Abstract
We recently identified physical exercise as a newly defined inducer of autophagy in vivo. Exercise induced autophagy in multiple organs involved in metabolic regulation, such as muscle, liver, pancreas and adipose tissue. To study the physiological role of exercise-induced autophagy, we generated mice with a knock-in nonphosphorylatable mutation in BCL2 (Thr69Ala, Ser70Ala and Ser84Ala) (BCL2 AAA) that are defective in exercise- and starvation-induced autophagy but not in basal autophagy. We found that BCL2 AAA mice could not run on a treadmill as long as wild-type mice, and did not undergo exercise-mediated increases in skeletal glucose muscle uptake. Unlike wild-type mice, the BCL2 AAA mice failed to reverse high-fat diet-induced glucose intolerance after 8 weeks of exercise training, possibly due to defects in signaling pathways that regulate muscle glucose uptake and metabolism during exercise. Together, these findings suggested a hitherto unknown important role of autophagy in mediating exercise-induced metabolic benefits. In the present addendum, we show that treadmill exercise also induces autophagy in the cerebral cortex of adult mice. This observation raises the intriguing question of whether autophagy may in part mediate the beneficial effects of exercise in neurodegeneration, adult neurogenesis and improved cognitive function.
Keywords: :
The relationship among autophagy, exercise and metabolic regulation has been a largely unexplored field. Physical exercise has numerous health benefits, such as life-span expansion, and protection against cardiovascular diseases, diabetes, cancer and neurodegenerative diseases.Citation1 Many of these health benefits overlap with known protective functions of the cellular pathway of macroautophagy (herein referred to as autophagy).Citation2,Citation3 Thus, we proposed that some of the health benefits of exercise may be due to autophagy activation.
To test this hypothesis, we exercised wild-type mice that transgenically express the fluorescent autophagy marker GFP-LC3Citation4 on a treadmill, using a running protocol with increasing speed at defined intervals.Citation5 We found that in both skeletal and cardiac muscle 30 min of exercise was sufficient to induce GFP-LC3 puncta (autophagosome) formation, which reached a plateau after 80 min. Using a combination of assays including GFP-LC3 puncta formation, LC3-II conversion and SQSTM1/p62 degradation, we showed that autophagy activity is induced by exercise in multiple organs, including skeletal muscle, heart, liver, pancreatic β cells and adipose tissue. These observations suggested a possible role of autophagy in metabolic regulation during exercise.
To study specific roles of exercise-induced autophagy, we utilized a mouse model that is defective in exercise-induced autophagy but maintains normal levels of basal autophagy. Previous reports have shown that loss of basal autophagy activity in cardiac or skeletal muscle leads to abnormal development, and cardiac failure and skeletal muscle atrophy, respectively;Citation6,Citation7 therefore, (inducible) tissue-specific knockout of autophagy genes in these organs would not be a suitable approach for examining the physiological effects of deficient exercise-induced autophagy. Thus, we generated a new mouse model (BCL2 AAA mice) with a knock-in mutation in the phosphorylation sites of the nonstructured loop of the anti-autophagy protein, BCL2.Citation8 In a previous study, we found that multisite phosphorylation of BCL2 is essential for its release from BECN1 (also known as Beclin 1) and for starvation-induced autophagy in vitro, and that nonphosphorylatable mutations in human BCL2 block starvation-induced, but not basal, autophagy.Citation9 As expected, we found that BCL2 AAA mice (which contain nonphosphorylatable mutations in the analogous sites in mouse BCL2) had normal muscle histology and normal levels of basal autophagy, but were defective in starvation- and exercise-induced autophagy. Accordingly, the BCL2 AAA mice serve as a useful model system to further study the functions of exercise-induced autophagy.
During acute exercise (single bout of treadmill running), compared with wild-type mice, BCL2 AAA mice had decreased exercise endurance and impaired increases in muscle glucose metabolism, including lower levels of decline in serum glucose and insulin levels, decreased plasma membrane relocalization of the SLC2A4 (also known as GLUT4) glucose transporter, decreased uptake of radiolabeled glucose, and decreased activation of AMP-activated protein kinase (AMPK). Similar findings were also observed in mice with mono-allelic loss of Becn1, and mice with hypomorphic expression of Atg16l1, suggesting that this phenotype was due to impaired autophagy activation, rather than an off-target effect of the BCL2 AAA mutation. These abnormalities in glucose metabolism during acute exercise in mice deficient in exercise-induced autophagy led us to investigate whether autophagy might contribute to some of the beneficial metabolic effects of chronic exercise training.
Exercise training protects against high-fat diet-induced type 2 diabetes in rodents and humans.Citation10-Citation13 We found that the induction of autophagy may be required for this protection, as only wild-type, but not BCL2 AAA, mice, reversed their dietary-induced glucose intolerance after 8 weeks of exercise training. We postulate that this mechanism may involve the impaired exercise-induced increase in muscle glucose uptake and AMPK activation that we observed during single bouts of forced treadmill exercise. Together, our findings suggested an unexpected role of autophagy in the regulation of glucose metabolism, AMPK activation, and exercise-mediated protection against type 2 diabetes. Given the central role of AMPK activation in treatment of diabetes and prevention of cancer,Citation14 this positive feedback loop (also reported by others in vitroCitation15) between autophagy and AMPK activation during exercise may have important implications for understanding the role of altered autophagy in metabolic diseases, cancer and aging.
Intriguingly, in addition to various peripheral organs involved in metabolism, such as muscle, liver, pancreas and adipose tissue,Citation5 we found that autophagy is also potently induced by acute exercise in the brain (). We exercised 8-week-old GFP-LC3 transgenic wild-type mice on the treadmill for 95 min, and examined biochemical markers of autophagy by western blot analyses, as well as GFP-LC3 puncta in brain sections after paraformaldehyde perfusion by fluorescence microscopy. We found that there was a 2-fold increase in numbers of GFP-LC3 puncta in the anterior cerebral cortex after exercise (). Importantly, we note that there is a very high level of auto-fluorescence in brain sections as compared with other tissues, which is perhaps why this fluorescent autophagy reporter has not been used extensively to study autophagy in the brain in prior studies (despite widespread use in other tissues). However, we found that the use of spectral unmixing (which separates the wavelengths of background auto-fluorescence from the wavelength of GFP) allowed us to detect authentic GFP-LC3 puncta. We also confirmed this finding by performing immunostaining with an anti-GFP antibody (). Of note, we did not detect significant increases in GFP-LC3 puncta in regions of the brain besides the cerebral cortex, including the olfactory bulb, hypothalamus, midbrain or cerebellum (data not shown). We cannot rule out the possibility that other methods of sample preparation, similar to those used in recent reports that have detected increased GFP-LC3 puncta in the brains of mice subjected to starvation,Citation16 might be more sensitive and detect autophagy induction in other regions of the brain.
Figure 1. Autophagy is induced by exercise in the brain. (A) Representative images of GFP-LC3 puncta in the cerebral cortex from wild-type GFP-LC3 mice at rest and after 95 min of exercise. The top panel shows the GFP-LC3 puncta in the brain after the auto-fluorescent signals were removed by spectral unmixing analyses. For spectral unmixing, images were acquired under a Zeiss 63x 1.4 NA PLAN-APOCHROMAT objective using a Zeiss LSM510 microscope equipped with a META multispectral detector at excitation of 488 nm. Linear unmixing was performed using the Zeiss LSM software by subtracting background (no GFP) peaks of 533 nm and 575 nm from the GFP spectrum, which peaked at 520 nm in tissue sections from GFP-LC3 mice. The bottom panel shows representative images of GFP immunofluorescence staining in cerebral crotex from the same wild-type GFP-LC3 at rest and after 95 min of exercise. Brain sections were stained with an anti-GFP antibody and an Alexa Fluor 594-conjugated secondary antibody. Scale bar: 20 μm. (B) Quantification of GFP-LC3 puncta shown in (A). A minimum of 8 fields per mouse was analyzed. Results shown represent mean ± s.d. for 3 mice per experimental group. (C and D) Biochemical analysis of autophagy induction in extracts of whole brain (C) or cerebral cortex (D) from wild-type (WT) and BCL2 AAA mice at rest (-) or after 95 min of exercise by western blot detection of SQSTM1/p62 and LC3-II conversion. Actin was used as a loading control. Three mice per experimental group are shown.
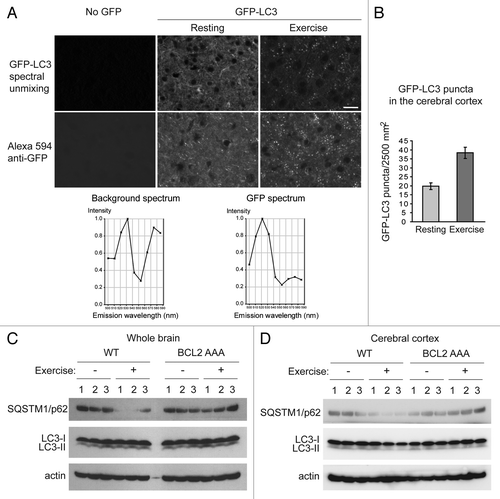
In addition to increased GFP-LC3 puncta, we detected a marked decrease in SQSTM1/p62 levels in whole brain lysates and cerebral cortex lysates from wild-type mouse brains after exercise, which was not observed in the brains of BCL2 AAA mice after an identical duration and intensity of treadmill exercise ( and ). These data suggest that autophagic flux in brain is increased after exercise in wild-type mice but not in BCL2 AAA mice. We did not detect changes in LC3-II conversion pre- and post- exercise in either genotype in whole brain (), or in cerebral cortex (); this may reflect decreased sensitivity of the LC3-II conversion assay as compared with measurements of SQSTM1/p62 degradation or GFP-LC3 puncta. It is also possible that there are other Atg8 homologs in the brain that may play more important roles than LC3B in autophagy, and that the anti-LC3B antibody used for western blot analysis in our study therefore did not detect conversion of the biologically relevant Atg8 homolog. Further studies will be required to elucidate in more detail the role of lipidation of specific mammalian Atg8 homologs in exercise-induced autophagy in the brain.
Taken together, we conclude that exercise induces autophagy in the cerebral cortex of the brain. More detailed analyses will be required to determine the precise subpopulations of neurons that upregulate autophagy in response to exercise. Previous studies have shown that treadmill exercise upregulates sirtuin 1 levels and AMPK activation in rat brain.Citation17 As both of these factors function as positive regulators of autophagy,Citation18 one obvious question is whether they may contribute to the upregulation of autophagy that we observed in the brains of wild-type mice after exercise.
There are numerous additional questions that remain to be answered. For example, the precise molecular mechanisms that cause altered glucose metabolism in autophagy-deficient mice remain unknown. Another open question is whether exercise-induced autophagy contributes to the beneficial effects of exercise on diseases other than diabetes, such as aging, cancer, cardiovascular diseases, inflammatory diseases and neurodegenerative diseases. Based on our new data that exercise can induce autophagy in certain regions of the brain, it will be important to investigate the physiological consequences of this phenomenon. Of note, autophagy is an important “housekeeping” mechanism that eliminates protein aggregates and damaged organelles in neurons,Citation19,Citation20 and exercise is an intervention that improves neuronal synaptic plasticity, promotes adult neurogenesis, prevents cognitive decline in aging, and delays the onset of neurodegenerative diseases.Citation21 Thus, a crucial question is whether exercise-induced autophagy in the brain mediates some of these neuroprotective effects. Another intriguing question is whether the signals that trigger exercise-induced autophagy in the brain are cell-autonomous or derived from extrinsic systemic cues.
References
- Handschin C, Spiegelman BM. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 2008; 454:463 - 9; http://dx.doi.org/10.1038/nature07206; PMID: 18650917
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101 - 11; http://dx.doi.org/10.1091/mbc.E03-09-0704; PMID: 14699058
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012; 481:511 - 5; http://dx.doi.org/10.1038/nature10758; PMID: 22258505
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab 2009; 10:507 - 15; http://dx.doi.org/10.1016/j.cmet.2009.10.008; PMID: 19945408
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007; 13:619 - 24; http://dx.doi.org/10.1038/nm1574; PMID: 17450150
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927 - 39; http://dx.doi.org/10.1016/j.cell.2005.07.002; PMID: 16179260
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678 - 88; http://dx.doi.org/10.1016/j.molcel.2008.06.001; PMID: 18570871
- Henriksen EJ. Invited review: Effects of acute exercise and exercise training on insulin resistance. J Appl Physiol 2002; 93:788 - 96; PMID: 12133893
- Tuomilehto J, Lindström J, Eriksson JG, Valle TT, Hämäläinen H, Ilanne-Parikka P, et al, Finnish Diabetes Prevention Study Group. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001; 344:1343 - 50; http://dx.doi.org/10.1056/NEJM200105033441801; PMID: 11333990
- Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al, Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393 - 403; http://dx.doi.org/10.1056/NEJMoa012512; PMID: 11832527
- Duncan GE, Perri MG, Theriaque DW, Hutson AD, Eckel RH, Stacpoole PW. Exercise training, without weight loss, increases insulin sensitivity and postheparin plasma lipase activity in previously sedentary adults. Diabetes Care 2003; 26:557 - 62; http://dx.doi.org/10.2337/diacare.26.3.557; PMID: 12610001
- Hardie DG. Adenosine monophosphate-activated protein kinase: a central regulator of metabolism with roles in diabetes, cancer, and viral infection. Cold Spring Harb Symp Quant Biol 2011; 76:155 - 64; http://dx.doi.org/10.1101/sqb.2011.76.010819; PMID: 22071265
- Malik SA, Orhon I, Morselli E, Criollo A, Shen S, Mariño G, et al. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene 2011; 30:3918 - 29; http://dx.doi.org/10.1038/onc.2011.104; PMID: 21460857
- Alirezaei M, Kemball CC, Flynn CT, Wood MR, Whitton JL, Kiosses WB. Short-term fasting induces profound neuronal autophagy. Autophagy 2010; 6:702 - 10; http://dx.doi.org/10.4161/auto.6.6.12376; PMID: 20534972
- Bayod S, Del Valle J, Canudas AM, Lalanza JF, Sanchez-Roige S, Camins A, et al. Long-term treadmill exercise induces neuroprotective molecular changes in rat brain. J Appl Physiol 2011; 111:1380 - 90; http://dx.doi.org/10.1152/japplphysiol.00425.2011; PMID: 21817108
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280 - 93; http://dx.doi.org/10.1016/j.molcel.2010.09.023; PMID: 20965422
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/10.1038/nature04723; PMID: 16625205
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204
- Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci 2007; 30:464 - 72; http://dx.doi.org/10.1016/j.tins.2007.06.011; PMID: 17765329