Abstract
The very high mortality rate of gliomas reflects the unmet therapeutic need associated with this type of brain tumor. We have discovered that the plasma membrane fulfills a critical role in the propagation of tumorigenic signals, whereby changes in membrane lipid content can either activate or silence relevant pathways. We have designed a synthetic fatty acid, 2-hydroxyoleic acid (2OHOA), that specifically activates sphingomyelin synthase (SGMS), thereby modifying the lipid content of cancer cell membranes and restoring lipid levels to those found in normal cells. In reverting, the structure of the membrane by activating SGMS, 2OHOA inhibits the RAS-MAPK pathway, which in turn fails to activate the CCND (Cyclin D)-CDK4/CDK6 and PI3K-AKT1 pathways. The overall result in SF767 cancer cells, a line that is resistant to apoptosis, is the sequential induction of cell cycle arrest, cell differentiation and autophagy. Such effects are not observed in normal cells (MRC-5) and thus, this specific activation of programmed cell death infers greater efficacy and lower toxicity to 2OHOA than that associated with temozolomide (TMZ), the reference drug for the treatment of glioma.
Gliomas are CNS tumors that are resistant to apoptosis and that are associated with high mortality. The high proliferation rate of gliomas and other cancer cells is a key (upstream) event in their tumorigenic transformation, which we have shown to be associated with very low levels of sphingomyelin (SM) and a high phosphatidylethanolamine (PE) content in the plasma membrane. 2OHOA is a compound that specifically activates SGMS, restoring the SM and PE levels in cancer cell membranes to those found in normal cells. This effect on membrane lipid structure changes the type of proteins that interact with the membrane, and influences other protein-protein interactions, thereby inducing cell cycle arrest, cancer cell differentiation and autophagy. Indeed, inhibiting SGMS in part reverses the antiproliferative effects of 2OHOA, demonstrating the specificity of this effect. By contrast, 2OHOA does not alter the lipid profile of normal cells, in which the relatively high levels of SM, the product of SGMS, and the low levels of PE, the substrate of SGMS, maintain the activity of SGMS at a low level. This regulatory influence on the lipid composition of the glioma cell membrane causes RAS to translocate to the cytoplasm and the inactivation of the MAPK pathway, as well as PRKC/PKC translocation to the membrane associated with the concomitant induction of the CDK inhibitors, CDKN1A/p21Cip1 and CDKN1B/p27Kip1. As a result, the formation of CCND-CDK4/CDK6 complexes is impaired causing hypophosphorylation of the retinoblastoma protein (RB1/pRb), E2F1 inhibition and knockdown of DHFR. In addition, 2OHOA provokes the inhibition of the PI3K-AKT1 pathway, probably due to crosstalk with the receptor tyrosine kinase (RTK)-RAS-MAPK pathway ().
Figure 1. The induction of autophagy in glioma cells treated with 2OHOA. The illustration on the left depicts the membrane structure and the active (black) or inactive (red) cell signals in glioma cells. High PE and low SM levels favor the activation of the MAPK pathway, which in turn induces the activity of the cyclin-CDK and PI3K pathways. The illustration on the right depicts the membrane lipid status in 2OHOA-treated cells, with more SM and less PE than untreated glioma cells, which causes the uncoupling of RAS and the concomitant inactivation of the MAPK and related pathways. Activation of FOXO1, overexpression of CDKN1B, hypophosphorylation of RB1 and inactivation of AKT1, contributes to the molecular scenario associated with the activation of autophagy in glioma but not normal cells. In this context, the membrane lipid structure appears to be the most upstream event that controls cell proliferation, differentiation and autophagy in glioma cells, SGMS thereby representing a new target for anticancer therapies.
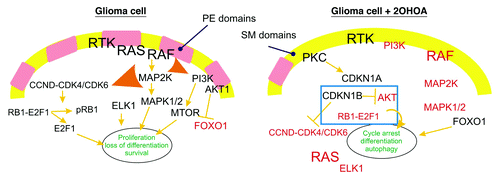
In light of these data, the membrane lipid composition appears to be critical for the increased proliferation, loss of differentiation and the evasion of cell death that is typical of tumor cells, and it offers a molecular explanation for the mode of action of 2OHOA in combating cancer. We have shown how membrane lipids regulate the binding of peripheral signaling proteins and protein-protein interactions at the membrane, defining microdomains with distinct affinities for specific proteins. Accordingly, RAS is preferentially bound to the membrane of many cancer cells, while in normal cells and following exposure of cancer cells to 2OHOA, it preferentially accumulates in the cytoplasm. Since the presence of RAS at the membrane is necessary to propagate signals from RTKs to RAF, the detachment of RAS from the membrane inactivates the MAPK pathway ().
The first event induced by 2OHOA is cell cycle arrest (initiated within 24 h of exposure), later also inducing glioma cell differentiation (ca. 48–72 h). Cancer cells are characterized by rapid growth, as well as their dissemination and the invasion of other tissues. In this context, the changes caused by 2OHOA in glioma cell proliferation and differentiation possibly induce a molecular conflict that triggers cancer cell death. From the molecular point of view, this conflict could be produced when cells such as SF767 glioma cells accumulate high levels of CDKN1B, hypophosphorylated RB1, and when AKT1 is inhibited and FOXO1 activated, precisely the conditions promoted by 2OHOA (). Interestingly, autophagy appears to be delayed with respect to cell cycle arrest and the induction of differentiation. Indeed, significant increases in the appearance of the markers of autophagy, LC3B and ATG5 were only observed 72 h after treatment with 2OHOA. Accordingly, it appears that the regulatory scenario imposed by the membrane structure is incompatible with the mutations present in many cancer cell lines. Finally, we have seen that the specific cell death induced by 2OHOA in glioma cells is associated with ER stress and sphingolipidosis caused by an increase in SM and concomitant alterations in sphingolipid metabolism. These phenomena could be directly related to cancer cell death and they could cause autophagy or other types of programmed cell death (e.g., apoptosis) observed in other types of cancer cells. In addition, sphingolipidosis/ER stress could cause the various morphological/cellular alterations observed upon 2OHOA treatments in cancer cells.
A number of conclusions can be drawn from our studies. First, low SM and high PE levels in the plasma membrane appear to be necessary to induce cancer cell proliferation, along with oncogene activation and tumor suppressor inactivation, representing the third basic requirement for the expression of a malignant phenotype. Moreover, the structural status of the membrane appears to lie upstream of the activation of certain oncogenes (e.g., RAS), therefore being a cellular switch for cell proliferation. Thus, normalization of the membrane lipid composition/structure by 2OHOA induces cell cycle exit and initiation of a program of autophagy. Indeed, when glioma (SF767) cells stop proliferating and start differentiating, the program of autophagy (or other programs of cell death) commences. Micrographs showing the presence of autophagosomes, dramatic cell fragmentation and the appearance of the autophagosome marker LC3B along with ATG5, clearly indicate the relatively late (time and concentration dependent) onset of autophagy in glioma cells. Second, we have defined SGMS as a new anticancer drug target for which 2OHOA is a first-in-class SGMS activator. Third, 2OHOA clearly induces glioma cell differentiation. The greater efficacy and lack of tumor relapse after 2OHOA administration with respect to TMZ (the reference drug for the treatment of glioma) constitutes proof-of-relevance and demonstrates the great potential of the specific induction of autophagy to combat cancer. Normal cells already have high levels of SM, and 2OHOA does not induce a further rise in the membrane SM content and the ensuing events observed in cancer cells, including autophagic cell death. Hence, the IC50 of 2OHOA in normal human embryonic cells is about 100-fold greater than that observed in cancer cells. This very high specificity for cancer cells is associated with the absence of side effects associated with 2OHOA.