Abstract
Atherosclerosis commonly causes coronary and cerebrovascular diseases, which are major morbidities worldwide. Controlling these conditions remains a challenge owing to an incomplete understanding of underlying molecular mechanisms. We have recently shown that PPM1D/WIP1 phosphatase plays a crucial role in regulating atherosclerosis in mice. Deletion of Ppm1d results in the suppression of lipid droplet accumulation in macrophages, which prevents the formation of foam cells, and ultimately the development of atherosclerotic plaques. This process is controlled by the ATM-MTOR pathway and depends on the activation of selective autophagy to regulate cholesterol efflux from macrophage foam cells. Our data suggest that modulating autophagy through the PPM1D-ATM-MTOR pathway may be beneficial at both early and advanced stages of atherosclerosis.
Atherosclerosis is the major underlying cause of serious cardiovascular diseases such as heart attack and stroke. Macrophage conversion into foam cells plays a crucial role in the early stages of atherosclerosis. Uptake of oxidized low-density lipoproteins (oxLDLs) by macrophages leads to intracellular cholesterol accumulation in the form of lipid droplets (LDs), and in turn the formation of foam cells. Because macrophages are incapable of processing oxLDLs, they grow and ultimately rupture and deposit additional cholesterol into arterial intima, resulting in chronic inflammation and narrowing arteries.
PPM1D phosphatase is emerging as a potent regulator of tumorigenesis. Its deletion results in a profound tumor-resistant phenotype in mice; in many instances, this effect is mediated through the PPM1D-dependent regulation of apoptosis. Ppm1d deletion enhances the apoptosis of B lymphocytes, thereby delaying the onset of tumor formation in a model of MYC-driven lymphomagenesis. Likewise, PPM1D deficiency inhibits polyp formation and increases mouse survival by enhancing the apoptosis of cancer-initiating intestinal stem cells in an adenomatous polyposis coli (APC)-driven colorectal cancer model. At the molecular level, PPM1D negatively regulates ATM, with deletion or knockdown of Ppm1d resulting in activation of the ATM‐TP53 tumor suppressor pathway. In tumorigenesis, TP53 is an important downstream target of ATM; Tp53 deletion results in the reversal of many (if not all) tumor-resistant phenotypes of PPM1D-deficient mice. Moreover, TP53 appears to be centrally involved in regulating the PPM1D-mediated decline in the proliferation of neural stem cells and progenitor cells, further supporting its role as a key downstream target of PPM1D.
Both ATM and TP53 have recently been shown to be associated with metabolic disorders such as excessive adiposity and atherosclerosis. We noticed that PPM1D-deficient mice are lean and exhibit reduced age-dependent weight gain. Therefore, we decided to explore further whether PPM1D is involved in obesity and atherosclerosis. Using intercrossed Ppm1d−/− and atherosclerosis-prone Apoe−/− mice, we found that PPM1D promotes diet-induced weight gain and fat accumulation in mice. Most intriguingly, PPM1D-deficient mice exhibit a reduced number and size of atherosclerotic lesions. By conducting additional crosses of Ppm1d−/− with Atm+/− and Tp53−/− mice, we demonstrated that deleting Atm, but not Tp53, mediates PPM1D-dependent resistance to weight gain, fat accumulation, high blood pressure and atherosclerosis.
We found that the accumulation of cholesteryl esters (and thus foam cell formation) is strongly dependent on the presence of PPM1D. Recent studies discovered that lipid metabolism can be regulated by autophagy when LDs are engulfed by autophagic machinery and delivered to lysosomes for breakdown, a process called lipophagy. To evaluate whether PPM1D is involved in lipophagy, we analyzed autophagy markers and observed that autophagic flux was activated in PPM1D-deficient cells, suggesting that Ppm1d deletion results in the activation of both the initiation and the progression of autophagy in lipid-loaded macrophages. In turn, siRNA-mediated knockdown of autophagy effectors ATG5 and ATG7 leads to the accumulation of cholesteryl esters and increased foam cell formation in Ppm1d−/− macrophages, confirming that autophagy plays a crucial role in LD deposition in PPM1D-deficient macrophages. In PPM1D-deficient macrophages, we also observed that MTOR kinase is downregulated through the activation of AMPK, and knockdown of TSC2 reverses the inhibitory effect of PPM1D deficiency on foam cell formation.
In agreement with a recent study, we observed that cholesterol efflux is significantly enhanced in PPM1D-deficient macrophages. Autophagy-dependent cholesterol efflux in PPM1D-deficient macrophages relies on the activity of lysosomal acid lipase to generate free cholesterol for efflux, and this mechanism may be particularly relevant to the reversal of early atherosclerotic lesions. At early stages, cholesterol is predominantly accumulated in cytosolic LDs. These cytosolic LDs can be subsequently fused with autophagosomes and lysosomes to produce free cholesterol for efflux if autophagy is activated, as in PPM1D-deficient macrophages (). PPM1D deficiency attenuates early foam cell formation, thereby delaying atherosclerosis, even in mice that have been on a high-fat diet for as long as 14 weeks.
Figure 1. A role for PPM1D in atherosclerosis. In early atherosclerosis, macrophages take up oxLDL, process it and deposit cholesterol esters in the form of cytoplamic LDs as shown in (A). If autophagy is activated through the PPM1D-ATM-MTOR pathway, LDs are fused with autophagosome and subsequently with lysosomes, free cholesterol is produced, and it is subsequently effluxed from the cell. In advanced stages of atherosclerosis (B), cholesterol is trapped in lysosomes of foam cells. The prediction is that the PPM1D/ATM‐dependent inhibition of the MTOR pathway at this stage could also lead to activation of autophagy-dependent cholesterol efflux as well as LC3-associated clearance of dead cells.
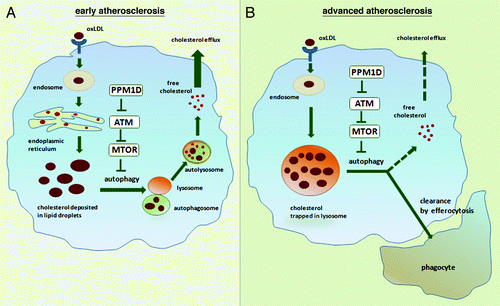
In light of recent data revealing features of defective autophagy in advanced atherosclerotic lesions, it would be interesting to know if PPM1D inactivation (conditionally or with a drug) would have any protective effect during later stages of atherosclerosis. In contrast to the early stages of atherosclerosis, in advanced lesions cholesterol is primarily trapped in lysosomes of foam cells (). Our results suggest that PPM1D might play a role in the progression of advanced lesions through its ability to modulate autophagy through ATM and MTOR signaling pathways. Even in the absence of noticeable LD accumulation, as with chemical inhibition of cholesterol esterification, there is an autophagy-attributable efflux to APOA1. Chemical inhibition of cholesterol esterification, which takes place in the endoplasmic reticulum (ER), prevents cholesterol deposition in LDs; however, a similar outcome can also be achieved through the autophagy-dependent degradation of the ER itself, a process called reticulophagy. In this scenario, activation of autophagy would redirect ER-enriched cholesterol to fuse with autophagosomes, and subsequently with lysosomes for degradation, thus ultimately reducing LD accumulation and foam cell formation.
In advanced atherosclerotic lesions, lysosomes with trapped unprocessed cholesterol are likely to become functionally inactive. However, it is not known whether this altered non-functional lysosomal storage of cholesterol can be redirected for autophagy-dependent degradation. If this process is possible, it might provide further justification to activate autophagy in advanced stages of atherosclerosis. In addition, the in vivo protective effects of autophagy in advanced atherosclerotic lesions may be due to autophagy-dependent reduction in apoptosis and simultaneous increase in LC3-associated phagocytic clearance of the dead cells (). In vivo activation of autophagy will delay cell death if repair is possible, but otherwise will also ensure that the cells are efficiently cleared by phagocytes, a mechanism that would be particularly critical in later stages of atherosclerosis. In addition, numerous reports have suggested that the systemic administration of MTOR inhibitors attenuates plaque progression at later stages of atherosclerosis, likely through the regulation of autophagy; however, additional mechanisms cannot be excluded.
In summary, our study revealed an important role for PPM1D phosphatase in the regulation of autophagy-dependent cholesterol efflux through the ATM-MTOR signaling pathway, and therefore in macrophage foam cell formation. These findings may provide the basis to design novel therapeutic strategies for efficient cholesterol removal from foam cells, and thereby reduce lipid load in early and advanced atherosclerotic plaques. Hence, macrophage-specific activation of autophagy through the regulation of the PPM1D-ATM-MTOR pathway may provide new prospects for developing drugs to treat atherosclerosis.