Abstract
SNCA (α-synuclein) misfolding and aggregation is strongly associated with both idiopathic and familial forms of Parkinson disease (PD). Evidence suggests that SNCA has an impact on cell clearance routes and protein quality control systems such as the ubiquitin-proteasome system (UPS) and autophagy. Recent advances in the key role of the autosomal recessive PARK2/PARKIN and PINK1 genes in mitophagy, highlighted this process as a prominent new pathogenic mechanism. Nevertheless, the role of autophagy/mitophagy in the pathogenesis of sporadic and autosomal dominant familial forms of PD is still enigmatic. The yeast Saccharomyces cerevisiae is a powerful “empty room” model that has been exploited to clarify different molecular aspects associated with SNCA toxicity, which combines the advantage of being an established system for aging research. The contribution of autophagy/mitophagy for the toxicity induced by the heterologous expression of the human wild-type SNCA gene and the clinical A53T mutant during yeast chronological life span (CLS) was explored. A reduced CLS together with an increase of autophagy and mitophagy activities were observed in cells expressing both forms of SNCA. Impairment of mitophagy by deletion of ATG11 or ATG32 resulted in a CLS extension, further implicating mitophagy in the SNCA toxicity. Deletion of SIR2, essential for SNCA toxicity, abolished autophagy and mitophagy, thereby rescuing cells. These data show that Sir2 functions as a regulator of autophagy, like its mammalian homolog, SIRT1, but also of mitophagy. Our work highlights that increased mitophagy activity, mediated by the regulation of ATG32 by Sir2, is an important phenomenon linked to SNCA-induced toxicity during aging.
Introduction
SNCA is a ubiquitously expressed protein in the brain with an intrinsic ability to bind to lipids and membranes. The function of SNCA is still unclear, but it regulates synaptic plasticity, dopamine neurotransmission, endoplasmic reticulum-Golgi trafficking, and acts as a molecular chaperone.Citation1 Overexpression or mutation of SNCA results in its misfolding and the formation of oligomeric and aggregated species that are associated with autosomal dominant forms of Parkinson disease.Citation2,Citation3 Impairment and deregulation of protein quality control systems has emerged as a central, and common, pathogenic event in different models of synucleinopathy.Citation4 Cell proteostasis is maintained by a precisely regulated protein quality control system, consisting of a balance between the folding and refolding of misfolded proteins, by molecular chaperones, and the proteolytic systems, by the UPS and autophagy.Citation5 Soluble SNCA is degraded by the UPS pathwayCitation6,Citation7 and by chaperone-mediated autophagy (CMA).Citation8 Nevertheless, studies in yeast and cell line models show that insoluble SNCA and SNCA aggregates interact with the proteasome leading to its inhibition,Citation7,Citation9-Citation11 an observation also corroborated by assays performed in the substantia nigra of PD brains.Citation12 Mutant and dopamine-oxidized forms of SNCA also impair the CMA pathway leading to an upregulation of macroautophagy (autophagy).Citation4,Citation13 Thus, dysfunctional clearance of SNCA and, consequently, impairment of protein quality control systems, particularly autophagy, has become a prominent pathogenic mechanism underlying neurodegeneration.Citation4 Mitochondrial dysfunction and oxidative stress have also been implicated in the pathogenesis of PD. Dysfunctional mitochondria are observed in the substantia nigra of PD patients; it was recently proposed that this could be a consequence of the loss of mitochondria due to overactivation of autophagy.Citation14 However, little is known about the factors that predispose mitochondria for degradation by mitophagy. Studies on genes associated with autosomal recessive forms of PD demonstrate that PINK1 and PARK2 collaborate in mitochondrial quality control systems.Citation15-Citation17 PARK2 is targeted to mitochondria to induce autophagy of damaged mitochondriaCitation16 and PINK1 is a mitochondria-targeted serine-threonine kinase that assists in the translocation of PARK2 into mitochondria.Citation15 Hence, defects in mitophagy have recently emerged as a central pathogenic hypothesis in PD.Citation13, Citation18,Citation19 Nevertheless, the precise role of autophagy and mitophagy in the pathogenesis of PD still remains controversial.Citation18 Although existing data suggests that SNCA inhibits mitochondrial fusion,Citation20 mitochondrial functionCitation21 and autophagy efficiencyCitation22 while increasing mitophagy,Citation21 several additional questions arise. Therefore, it is crucial to understand the mechanisms underlying autophagy and mitophagy in relation to SNCA toxicity. Here, using the simple and powerful S. cerevisiae model organism, we aimed to elucidate the involvement of autophagy and mitophagy in the toxicity induced by wild-type SNCA (SNCA WT) or the A53T mutant during yeast chronological life span. Our data show that SNCA-induced toxicity stimulates mitophagy and shortens yeast CLS. Deletion of the mitophagy-specific gene ATG32, which leads to mitophagy impairment,Citation23 rescues cells from SNCA-induced toxicity. Our data strongly suggest that SNCA toxicity is dependent on Sir2, the yeast homolog of mammalian SIRT1, which specifically stimulates autophagy/mitophagy in aged cells. Altogether, our study indicates that Sir2 is an essential mediator of SNCA toxicity through its control of autophagy/mitophagy.
Results
Autophagy modulation rescues aged cells from SNCA-induced toxicity
Yeast cells have been extensively used as a model to study the cytotoxic effects of SNCA.Citation24 Previous data show SNCA-mediated cell death in yeast through an age- and mitochondria-dependent process,Citation36 which is in good agreement with the late onset of the vast majority of PD cases. Nevertheless, it is still not clear how age traits potentiate the cytotoxic effects of SNCA. To address this question, SNCA expression was induced from a Tet-On promoter in different phases of yeast growth (exponential, diauxic or stationary) and chronological life span was determined. As expected, CLS analysis demonstrated that the toxic effects of SNCA expression were dependent on the growth phase (). A significant CLS reduction was observed after SNCA expression, ranging from 25% in exponential phase cells to 28.60% and 41.66% in diauxic and stationary phase cells, respectively.
Figure 1. Autophagy modulation rescues aged cells from SNCA-induced toxicity. Chronological life span of wild-type cells expressing SNCA WT, under the control of a Tet-On promoter, or harboring the vector control. SNCA WT expression was induced at (A) exponential, (B) diauxic or (C) stationary growth phases and cell viability was measured at 2–3 d intervals. SNCA WT levels in wild-type cells in which expression was induced at the different growth phases, (D) exponential, (E) diauxic or (F) stationary. (G) Chronological life span of stationary wild-type cells expressing SNCA WT in the presence or absence of chloroquine (CQ), an inhibitor of autophagy. The data represent mean ± SEM of three biological independent replicas. Significance of the chronological life span curves was determined by two-way ANOVA (***p < 0.001).
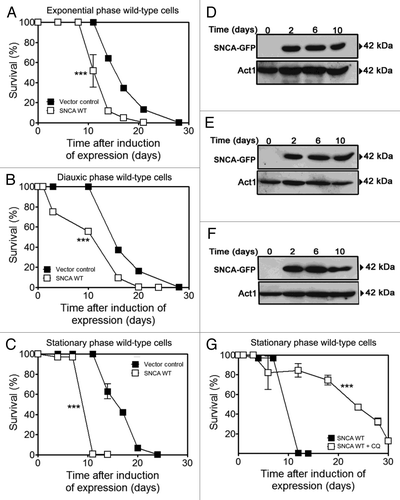
Numerous studies propose that different signaling pathways involved in regulating aging converge on autophagy due to the inability of post-mitotic cells to get rid of the “metabolic waste.”Citation26 Although autophagy also plays a relevant role in SNCA-induced toxicity in different models, the question of whether autophagy induction would have a cytoprotective role remains to be elucidated.Citation14,Citation27 Thus, we set out to determine whether autophagy modulation in aged yeast cells expressing SNCA WT would have an impact on SNCA toxicity and longevity. To this end, SNCA expression was induced in stationary phase cells (day 0 of CLS) and autophagy was pharmacologically inhibited by adding chloroquine (CQ) that acts at the terminal step of the autophagic pathway by increasing lysosomal/vacuolar pH and inhibiting lysosome/vacuole-autophagosome fusion.Citation28 The results obtained showed that autophagy inhibition in aged yeast cells resulted in decreased SNCA toxicity as reflected by CLS extension (), thereby demonstrating an unexpected relationship between autophagy and SNCA increased toxicity in aged yeast cells.
SNCA-induced toxicity in aged cells is associated with selective degradation of mitochondria
To further clarify the role of autophagy on the SNCA-induced toxicity in aged yeast cells, we decided to determine the levels of selected ATG genes, as well as the autophagic activity in SNCA models with different toxicity levels. SNCA can be expressed from either constitutive or regulable promoters resulting in different levels of expression and toxicity.Citation29 Here, we used two different systems: (1) a high-toxicity model that consists of the expression of SNCA under the control of a GAL1-inducible promoter in stationary growth phase cells, with two copies of the construct integrated in the genome; and (2) a moderate-toxicity model in which SNCA is constitutively expressed under the control of an endogenous triose phosphate isomerase (TPI1) promoter from a single integrated copy (see Materials and Methods section).
As expected, the expression of SNCA WT or the familial mutant SNCA A53T by the high toxicity system leads to only 10% of survival, compared with about 100% survival of cells harboring the vector control or the SNCA A30P nontoxic mutantCitation30 after 30 h of induction (). The analysis of the relative mRNA expression levels of the ATG8 gene, encoding a protein that remains attached to autophagosome structures,Citation31 and that is transcriptionally regulated in yeast,Citation32,Citation33 revealed that mRNA expression levels were already 2-fold increased after 12 h of SNCA WT or A53T expression when compared with cells expressing the vector control or the nontoxic A30P (). At 24 h of SNCA WT or A53T expression the ATG8 mRNA levels were even higher (), showing that the toxicity and CLS reduction promoted by expression of SNCA WT or A53T are associated with an increase of ATG8 mRNA levels.
Figure 2. SNCA-induced toxicity in aged cells is associated with selective degradation of mitochondria. (A) Chronological life span and SNCA levels of wild-type cells expressing the vector control, the SNCA WT, the A53T or the A30P mutant under the control of the GAL1 promoter, a high toxicity model. (B and D) ATG8 and ATG32 mRNA relative expression levels at 0, 12 and 24 h after SNCA-induced expression. Three reference genes [ACT1 (actin), PDA1 (α subunit of pyruvate dehydrogenase) and TDH2 (isoform 2 of glyceraldehyde-3-phosphate dehydrogenase)] were used as internal standards and for the normalization of mRNA expression levels. (C) The splicing activation of the HAC1 mRNA at 0, 12 and 24 h after SNCA-induced expression. (E) Chronological life span and SNCA levels of wild-type cells expressing the vector control, the SNCA WT, the A53T or the A30P mutant under the control of the constitutive TPI1 promoter, a moderate toxicity model. The data represent mean ± SEM of six biological independent replicas. Cell viability was measured at 2–3 d intervals beginning at the day that cultures achieved stationary phase (day 0) and is expressed as % survival compared with survival at day 0 (100%). Autophagic and mitophagic activity were measured through the alkaline phosphatase assay that was performed in the wild-type cells expressing the vector control, the SNCA WT, the A53T or the A30P mutant and co-harboring a plasmid expressing the inactive Pho8 proenzyme targeted to (F) the cytosol or (G) to the mitochondrial matrix. The error bars represent the standard error of the mean (SEM). Significance of the data was determined by two-way ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001).
![Figure 2. SNCA-induced toxicity in aged cells is associated with selective degradation of mitochondria. (A) Chronological life span and SNCA levels of wild-type cells expressing the vector control, the SNCA WT, the A53T or the A30P mutant under the control of the GAL1 promoter, a high toxicity model. (B and D) ATG8 and ATG32 mRNA relative expression levels at 0, 12 and 24 h after SNCA-induced expression. Three reference genes [ACT1 (actin), PDA1 (α subunit of pyruvate dehydrogenase) and TDH2 (isoform 2 of glyceraldehyde-3-phosphate dehydrogenase)] were used as internal standards and for the normalization of mRNA expression levels. (C) The splicing activation of the HAC1 mRNA at 0, 12 and 24 h after SNCA-induced expression. (E) Chronological life span and SNCA levels of wild-type cells expressing the vector control, the SNCA WT, the A53T or the A30P mutant under the control of the constitutive TPI1 promoter, a moderate toxicity model. The data represent mean ± SEM of six biological independent replicas. Cell viability was measured at 2–3 d intervals beginning at the day that cultures achieved stationary phase (day 0) and is expressed as % survival compared with survival at day 0 (100%). Autophagic and mitophagic activity were measured through the alkaline phosphatase assay that was performed in the wild-type cells expressing the vector control, the SNCA WT, the A53T or the A30P mutant and co-harboring a plasmid expressing the inactive Pho8 proenzyme targeted to (F) the cytosol or (G) to the mitochondrial matrix. The error bars represent the standard error of the mean (SEM). Significance of the data was determined by two-way ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001).](/cms/asset/f6072eec-3535-45c8-83e9-fae538abadc9/kaup_a_10921275_f0002.gif)
Previous data have demonstrated that endoplasmic reticulum (ER) stress upregulates the transcription of numerous genes, including genes related to autophagy, like ATG8 and ATG14,Citation34 and that autophagy is important for survival during a sustained unfolded protein response (UPR).Citation35 Due to the observed induction of the ATG8 mRNA levels, we also decided to evaluate the state of the Ire1-Hac1 signaling pathway by assessing the splicing activation of the HAC1 mRNA after the induction of heterologous expression of SNCA. The data showed that at 12 and 24 h, the cells expressing SNCA WT or A53T displayed HAC1 mRNA splicing (). These results indicate that SNCA toxicity induced a prolonged and sustained UPR response that could underlie the increase in ATG8 mRNA levels.
Oxidative stress and mitochondrial dysfunction are hallmarks of SNCA-induced toxicity in aged cellsCitation36 and mitophagy seems to be a crucial route to eliminate the dysfunctional mitochondria.Citation37 Thus, we also determined the relative mRNA levels of ATG32, which encodes a protein specifically involved in mitophagy.Citation38 The results showed that ATG32 mRNA expression levels were significantly increased (2-fold) at 12 and 24 h in aged cells expressing SNCA WT or A53T () suggesting that the expression of SNCA leads to autophagy induction, specifically to mitochondria-selective degradation.
To further determine whether the increased ATG8 and ATG32 mRNA levels result in increased autophagy and mitophagy activities, we used an expression system with moderate toxicity to perform additional functional analyses (). For that purpose, PHO8 mutant cells were transformed with a plasmid expressing an inactive Pho8 proenzyme targeted to the cytosol, to assess autophagy, or carrying an inactive Pho8 proenzyme targeted to the mitochondrial matrix to determine mitophagy.Citation39,Citation40 The data from the alkaline phosphatase (ALP) assay showed that the heterologous expression of SNCA WT or A53T induced an increase of the autophagic activity over time when compared with vector control cells or cells expressing the nontoxic A30P (). The same profile was observed with the ALP assay directly correlated with mitophagy activity ().
Altogether, the results obtained with the different SNCA expression and toxicity systems suggested that the expression of SNCA WT and A53T results in a sustained UPR response that is correlated with an activation of autophagy with selective degradation of mitochondria, as demonstrated by the mitophagy ALP assay and the increased ATG32 mRNA levels.
SNCA-induced toxicity is mitophagy dependent
The role of mitophagy in the SNCA-induced toxicity is still controversial.Citation4 To get new insight into the role of mitophagy in SNCA-induced toxicity we extended our analysis to the evaluation of the CLS in cells lacking genes associated with specific types of autophagy, namely, ATG11 and ATG32. Atg11 is a scaffold protein that directs receptor (Atg19)-bound cargo to the phagophore assembly site and that participates in all specific types of autophagy.Citation41 Atg32, is a mitochondrial protein that confers selectivity to the sequestration and recruitment of mitochondria by the autophagy machinery.Citation42
In atg11Δ cells, the expression of SNCA WT and A53T no longer had an effect on longevity, since the CLS curves overlapped with the curve obtained for vector control cells or even presented an increased survival percentage as shown for SNCA A53T (). The lack of effect of SNCA WT and A53T expression in atg11Δ cell survival was also confirmed by different viability assays such as plasma membrane integrity, assessed by the exclusion of propidium iodide (PI), and by the metabolic activity evaluated by FUN1 (Fig. S1). This clearly demonstrated that selective autophagy, the process abolished in atg11Δ cells, is associated with the toxicity of SNCA in aged yeast cells. Interestingly, expression of the nontoxic SNCA A30P induced a slight decrease of longevity, compared with atg11Δ cells expressing the vector control, particularly when viability was evaluated by PI exclusion or FUN1 processing (; Fig. S1B and S1F). This most likely relates to the previously made observations that the A30P mutant is predominantly cytoplasmicCitation43 and subject to vacuolar degradation, in contrast to SNCA WT and A53T.Citation44 It is conceivable that the increased toxicity of the A30P mutant in ATG11 deleted cells is associated with a general defect in cargo recognition given the role of Atg11 in connecting the cargo molecules with components of the vesicle-forming machinery.Citation45,Citation46
Figure 3. SNCA-induced toxicity is mitophagy dependent. (A and B) Chronological life span and (C and D) SNCA levels of atg11Δ and atg32Δ cells, respectively, expressing the vector control, the SNCA WT, the A53T or the A30P mutant under the control of the constitutive TPI1 promoter. Cell viability was measured at 2–3 d intervals beginning at the day that cultures achieved stationary phase (day 0) and is expressed as % survival compared with survival at day 0 (100%). The data represents mean ± SEM of six biological independent replicas. (E and F) Mean (50% survival) and maximum (10% survival) chronological life spans were determined from curve fitting of the survival data (A and B) (from pair matched, pooled experiments) with the statistical software Prism (GraphPad Software). Significance was determined between wild-type cells and atg11Δ or atg32Δ cells expressing vector control or SNCA variants (*). The significance determined between cells expressing the vector control or SNCA variants within each strain (wild-type, atg11Δ or atg32Δ) was also determined (#). The alkaline phosphatase assay was performed to assess (G and H) autophagy or (I and J) mitophagy in atg11Δ or atg32Δ cells, respectively. The error bars represent the standard error of the mean (SEM). Significance of the data was determined by two-way ANOVA (#p < 0.05; *p < 0.05; **p < 0.01; ***p < 0.001).
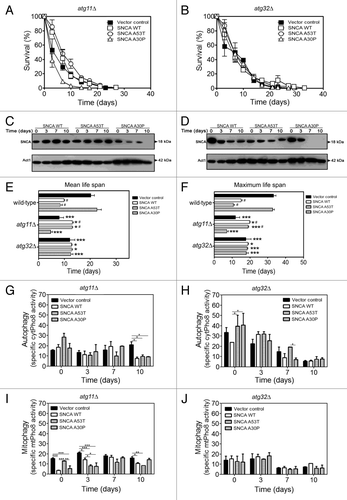
Consistently, and as expected, the ALP assay indicated that atg11Δ cells displayed slightly higher levels of autophagic activity, as demonstrated by the ALP assay of vector control atg11Δ cells (), compared with vector control wild-type cells (). Nevertheless, the observed induction of autophagy in wild-type cells () upon expression of the different SNCA variants was abrogated to basal levels in atg11Δ cells (). Importantly, mitophagy upon expression of the different SNCA variants was abrogated in atg11Δ cells ().
Next, we addressed the effect of SNCA expression in CLS of atg32Δ cells. Similarly to what we observed for the atg11Δ cells, the longevity of atg32Δ cells harboring the vector control or expressing A30P was reduced when compared with wild-type cells (compare and ), and the expression of SNCA WT or A53T no longer triggered a toxic effect (). These data were also confirmed by PI and FUN1 staining (Fig. S1). Although, as expected due to the inhibition of mitophagy, atg32Δ cells displayed higher autophagic activity and these levels decreased during aging both in vector control and in cells expressing the different SNCA variants ( and ). Furthermore, the ALP assay indicated that expression of the different SNCA variants in atg32Δ cells no longer induced mitophagy ().
The analysis of cell survival by different methods (CFUs, PI and FUN1 staining) allowed the determination of the mean (50% survival) and maximum (10% survival) chronological life spans of wild-type and the mutant strains. The results showed that the expression of SNCA WT and A53T in atg11Δ and atg32Δ cells promoted a statistically significant increase of the mean and maximum life span when compared with wild-type cells (). To test the hypothesis that lower toxicity of SNCA WT and A53T expression may be not detected due to an inherent mitochondrial dysfunction of atg11Δ and atg32Δ cells, we determined the index of respiratory competence (IRC).Citation47 The results obtained showed that atg32Δ cells are able to grow on glycerol and present an IRC similar to wild-type cells (Table S1). atg11Δ cells also displayed an IRC similar to wild-type cells, nevertheless, mitochondrial functionality of these mutant cells started to be affected at 7 d of CLS (Table S1). Moreover, the data also revealed that SNCA WT and A53T expression did not negatively affect the IRC of atg11Δ and atg32Δ cells but, instead, increased their IRC when compared with wild-type cells (Table S1). Altogether, the results strongly suggest that the lack of SNCA WT and A53T toxicity is not associated with mitochondrial dysfunction of atg11Δ and atg32Δ cells.
When combined, our data confirm that the toxicity of SNCA WT and the A53T mutant as observed in wild-type cells was dependent on Atg11 and Atg32, two factors required for the induction of mitophagy.
To further corroborate the biochemical data obtained with the ALP assay, we assessed mitophagy by microscopy visualization of the process. Wild-type, atg11∆ and atg32∆ mutant cells expressing SNCA toxic forms, GFP-Atg8, mtDsRed (mitochondrially-targeted DsRed) and immunostained for SNCA, were analyzed by confocal microscopy. In wild-type cells harboring the vector control, GFP-Atg8 was distributed in the cytosol with a punctate pattern, and mtDsRed labeled a typical mitochondrial network (). The expression of SNCA WT resulted in the accumulation of GFP in the vacuole in the vast majority of the cells, indicating that GFP-Atg8 was shuttled to the vacuole and that autophagy was induced. Furthermore, the vast majority of the cells did not present the mitochondrial mtDsRed signal, and in those cases where red fluorescence was observed, the typical mitochondrial network was abolished (). The same results were obtained for yeast cells expressing the A53T mutant (data no shown). In contrast, in atg11Δ and atg32Δ cells expressing the SNCA toxic forms, the GFP-Atg8 was essentially distributed in the cytosol, and the mitochondrial network remained detectable although we observed some mitochondrial fragmentation (). The cells expressing the nontoxic A30P mutant produced similar results as those obtained with the cells harboring the vector control (data no shown). Cells expressing SNCA WT or A53T displayed the typical formation of SNCA foci (), and the number of foci seemed to be independent of the abrogation of mitophagy by the deletion of the ATG11 and ATG32 genes, and this was despite the decreased toxicity of SNCA in these mutant cells ().
Figure 4. Mitophagy is involved in the SNCA-induced toxicity. Wild-type, atg11Δ and atg32Δ cells expressing, (A) mtDsRed and GFP-Atg8 and (B) SNCA WT were analyzed for mitophagy and autophagy by confocal fluorescence microscopy. SNCA WT was analyzed after incubation with antibody against SNCA as the primary and the Pacific Blue conjugated goat anti-rabbit secondary antibody. Single confocal planes are shown. Scale bars: 5 µm. (C) Foci number was quantified in wild-type, atg11Δ and atg32Δ cells expressing SNCA WT. The error bars represent the standard error of the mean (SEM).
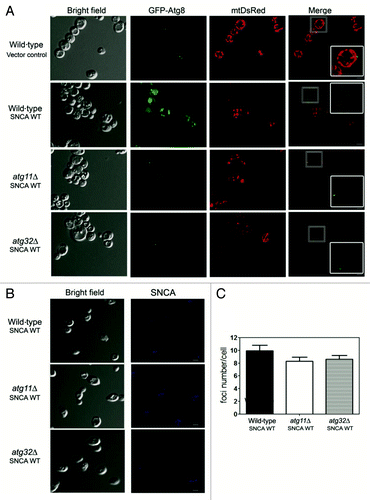
Hence, the results indicate that in control conditions mitophagy is important to maintain the homeostasis of aged cells. In contrast, under conditions of SNCA toxicity, mitophagy appears to result in increased toxicity rather than in cellular protection for the aged cells. Thus, mitophagy mediates SNCA-induced toxicity contributing to the reduction of CLS.
Superoxide anion accumulation under SNCA-induced toxicity is decreased in cells with impaired mitophagy
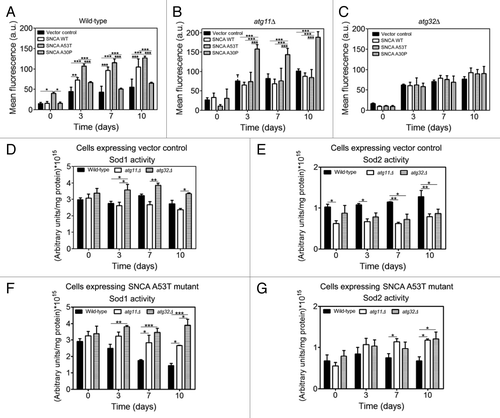
Previously it was shown that SNCA expression triggers intracellular accumulation of reactive oxygen species (ROS) and mitochondrial dysfunction in yeast.Citation36 ROS are signaling molecules implicated in several cellular programs during both physiological and pathological conditions.Citation48 Recent findings established a role for superoxide anion (O22−), in the regulation of autophagy,Citation49 particularly in conditions of prolonged starvation.Citation48 Here, we examined whether induction of autophagy/mitophagy, due to expression of SNCA variants, was mediated by the accumulation of O22−. As such, we detected the accumulation of O22− during CLS by using the fluorescent probe dihydroethidium (DHE).Citation50 Flow cytometry analysis revealed that wild-type cells expressing SNCA WT or A53T displayed a significant increase of DHE fluorescence intensity over time, as compared with the wild-type cells harboring the vector control or expressing the nontoxic A30P mutant (). In contrast, the expression of SNCA WT or A53T in cells lacking either the ATG11 or ATG32 gene did not result in such increased accumulation of O22− (), although elevated levels of DHE fluorescence were observed in the atg11∆ cells expressing the nontoxic A30P, consistent with the reduced CLS of these cells ( and ). Consistently, in vector control cells, corresponding to 2% glucose grown cells, a condition known to induce the accumulation of O22−, the abrogation of ATG11 or ATG32 resulted in increased accumulation of O22− (). To further explore the reasons underlying the decrease in O22− accumulation displayed by cells with an impairment of mitophagy, we have determined the activity of superoxide dismutases, both cytosolic Cu/Zn-dependent (Sod1) and the mitochondrial Mn-dependent superoxide dismutases (Sod2), known to inhibit the accumulation of superoxide anions. The results showed that while atg11Δ cells expressing the vector control displayed Sod1 activity comparable to wild-type cells, atg32Δ cells presented a significant increase in Sod1 activity (). Nevertheless, both atg11Δ and atg32Δ cells had lower basal levels of Sod2 activity, that do not increase over the CLS (). The expression of SNCA A53T in wild-type cells led to a decrease of Sod1 and a maintenance of Sod2 activity over the CLS time evaluated (). In contrast, the expression of SNCA A53T did not affect the activity of Sod1 in atg32Δ cells, and promoted a slight increase of this activity in atg11Δ cells, while it increased the Sod2 activity (). Hence, the ability of atg11Δ and atg32Δ cells to maintain Sod1 activity and to increase Sod2 over the CLS when expressing SNCA WT (data not shown) or A53T () could be responsible for the decreased accumulation of O22− observed (). Note that these data on the oxidative status of atg11Δ and atg32Δ cells are in agreement with the respiratory competence displayed by these cells (Table S1).
Figure 5. Superoxide anion accumulation under SNCA-induced toxicity is decreased in cells with impaired mitophagy. During chronological life span of (A) wild-type, (B) atg11Δ and (C) atg32Δ cells, expressing the vector control, the SNCA WT, the A53T or the A30P mutant forms under the control of the constitutive TPI1 promoter, the accumulation of superoxide anion was evaluated by flow cytometry, using the fluorescent probe dihydroethidium (DHE). The error bars represent the standard error of the mean (SEM). (D–G) Superoxide dismutase activity was analyzed in native gels. Bands corresponding to Sod1 activity of cells expressing vector control (D) or SNCA A53T mutant (F) and Sod2 activity of cells expressing vector control (E) or SNCA A53T mutant (G) were measured with a densitometer and the relative intensities calculated (Quantity One software, BioRad). Values indicate mean ± SEM from three independent experiments. Significance of the values between yeast strains was determined by two-way ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001).
SIR2 mediates autophagy/mitophagy induction under SNCA-induced toxicity
Autophagy induction can be mediated by several pathways. Earlier studies in mammalian cells showed that the human homolog of yeast sirtuin 2 (Sir2), SIRT1, is required to sustain autophagy in response to nutrient starvation through deacetylation of autophagic regulators, including ATG5, ATG7, ATG8 and ATG12.Citation51 Recent studies have also shown that in yeast cells SNCA toxicity is Sir2 dependent.Citation25 Altogether, these findings led us to test if Sir2 promoted autophagy/mitophagy in response to SNCA toxicity. In agreement with the observations made previously,Citation25 we detected a CLS extension of sir2Δ cells expressing SNCA WT or A53T as compared with wild-type cells expressing the same proteins (). The CLS extension of sir2Δ cells expressing SNCA WT or A53T was also validated by the analysis of PI exclusion and FUN1 metabolic processing (Fig. S1). Interestingly, the deletion of SIR2 in cells expressing the A30P nontoxic mutant resulted in a shorter CLS, which is in accordance with the data obtained with the atg11Δ cells. For the cells carrying the vector control, the SIR2 deletion did not alter the CLS in comparison to the wild-type cells. These results are in agreement with the previous report showing that SIR2 deletion suppresses the toxicity induced by SNCA.Citation25 Thus, the next question was whether Sir2 mediates autophagy/mitophagy induction upon SNCA expression. The results showed that SIR2 deletion ameliorates SNCA toxicity due to an autophagy/mitophagy inhibition as reflected by the abolishment of the ALP activity in sir2Δ cells (). These results were confirmed by the visual assay for mitophagy in sir2Δ cells where GFP-Atg8 was essentially found distributed in the cytosol independently of the expression of SNCA variants, and mitochondrial mtDsRed marked a mitochondrial network (). Furthermore, analysis of the ATG8 and ATG32 relative mRNA levels in sir2Δ cells, expressing SNCA WT or A53T, revealed that their expression is strongly dependent on Sir2 in stationary phase cells, particularly the expression of ATG32 (). Thus, our data strongly supports the idea that SNCA toxicity is dependent on Sir2, an essential mediator of autophagy/mitophagy through the transcriptional regulation of ATG8 and ATG32 in stationary phase cells expressing SNCA toxic variants.
Figure 6. Sir2 mediates autophagy/mitophagy induction under SNCA-induced toxicity. (A) Chronological life span and (B) SNCA levels of sir2Δ cells expressing the vector control, the SNCA WT, the A53T or the A30P mutant under the control of the constitutive TPI1 promoter. Cell viability was measured at 2–3 d intervals beginning at the day that cultures achieved stationary phase (day 0) and is expressed as % survival compared with survival at day 0 (100%). The alkaline phosphatase assay was performed to assess (C) autophagy or (D) mitophagy, respectively. (E) sir2Δ cells expressing mtDsRed and GFP-Atg8 were analyzed for mitophagy and autophagy by confocal fluorescence microscopy. Single confocal planes are shown. Scale bars: 5 µm. The error bars represent the standard error of the mean (SEM). (F–I) Relative ATG8 and ATG32 mRNA levels in stationary phase wild-type and sir2Δ cells expressing SNCA at day 0, 3, 7 and 10. Three reference genes [ACT1 (actin), PDA1 (α subunit of pyruvate dehydrogenase) and TDH2 (isoform 2 of glyceraldehyde-3-phosphate dehydrogenase)] were used as internal standards and for the normalization of mRNA expression levels. Significance of the data was determined by two-way ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001).
![Figure 6. Sir2 mediates autophagy/mitophagy induction under SNCA-induced toxicity. (A) Chronological life span and (B) SNCA levels of sir2Δ cells expressing the vector control, the SNCA WT, the A53T or the A30P mutant under the control of the constitutive TPI1 promoter. Cell viability was measured at 2–3 d intervals beginning at the day that cultures achieved stationary phase (day 0) and is expressed as % survival compared with survival at day 0 (100%). The alkaline phosphatase assay was performed to assess (C) autophagy or (D) mitophagy, respectively. (E) sir2Δ cells expressing mtDsRed and GFP-Atg8 were analyzed for mitophagy and autophagy by confocal fluorescence microscopy. Single confocal planes are shown. Scale bars: 5 µm. The error bars represent the standard error of the mean (SEM). (F–I) Relative ATG8 and ATG32 mRNA levels in stationary phase wild-type and sir2Δ cells expressing SNCA at day 0, 3, 7 and 10. Three reference genes [ACT1 (actin), PDA1 (α subunit of pyruvate dehydrogenase) and TDH2 (isoform 2 of glyceraldehyde-3-phosphate dehydrogenase)] were used as internal standards and for the normalization of mRNA expression levels. Significance of the data was determined by two-way ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001).](/cms/asset/dea99e17-be2c-4fc5-9455-264a77a97b70/kaup_a_10921275_f0006.gif)
Discussion
It has long been puzzling why protein aggregation and neurotoxicity develop late in life, even in familial forms of neurodegenerative disease where mutant proteins are present throughout life. To address this issue, we expressed human SNCA in yeast cells, a powerful model for investigating the cellular and molecular basis of SNCA toxicity.Citation24 In addition, this model has the advantage of being an established system for aging research.Citation52 Here, we present data showing that expression of SNCA WT or A53T mutant in aged yeast cells results in shortening of CLS and increased SNCA toxicity. Aging is, by far, the strongest risk factor for developing neurodegenerative diseases.Citation53 This is thought to be due to the age-related decline and dysfunction of fundamental housekeeping processes that are crucial for maintaining cellular homeostasis, such as autophagy,Citation54,Citation55 which contributes to the accumulation of misfolded proteins.Citation56 Our data show that pharmacological inhibition of autophagy, with chloroquine, rescues aged yeast cells from SNCA-induced toxicity. Thus, we hypothesized that the decline of autophagy, its function and/or selectivity, during aging might aggravate SNCA toxicity. Our findings confirmed that cells expressing PD-causing variants of SNCA displayed increased autophagic activity, which strongly supports our hypothesis and establishes an association between increased autophagic activity and SNCA toxicity in aged cells. Notably, it was demonstrated that SNCA expression in mammalian cells impairs chaperone-mediated autophagy and induces the upregulation of macroautophagy, which seems to contribute to neuronal cell death.Citation57 Although increased autophagic activity can mediate SNCA clearance in functionally competent cellsCitation58 and thereby have anti-aging effects,Citation59 it might also affect autophagy efficiency and selectivity in aged cells.Citation4 The controversial role of autophagy is further accompanied by oxidative stress and mitochondrial dysfunction, which play a central role in SNCA-induced toxicity.Citation36,Citation60 Although the relevance of mitophagy, induced by mitochondria-damaging agents, for the etiology of PD remains debatable,Citation18 accumulating evidence on the key role of the PD-associated proteins PINK1 and PARK2 in targeting specific dysfunctional mitochondria for degradation by mitophagy,Citation4 supports the interconnection of these cellular processes and their involvement in the pathogenesis of PD.
Based on those findings, we wondered if dysfunctional mitochondria were being targeted for degradation by the autophagic machinery. Our results showed that SNCA expression induces mitophagy and that deletion of ATG11 or ATG32, both required for mitophagy, prevents SNCA-induced toxicity as revealed by increased mean and maximum chronological life spans. Furthermore, the decreased SNCA toxicity observed in cells with an impairment of mitophagy was not due to an inherent mitochondria dysfunction of ATG11 or ATG32 mutant cells, although these mutant cells display mitochondria dysfunction when subjected to severe nitrogen starvation.Citation61
To elucidate the pathways/molecules associated with SNCA-induced autophagy/mitophagy activation during aging, the unfolded protein response and reactive oxygen species were investigated. Our findings demonstrate that a sustained UPR, elicited by SNCA, could underlie the observed stimulation of autophagy/mitophagy, which is reminiscent of observations made in mouse models of synucleinopathy.Citation62 ROS, particularly superoxide anions, are crucial signaling molecules implicated in the control and regulation of autophagy and aging progression. Our work does not rule out superoxide anions as mediators of autophagy/mitophagy due to SNCA accumulationCitation63 and aging.Citation64 In addition, our data suggest that the sustained superoxide dismutase activities observed over CLS in atg11∆ or atg32∆ cells could be the mechanism behind the decreased superoxide anion levels displayed by these cells.
Mammalian SIRT1 was recently described as an inducer of autophagy under starvation conditions.Citation65 Here, we investigated such a role for the yeast homolog, Sir2. Our findings revealed that deletion of the SIR2 gene not only alleviated SNCA toxicity, as previously described,Citation25 and as evidenced by the increased CLS, but also that this phenomenon is linked to a drastic inhibition of autophagy and mitophagy as demonstrated by ALP assay. Notably, Sir2 was shown to be essential for the regulation of ATG8, and particularly, ATG32 mRNA levels in stationary phase cells expressing SNCA toxic variants.
In summary, our data show that increased autophagy/mitophagy, promoted by SNCA WT and A53T mutant, has a deleterious effect in aged cells. This appears counterintuitive as autophagy and mitophagy are considered to represent prosurvival processes. Nevertheless, several explanations could be envisioned to explain how impaired mitophagy due to the presence of toxic SNCA may extend CLS. One possible explanation is that aged cells have a reduced ability to simultaneously upregulate anabolic processes in order to compensate for the loss of cellular material by increased mitophagy. Another conceivable hypothesis is that the selectivity of various types of autophagy, such as mitophagy, is lost and one or more factors, commonly required for various types of autophagy, may become limiting when mitophagy is upregulated. The loss of selectivity could also result in the degradation of functional competent mitochondria, crucial to sustain cell survival of postmitotic cells, contributing to the toxicity observed. Our findings further reveal that autophagy and mitophagy are mainly regulated through Sir2, in agreement with the role that alterations on the acetylproteome have on the regulation of autophagy.Citation66,Citation67 Future studies will have to dissect how different pathways of cellular quality control influence each other and how they are regulated in a coordinated manner.
Material and Methods
Strains and media
The yeast strains and plasmids used in this study are listed in and , respectively. All yeast cultures were inoculated in selective SC medium, containing 0.67% (w/v) yeast nitrogen base (Difco Laboratories, 291940), 2% (w/v) glucose (Fagron, 3094-12) as carbon source, supplemented with the appropriate amino acids: 100 mg/L uracil (Sigma, U0750), 300 mg/L leucine (Sigma, L8000), 50 mg/L histidine (Sigma, H8000),100 mg/L adenine (Sigma, A8626), 100 mg/L tryptophan (Sigma, T0254) or 100 mg/L methionine (Sigma, M9625). Depending on the expression model used, different manipulations were performed. For the Tet-On models, cells were grown at 26°C and 150 rpm, until exponential, diauxic and stationary phases, and then doxycycline (Sigma, D9891) was added to the medium at a final concentration of 2 µg/ml in order to induce the expression of SNCA. To perform the autophagy inhibition assay using the Tet-On models, the cells were grown until stationary phase and doxycycline was added together with 0.50 mM chloroquine (Sigma, C6628). For the galactose-inducible (high toxicity) models, cells were grown until stationary phase, harvested by centrifugation, washed twice with sterile water and then transferred to a SC medium containing 2% (w/v) galactose (Applichem, A113) to induce SNCA expression. Finally, the cells expressing SNCA under the control of the constitutive TPI1 promoter (moderate toxicity model) were grown until stationary phase.
Table 1. Yeast strains used in this study
Table 2. Plasmids used in this study
The survival of the cells from the inducible expression systems, Tet-On and GAL1, was assessed by counting colony-forming units (CFUs) after incubation of culture aliquots for 2 d at 30°C on YEPD agar plates. Regarding the moderate expressing system (constitutive TPI1 promoter), cultures reached stationary phase two days later and this was considered day 0 of chronological life span (CLS). Survival was assessed by CFUs beginning at day 0 of CLS (when viability was considered to be 100%), and then again every 2–3 d until less than 0.01% of the cells in the culture were viable.
Index of respiratory competence (IRC)
The IRC reflects the respiratory status of mitochondria and thus mitochondrial functionality. It can be defined as the ability of cells to grow on nonfermentable substrates such as glycerol (Merck, 104094). The IRC was calculated by the ratio between the number of CFUs on nonfermentable carbon source (yeast extract (Difco Laboratories, 212750)/peptone (Difco Laboratories, 211677) 2% glycerol, YPG) and the colonies on a fermentable carbon source (YEPD) agar plates.Citation47
Superoxide dismutase assays
For determination of superoxide dismutase activities, yeast extracts were prepared in 25 mM Tris buffer (pH 7.4) (Sigma, T1503) containing a cocktail of protease inhibitors (Sigma, P8215). Protein content of cellular extracts was estimated by the method of Bradford (Bio-Rad, 500-0006) using bovine serum albumin (Sigma, A7906) as a standard. Superoxide dismutase activities were measured based on their ability to inhibit reduction of nitro blue tetrazolium (Sigma, N6876) to formazan in nondenaturing polyacrylamide gels.Citation68 Sod2 activity was distinguished from Sod1 activity based on the ability of 2 mM cyanide (Sigma, 60178) to inhibit Sod1, but not Sod2. Quantification of band intensities was performed by densitometry using Quantity One Basic Software from Bio-Rad.
Quantitative mRNA expression
The quantitative mRNA expression analysis was performed according to the MIQE guidelines (Minimum Information for publication of Quantitative real-time PCR ExperimentsCitation69). Briefly, quantitative real-time PCR (qPCR) was used to measure the mRNA transcripts of the ATG8 and ATG32 genes. Three reference genes [ACT1 (actin), PDA1 (α subunit of pyruvate dehydrogenase) and TDH2 (isoform 2 of glyceraldehyde-3-phosphate dehydrogenase)] were selected due to their stable expression and were tested in the same experimental conditions allowing expression normalization.
Yeast samples for qPCR quantification were centrifuged (4,500 rpm, 4°C, 5 min) and the pellets were immediately stored at -80°C until RNA extraction. The RNA extraction was performed as previously described.Citation69 The RNA quality was assessed by agarose (Bioron, 604005) gel electrophoresis. Primers for qPCR were constructed, by in silico analysis, using Beacon Designer 7.90 software (PremierBiosoft International), and are listed in . Total RNA (300 ng) was reverse-transcribed into cDNA in a 20 μL reaction mixture using the iScript™ cDNA synthesis kit (Bio-Rad, 170-8841). Then, 22.5 ng of cDNA of each sample was tested in duplicate in a 96-well plate (Bio-Rad), in a 20-μL reaction mixture using the SsoFast Evagreen Supermix™ kit (Bio-Rad, 172-5201) and processed according to the manufacturer’s instructions in a CFX96™ Real Time System (Bio-Rad). A blank (no template control) was also incorporated in each assay. The thermocycling program consisted of one hold at 95°C for 1 min, followed by 39 cycles of 15 min at 95°C, 20 sec at 57°C and 20 sec at 72°C. After completion of these cycles, melting-curve data were collected to verify PCR specificity, contamination and the absence of primer dimers. The PCR efficiency of each primer pair (Eff in ) was evaluated by the dilution series method using a mix of sample cDNAs as the template and was determined from calibration curves using the Equation 10(−1/slope). Relative expression levels were determined with efficiency correction, which considers differences in the efficiencies between target and reference genes, using the gene expression module of the CFX manager Software (Bio-Rad).
Table 3. List of primers for quantification of mRNA expression and their efficiency (Eff)
Determination of HAC1 mRNA splicing
To determine the HAC1 mRNA splicing, real time reactions were performed in the same manner as those used for quantification of mRNA expression, using the primer pair: F: ATGACTGATTTTGAACTAACTAG and R: CAATTCAAATGAATTCAAACCTG (NCBI Gene ID: 850513). Afterward, the respective fragments were fractionated by electrophoresis on a 1% agarose gel.
Autophagy and mitophagy activity
The monitoring of autophagy and mitophagy was performed according to the protocol described by Mendl and co-authorsCitation39 and Noda and Klionsky.Citation40 Briefly, strains deleted for PHO8 were transformed with cytPho8- or mtPho8-expressing plasmids. At specific time points, 5 × 108 cells were collected, harvested, washed in 2 ml of ice-cold water containing 0.85% NaCl (Panreac, 131659.1211), 1 mM PMSF (Sigma, P7626), and resuspended in 300 µL lysis buffer (20 mM PIPES (Sigma, 80635), 0.5% Triton X-100 (Sigma, X100), 50 mM KCl (Merck, 104936), 100 mM potassium acetate (Sigma, P1190), 10 mM MgSO4 (Merck, 105886), 10 µM ZnSO4 (Sigma, Z4750) and 1 mM PMSF). An equal volume of acid-washed glass beads was added and the cells were lysed by vortexing for 7 min. To start the assay, 100 µl of extract was added to 400 µL reaction buffer [250 mM TRIS-HCl, pH 8.5, 0.4% Triton X-100, 10 mM MgSO4, 4 mM nitrophenyl phosphate (Sigma, N9389)], and samples were incubated 15 min at 37°C before terminating the reaction by adding 500 µL of stop buffer [2 M glycine (Sigma, 33226), pH 11]. Generation of nitrophenol was monitored by measuring absorbance at 405 nm using a microplate reader Model680 (Bio-Rad), and each sample was corrected with the time 0 blank. Protein concentration in the extracts was measured with the Bradford (Bio-Rad) according to the manufacturer’s instructions.
Confocal microscopy
For mitochondrial morphology and Atg8 analysis, strains expressing the SNCA or carrying the vector control, were transformed with pYX222-mtDsRed and pRS416-GFPAtg8. At specific time points, cells were collected and imaged using a confocal Olympus FLUOVIEW microscope. To visualize the SNCA foci, the cells were collected and fixed in 4% paraformaldehyde (Merck, 104005) during 15 min. After washing, first with 100 mM cacodylate (Sigma, C0250) (pH 7.4) and then with TDES [(100 mM Tris, pH 7.5, 25 mM DTT (Fermentas, R0862), 5 mM EDTA (Merck, 108418) and 1.2 M sorbitol (Sigma, 51876)], cells were incubated 10 min with TDES, at room temperature, to soften the cell wall, followed by a wash step with 100 mM phoscitrate buffer [(100 mM K2HPO4 (Merck, 105104), 100 mM citric acid (Sigma, C1909)]: 1 M sorbitol. Cells were then incubated 30 min with 100 mM phoscitrate buffer: 1 M sorbitol containing 50 µl of β-glucuronidase (Perkin Elmer, NEE154001EA) and 25 µl of 10 mg/ml zymolyase (Seikagaku Biobusiness, 12049) was added to produce spheroplasts. This was followed by a wash step with 100 mM cacodylate: 5 mM CaCl2: 1 M sorbitol. Next, cells were permeabilized with 0.1% Triton-X100 contained in 1X with phosphate-buffered saline (PBS), during 10 min. After 5 min rinsing with 0.1% BSA in PBS [137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4 (Merck, 106580), 1.8 mM KH2PO4 (Merck, 104873)], cells were incubated 2 h with the antibody anti-α-syn (Cell signaling, 2642) (1:400), followed by rinsing with 0.1% BSA in PBS for 15 min. Next cells were incubated with the Pacific Blue conjugated goat anti-rabbit secondary antibody (Molecular Probes, P10994) (1:100) for 4 h. Finally, cells were rinsed again with 0.1% BSA in PBS for 15 min, resuspended in PBS and visualized, at room temperature, by confocal microscopy. Images were acquired in a confocal Olympus FLUOVIEW microscope with an Olympus PLAPON 60×/oil objective, with a numerical aperture of 1.35. GFP and DsRed were excited with and argon laser and a helium-neon laser (GFP: 488 nm excitation; DsRed: 559 nm excitation). Pacific blue was excited with a UV laser (Pacific blue: 405 nm excitation). Background reduction was performed with appropriate saturation levels using software FV1000 (Olympus) and Adobe Photoshop CS. Image stacks for analysis were acquired with sequential steps of 0.25 to 0.5 µm per plane in the z-direction and a total thickness of 4–6 µm. The acquired stacks were rendered with FV1000 software.
Assessment of intracellular superoxide anion accumulation
Free intracellular reactive oxygen species (ROS), specifically superoxide anions were measured using dihydroethidium (DHE) (Molecular Probes, D11347). Aliquots of cells were collected at indicated time points and DHE was added to a □nal concentration of 5 μM from a 5 mM stock in DMSO (Sigma, D5879). After incubation for 10 min at 30°C, cells were washed once with 0.5 ml PBS, resuspended in 50 μL PBS, and then transferred to 1 ml PBS. The DHE signals were measured using FACSCaliber2 flow cytometer (BD-Biosciences) with a 488 nm excitation laser. Signals from 30,000 cells/sample were captured in FL3 (> 670 nm) at a flow rate of 1,000 cells/s. Data collected with the FACSCaliber2 flow cytometer were processed with Flowjo software (Tree Star) and quantified with WinList software (Verity Software House).
Preparation of protein extracts and western blot analysis
For detection of protein levels by western blot, the total cellular extracts were collected at specific time points and extracted as previously described.Citation70 Briefly, cells were pretreated with 2 M lithium acetate (Sigma, L4158) for 5 min at room temperature. After lithium acetate removal, 0.4 M NaOH (Panreac, 131687.1211) was added for 5 min on ice. Next, the cell were resuspended in SDS-PAGE sample buffer (Bio-Rad, 161-0737) and boiled for 5 min. Of total protein, 20 μg were resolved on a 12% SDS-PAGE gel and transferred to a nitrocellulose membrane (Bio-Rad, 170-4159) during 7 min at 25V. The membranes were blocked with Tris-buffered saline (TBS) with 0.1% Tween 20 (Sigma, P1379) (TBST) containing 5% skim milk, followed by incubation with anti-α-syn (1:1000) (Cell Signaling, 2642S) primary antibody in TBST containing 1% skim milk and anti-actin (1:5000) (kindly provide by Dr. C. Gourlay) primary antibody. After washing with TBS, the membranes were incubated with the respective secondary antibody, HRP-conjugated anti-rabbit IgG or HRP-conjugated anti-mouse IgG at a dilution of 1:5000 and detected by enhanced chemiluminescence (Thermo Scientific, 34095).
Statistical analysis
The results shown are mean values and standard error of the mean of at least three independent assays. Statistical analyses were determined using two-way ANOVA. A p value of less than 0.05 was considered as a significant difference.
| Abbreviations: | ||
| ALP | = | alkaline phosphatase |
| Atg | = | autophagy-related |
| BSA | = | bovine serum albumin |
| CQ | = | chloroquine |
| CLS | = | chronological life span |
| CMA | = | chaperone-mediated autophagy |
| CFU | = | colony-forming units |
| DHE | = | dihydroethidium |
| IRC | = | index of respiratory competence |
| PBS | = | phosphate-buffered saline |
| PD | = | Parkinson disease |
| PI | = | propidium iodide |
| ROS | = | reactive oxygen species |
| Sir2 | = | sirtuin 2 |
| SNCA | = | α-synuclein |
| SOD | = | superoxide dismutase |
| UPS | = | ubiquitin-proteasome system |
| UPR | = | unfolded protein response |
Additional material
Download Zip (189.5 KB)Acknowledgment
This work was supported by FCT–Fundação para a Ciência e Tecnologia (PTDC/BIA-MIC/114116/2009 and PTDC/SAU-NEU/105215/2008). B.S.M. and S.T. have fellowships from FCT (SRFH/BD/41674/2007 and SFRH/BPD/35767/2007, respectively). T.F.O. was supported by a Marie Curie International Reintegration Grant (Neurofold), an EMBO Installation Grant and Fundação para a Ciência e Tecnologia, Portugal. A.S.R. was supported by the DFG grant RE1575-1/1, the Cluster of Excellence Frankfurt Macromolecular Complexes at the Goethe University Frankfurt DFG project EXC 115 and the BMBF, Germany, GerontoMitoSys project. V.F. and J.W. were supported by grants of KU Leuven and IWT-Vlaanderen (SBO-NeuroTarget). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/21275
References
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 2006; 52:33 - 8; http://dx.doi.org/10.1016/j.neuron.2006.09.026; PMID: 17015225
- Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 2006; 7:306 - 18; http://dx.doi.org/10.1038/nrg1831; PMID: 16543934
- Auluck PK, Caraveo G, Lindquist S. α-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu Rev Cell Dev Biol 2010; 26:211 - 33; http://dx.doi.org/10.1146/annurev.cellbio.042308.113313; PMID: 20500090
- Chu CT. Diversity in the regulation of autophagy and mitophagy: lessons from Parkinson’s disease. Parkinsons Dis 2011; 2011:789431; http://dx.doi.org/10.4061/2011/789431; PMID: 21603187
- Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 2010; 11:777 - 88; http://dx.doi.org/10.1038/nrm2993; PMID: 20944667
- Lim KL, Tan JM. Role of the ubiquitin proteasome system in Parkinson’s disease. BMC Biochem 2007; 8:Suppl 1 S13; http://dx.doi.org/10.1186/1471-2091-8-S1-S13; PMID: 18047737
- Zabrocki P, Pellens K, Vanhelmont T, Vandebroek T, Griffioen G, Wera S, et al. Characterization of alpha-synuclein aggregation and synergistic toxicity with protein tau in yeast. FEBS J 2005; 272:1386 - 400; http://dx.doi.org/10.1111/j.1742-4658.2005.04571.x; PMID: 15752356
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004; 305:1292 - 5; http://dx.doi.org/10.1126/science.1101738; PMID: 15333840
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003; 278:25009 - 13; http://dx.doi.org/10.1074/jbc.M300227200; PMID: 12719433
- Chen Q, Thorpe J, Keller JN. Alpha-synuclein alters proteasome function, protein synthesis, and stationary phase viability. J Biol Chem 2005; 280:30009 - 17; http://dx.doi.org/10.1074/jbc.M501308200; PMID: 15941712
- Zhang NY, Tang Z, Liu CW. alpha-Synuclein protofibrils inhibit 26 S proteasome-mediated protein degradation: understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J Biol Chem 2008; 283:20288 - 98; http://dx.doi.org/10.1074/jbc.M710560200; PMID: 18502751
- McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW. Altered proteasomal function in sporadic Parkinson’s disease. Exp Neurol 2003; 179:38 - 46; http://dx.doi.org/10.1006/exnr.2002.8050; PMID: 12504866
- Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med 2012; 2:a009357; PMID: 22474616
- Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, et al. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 2011; 286:10814 - 24; http://dx.doi.org/10.1074/jbc.M110.132514; PMID: 21252228
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8:e1000298; http://dx.doi.org/10.1371/journal.pbio.1000298; PMID: 20126261
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795 - 803; http://dx.doi.org/10.1083/jcb.200809125; PMID: 19029340
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 2010; 107:378 - 83; http://dx.doi.org/10.1073/pnas.0911187107; PMID: 19966284
- Imai Y, Lu B. Mitochondrial dynamics and mitophagy in Parkinson’s disease: disordered cellular power plant becomes a big deal in a major movement disorder. Curr Opin Neurobiol 2011; 21:935 - 41; http://dx.doi.org/10.1016/j.conb.2011.10.016; PMID: 22048001
- Vives-Bauza C, Przedborski S. Mitophagy: the latest problem for Parkinson’s disease. Trends Mol Med 2011; 17:158 - 65; http://dx.doi.org/10.1016/j.molmed.2010.11.002; PMID: 21146459
- Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J 2010; 29:3571 - 89; http://dx.doi.org/10.1038/emboj.2010.223; PMID: 20842103
- Chinta SJ, Mallajosyula JK, Rane A, Andersen JK. Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett 2010; 486:235 - 9; http://dx.doi.org/10.1016/j.neulet.2010.09.061; PMID: 20887775
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, et al. α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol 2010; 190:1023 - 37; http://dx.doi.org/10.1083/jcb.201003122; PMID: 20855506
- Kanki T, Klionsky DJ. Atg32 is a tag for mitochondria degradation in yeast. Autophagy 2009; 5:1201 - 2; http://dx.doi.org/10.4161/auto.5.8.9747; PMID: 19736522
- Franssens V, Boelen E, Anandhakumar J, Vanhelmont T, Büttner S, Winderickx J. Yeast unfolds the road map toward alpha-synuclein-induced cell death. Cell Death Differ 2010; 17:746 - 53; http://dx.doi.org/10.1038/cdd.2009.203; PMID: 20019751
- Büttner S, Delay C, Franssens V, Bammens T, Ruli D, Zaunschirm S, et al. Synphilin-1 enhances α-synuclein aggregation in yeast and contributes to cellular stress and cell death in a Sir2-dependent manner. PLoS One 2010; 5:e13700; http://dx.doi.org/10.1371/journal.pone.0013700; PMID: 21060871
- Sampaio-Marques B, Felgueiras C, Silva A, Rodrigues F, Ludovico P. Yeast chronological lifespan and proteotoxic stress: is autophagy good or bad?. Biochem Soc Trans 2011; 39:1466 - 70; http://dx.doi.org/10.1042/BST0391466; PMID: 21936835
- Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 2010; 5:e9313; http://dx.doi.org/10.1371/journal.pone.0009313; PMID: 20174468
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990 - 5; http://dx.doi.org/10.1126/science.1099993; PMID: 15528435
- Khurana V, Lindquist S. Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker’s yeast?. Nat Rev Neurosci 2010; 11:436 - 49; http://dx.doi.org/10.1038/nrn2809; PMID: 20424620
- Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003; 302:1772 - 5; http://dx.doi.org/10.1126/science.1090439; PMID: 14657500
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy 2007; 3:181 - 206; PMID: 17224625
- Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol 1999; 147:435 - 46; http://dx.doi.org/10.1083/jcb.147.2.435; PMID: 10525546
- Chan TF, Bertram PG, Ai W, Zheng XF. Regulation of APG14 expression by the GATA-type transcription factor Gln3p. J Biol Chem 2001; 276:6463 - 7; http://dx.doi.org/10.1074/jbc.M008162200; PMID: 11096087
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000; 101:249 - 58; http://dx.doi.org/10.1016/S0092-8674(00)80835-1; PMID: 10847680
- Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem 2006; 281:30299 - 304; http://dx.doi.org/10.1074/jbc.M607007200; PMID: 16901900
- Büttner S, Bitto A, Ring J, Augsten M, Zabrocki P, Eisenberg T, et al. Functional mitochondria are required for alpha-synuclein toxicity in aging yeast. J Biol Chem 2008; 283:7554 - 60; http://dx.doi.org/10.1074/jbc.M708477200; PMID: 18192273
- Vives-Bauza C, de Vries RL, Tocilescu M, Przedborski S. PINK1/Parkin direct mitochondria to autophagy. Autophagy 2010; 6:315 - 6; http://dx.doi.org/10.4161/auto.6.2.11199; PMID: 20200476
- Kanki T, Klionsky DJ. The molecular mechanism of mitochondria autophagy in yeast. Mol Microbiol 2010; 75:795 - 800; http://dx.doi.org/10.1111/j.1365-2958.2009.07035.x; PMID: 20487284
- Mendl N, Occhipinti A, Müller M, Wild P, Dikic I, Reichert AS. Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2. J Cell Sci 2011; 124:1339 - 50; http://dx.doi.org/10.1242/jcs.076406; PMID: 21429936
- Noda T, Klionsky DJ. The quantitative Pho8Delta60 assay of nonspecific autophagy. Methods Enzymol 2008; 451:33 - 42; http://dx.doi.org/10.1016/S0076-6879(08)03203-5; PMID: 19185711
- Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci 2005; 118:7 - 18; http://dx.doi.org/10.1242/jcs.01620; PMID: 15615779
- Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 2009; 17:98 - 109; http://dx.doi.org/10.1016/j.devcel.2009.06.014; PMID: 19619495
- Flower TR, Clark-Dixon C, Metoyer C, Yang H, Shi R, Zhang Z, et al. YGR198w (YPP1) targets A30P alpha-synuclein to the vacuole for degradation. J Cell Biol 2007; 177:1091 - 104; http://dx.doi.org/10.1083/jcb.200610071; PMID: 17576801
- Zabrocki P, Bastiaens I, Delay C, Bammens T, Ghillebert R, Pellens K, et al. Phosphorylation, lipid raft interaction and traffic of alpha-synuclein in a yeast model for Parkinson. Biochim Biophys Acta 2008; 1783:1767 - 80; http://dx.doi.org/10.1016/j.bbamcr.2008.06.010; PMID: 18634833
- Yorimitsu T, Klionsky DJ. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol Biol Cell 2005; 16:1593 - 605; http://dx.doi.org/10.1091/mbc.E04-11-1035; PMID: 15659643
- Shintani T, Huang WP, Stromhaug PE, Klionsky DJ. Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev Cell 2002; 3:825 - 37; http://dx.doi.org/10.1016/S1534-5807(02)00373-8; PMID: 12479808
- Parrella E, Longo VD. The chronological life span of Saccharomyces cerevisiae to study mitochondrial dysfunction and disease. Methods 2008; 46:256 - 62; http://dx.doi.org/10.1016/j.ymeth.2008.10.004; PMID: 18930829
- Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 2010; 6:838 - 54; http://dx.doi.org/10.4161/auto.6.7.12113; PMID: 20505317
- Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 2011; 36:30 - 8; http://dx.doi.org/10.1016/j.tibs.2010.07.007; PMID: 20728362
- Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med 1998; 25:826 - 31; http://dx.doi.org/10.1016/S0891-5849(98)00163-4; PMID: 9823548
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Kaeberlein M, Burtner CR, Kennedy BK. Recent developments in yeast aging. PLoS Genet 2007; 3:e84; http://dx.doi.org/10.1371/journal.pgen.0030084; PMID: 17530929
- Gitler AD. Beer and bread to brains and beyond: can yeast cells teach us about neurodegenerative disease?. Neurosignals 2008; 16:52 - 62; http://dx.doi.org/10.1159/000109759; PMID: 18097160
- Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, et al. Huntingtin localization in brains of normal and Huntington’s disease patients. Ann Neurol 1997; 42:604 - 12; http://dx.doi.org/10.1002/ana.410420411; PMID: 9382472
- Sarkar S, Rubinsztein DC. Huntington’s disease: degradation of mutant huntingtin by autophagy. FEBS J 2008; 275:4263 - 70; http://dx.doi.org/10.1111/j.1742-4658.2008.06562.x; PMID: 18637946
- Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy 2007; 3:663 - 6; PMID: 17622797
- Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One 2009; 4:e5515; http://dx.doi.org/10.1371/journal.pone.0005515; PMID: 19436756
- Yu WH, Dorado B, Figueroa HY, Wang L, Planel E, Cookson MR, et al. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol 2009; 175:736 - 47; http://dx.doi.org/10.2353/ajpath.2009.080928; PMID: 19628769
- Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell 2011; 146:682 - 95; http://dx.doi.org/10.1016/j.cell.2011.07.030; PMID: 21884931
- Sharma N, Brandis KA, Herrera SK, Johnson BE, Vaidya T, Shrestha R, et al. alpha-Synuclein budding yeast model: toxicity enhanced by impaired proteasome and oxidative stress. J Mol Neurosci 2006; 28:161 - 78; http://dx.doi.org/10.1385/JMN:28:2:161; PMID: 16679556
- Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T, et al. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J Biol Chem 2012; 287:3265 - 72; http://dx.doi.org/10.1074/jbc.M111.280156; PMID: 22157017
- Bellucci A, Navarria L, Zaltieri M, Falarti E, Bodei S, Sigala S, et al. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J Neurochem 2011; 116:588 - 605; http://dx.doi.org/10.1111/j.1471-4159.2010.07143.x; PMID: 21166675
- Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, et al. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol 2000; 157:401 - 10; http://dx.doi.org/10.1016/S0002-9440(10)64553-1; PMID: 10934145
- Kregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am J Physiol Regul Integr Comp Physiol 2007; 292:R18 - 36; http://dx.doi.org/10.1152/ajpregu.00327.2006; PMID: 16917020
- Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis 2010; 1:e10; http://dx.doi.org/10.1038/cddis.2009.8; PMID: 21364612
- Morselli E, Mariño G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol 2011; 192:615 - 29; http://dx.doi.org/10.1083/jcb.201008167; PMID: 21339330
- Mariño G, Morselli E, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, et al. Longevity-relevant regulation of autophagy at the level of the acetylproteome. Autophagy 2011; 7:647 - 9; http://dx.doi.org/10.4161/auto.7.6.15191; PMID: 21460620
- Flohé L, Günzler WA. Assays of glutathione peroxidase. Methods Enzymol 1984; 105:114 - 21; http://dx.doi.org/10.1016/S0076-6879(84)05015-1; PMID: 6727659
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 2009; 55:611 - 22; http://dx.doi.org/10.1373/clinchem.2008.112797; PMID: 19246619
- Zhang T, Lei J, Yang H, Xu K, Wang R, Zhang Z. An improved method for whole protein extraction from yeast Saccharomyces cerevisiae. Yeast 2011; 28:795 - 8; http://dx.doi.org/10.1002/yea.1905; PMID: 21972073