease has been extensively studied by the scientific community since 1981, and actually is considered to represent a multifactorial etiology, involving both genetic and environmental factors. There is currently an intense research that focuses on the study of the deregulation of processes of synthesis or degradation of proteins as a possible cause of this neurodegenerative disease.
The LRRK2 gene encodes a large multidomain protein with GTPase and kinase activity. In the structure of LRRK2, several mutations have been detected associated with autosomal-dominant PD, with the G2019S LRRK2 mutation being one of the most prevalent. This mutation replaces the amino acid glycine, localized at protein position 2019, by the amino acid serine, increasing the molecular kinase activity, and it has also been associated in many studies with the deregulation of degradative processes, specifically the autophagic process.
Using a cellular model of fibroblasts obtained from PD patients with the G2019S LRRK2 mutation, we performed a complete characterization of basal autophagy in these mutant cells compared with control cells, without this mutation ().
Figure 1. Table and illustration with techniques used for studying basal autophagy. Complete autophagic flux was studied in both cell types, analyzing each stage of autophagy using various techniques. We observed that G2019S LRRK2 mutant fibroblasts present higher basal autophagy levels than wild-type cells. TEM, transmission electron microscopy; CMFDA, 5-chloromethylfluorescein diacetate; LTR, LysoTracker Red.
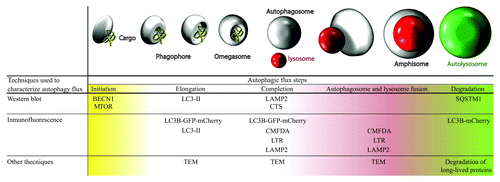
This study shows the presence of a higher number of autophagic structures in cells with the G2019S LRRK2 mutation compared with the control cells. To assess that this observation was due to blocked autophagy in its final step (which would prevent protein turnover within the lysosome), we designed several experiments to check protein degradation. All experiments of protein degradation, autophagy induction and blockage with different treatments indicated that in the G2019S LRRK2 mutant fibroblasts there was an increase of complete autophagic flux activity.
The second objective of the study was focused on the link between the deregulated autophagy observed in G2019S LRRK2 mutant fibroblasts and the MAPK1/3 pathway, opening an interesting way to elucidate the molecular mechanisms that render cells with this mutation more sensitive compared with those without it.
The G2019S mutation is localized in a residue of the activation site of the kinase domain of LRRK2, and helps to facilitate substrate access, increasing up to three times its kinase activity, which is associated directly with an increase in neurotoxicity and neuronal death. Deregulation in LRRK2 kinase activity is, therefore, an important objective to evaluate with regard to the possible causes of PD. An increase in kinase activity of any protein causes an increase in the phosphorylation of proteins that are direct or indirect downstream targets. One example of components whose activity depends on LRRK2 kinase activity is the MAPK1/3 pathway, which is overactivated in G2019S LRRK2 mutants. This fact, together with the participation of MAPK1/3 in the process of autophagy regulation, makes the study of the involvement of this pathway in the exacerbated autophagy observed in mutant fibroblasts very interesting.
After checking G2019S LRRK2 fibroblasts for the presence of the mutation and its exacerbated kinase activity, which corresponds to an increase in the levels of protein activity of MAPK1/3 at baseline, we used a treatment with an inhibitor of the MAPK1/3 pathway (1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene; U0126). The result observed in control fibroblasts was a drop in cell death and levels of autophagy as an adaptive response of the cell; however, in cells with the G2019S LRRK2 mutation we observed a decrease in cell death and autophagy basal levels after U0126 exposure. Therefore, the exacerbated autophagy and MAPK1/3 pathway can be related. To confirm that the exacerbated autophagy is responsible, at least in part, for most cellular sensitivity, we programmed a genetic inhibition of autophagy in both cell types, observing similar results to those obtained after U0126 treatment. Therefore, autophagy deregulation is responsible for changes in cell sensitivity. In summary, the damage observed in the G2019S LRRK2 mutant fibroblast is induced by the exacerbated autophagy levels observed in these cells by overactivation of the MAPK1/3 route, resulting in a negative effect in mutant cells ().
Figure 2. Diagram with the proposed mechanism by which G2019S LRRK2 fibroblasts present an exacerbated MAPK1/3-dependent autophagy activity. Fibroblasts with the G2019S LRRK2 mutation show exacerbated kinase activity, which corresponds to an increase in the phosphorylated levels of MAPK1/3 activity at baseline with respect to control cells. Also, they present an exacerbated autophagic flux that is reverted using an inhibitor of the MAPK1/3 pathway, decreasing in the same manner the cell death observed in mutant cells.
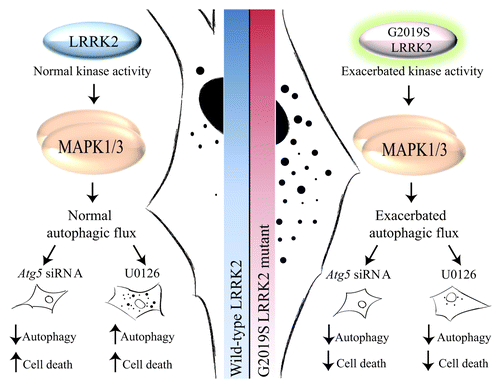
The increase in the degradation process of misfolded proteins may be explained by two possible hypotheses: it can be a direct effect by activation of a positive regulator of autophagy, such as is the MAPK1/3 pathway; however, this could also be an indirect effect induced by the accumulation of misfolded proteins, an effect that has also been associated with an overactivation of the same route. Whatever the reason, exacerbated autophagy causes significant cell damage, and it can sensitize the cell to certain extracellular stimuli. Therefore, and as occurs in most cases of parkinsonism, a genetic predisposition coupled to a particular negative environment triggers cell death that will cause the onset of disease symptoms. For this reason, it is of great importance to know the exact molecular mechanisms, by which the G2019S LRRK2 mutant cells have exacerbated autophagy, to try to reverse this abnormal situation and remove a major risk factor that contributes to developing the disease.
Acknowledgments
The work was supported by Junta de Extremadura predoctoral fellowship, ISCIII Ministerio de Economía y Competitividad, Spain (CP0800010, PI11/0040), FUNDESALUD (PRIS11014, PRIS11019), CIBERNED (CB06/05/004) and Consejería, Empleo, Empresa e Innovación Junta de Extremadura (GR10054).