Abstract
Channel activators (potentiators) of cystic fibrosis (CF) transmembrane conductance regulator (CFTR), can be used for the treatment of the small subset of CF patients that carry plasma membrane-resident CFTR mutants. However, approximately 90% of CF patients carry the misfolded ΔF508-CFTR and are poorly responsive to potentiators, because ΔF508-CFTR is intrinsically unstable at the plasma membrane (PM) even if rescued by pharmacological correctors. We have demonstrated that human and mouse CF airways are autophagy deficient due to functional sequestration of BECN1 and that the tissue transglutaminase-2 inhibitor, cystamine, or antioxidants restore BECN1-dependent autophagy and reduce SQSTM1/p62 levels, thus favoring ΔF508-CFTR trafficking to the epithelial surface. Here, we investigated whether these treatments could facilitate the beneficial action of potentiators on ΔF508-CFTR homozygous airways. Cystamine or the superoxide dismutase (SOD)/catalase-mimetic EUK-134 stabilized ΔF508-CFTR at the plasma membrane of airway epithelial cells and sustained the expression of CFTR at the epithelial surface well beyond drug withdrawal, overexpressing BECN1 and depleting SQSTM1. This facilitates the beneficial action of potentiators in controlling inflammation in ex vivo ΔF508-CFTR homozygous human nasal biopsies and in vivo in mouse ΔF508-CFTR lungs. Direct depletion of Sqstm1 by shRNAs in vivo in ΔF508-CFTR mice synergized with potentiators in sustaining surface CFTR expression and suppressing inflammation. Cystamine pre-treatment restored ΔF508-CFTR response to the CFTR potentiators genistein, Vrx-532 or Vrx-770 in freshly isolated brushed nasal epithelial cells from ΔF508-CFTR homozygous patients. These findings delineate a novel therapeutic strategy for the treatment of CF patients with the ΔF508-CFTR mutation in which patients are first treated with cystamine and subsequently pulsed with CFTR potentiators.
Introduction
Autophagy is a regulated pathway involving the lysosomal degradation of cytoplasmic organelles or cytosolic components.Citation1,Citation2 In the last few years, autophagy has emerged not just as a simply degradative process, but also as a cellular mechanism essential for the maintenance of cellular homeostasis and of the energetic balance.Citation3,Citation4 Thus, an ever-growing number of publications reveals that autophagic dysfunction is associated with several human pathologies. Disabled autophagy is directly involved in the pathogenesis of multiple diseases including cancer, viral infection, neurodegenerative diseases and chronic inflammatory disease.Citation5,Citation6 We and others demonstrated that human and mouse cystic fibrosis (CF) airways are autophagy deficient,Citation7-Citation9 exhibit highly reduced autophagosome formation, display a defect in mitochondrial membrane depolarization and accumulate SQSTM1/p62 (SQSTM1).Citation7,Citation8
CF is an autosomal recessive disorder, the most common lethal genetic disease in Caucasians, characterized by chronic lung disease, the main cause of morbidity and mortality, pancreatic dysfunction, raised electrolyte levels in sweat, and male infertility.Citation10-Citation12 CF is caused by mutations of one single protein, the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-regulated chloride channel that is primarily located at the apical membrane of epithelial cells.Citation13-Citation15 Although more than 1,500 different disease-associated mutations have been identified, a single codon deletion, ΔF508, occurs in about 90% of CF patients on at least one allele.Citation16 Due to its misfold, ΔF508-CFTR loses its essential ion channel activity at the plasma membrane (PM), thus provoking local inflammation, increased susceptibility to respiratory bacterial infections, and progressive pulmonary and digestive insufficiency.Citation10,Citation17,Citation18
Most candidate drugs are being tested to treat CF symptoms and complications, downstream of the loss-of-function of CFTR. Citation18-Citation21 However, an ideal CF drug should be capable of improving the upstream cause of CF and hence rescue a functional CFTR at the epithelial surface.Citation18 Therefore, over the last few years, several rationale mutation specific (or “CFTR-repairing”) therapeutic strategies have emerged.Citation22
Still more innovative strategies, such as manipulation of proteostasis have recently been proposed for the treatment of CF.Citation22,Citation23 We previously reported that defective CFTR leads to the inhibition of autophagy in the epithelial cells of human and mouse CF airways.Citation7,Citation8 Increased levels of reactive oxygen species (ROS), induced by defective CFTR function, lead to tissue transglutaminase-2 (TGM2) activationCitation24,Citation25 driving cross-linking and aggresome accumulation of several TGM2-substrate proteins,Citation24 including the sequestration of the essential autophagy protein BECN1.Citation7 The functional sequestration of BECN1 dislodges the PtdIns3K complex III away from the endoplasmic reticulum (ER), thus inhibiting autophagosome formation.Citation7 Defective autophagy was also confirmed in CF macrophages.Citation9 Rescuing autophagy is an emerging approach to treat several human conformational diseases.Citation6 Restoring BECN1 and autophagy, either by transfection-enforced BECN1 overexpression or by means of TGM2 inhibitors (e.g., cystamine) or antioxidants (e.g., N-acetyl-cysteine or the superoxide dismutase (SOD)/catalase-mimetic EUK-134), blunts inflammation in ΔF508-CFTR homozygous airways, both in mice in vivo and in human tissues, in vitro.Citation7,Citation8
A still partially functional ΔF508-CFTR can be rescued at the plasma membrane (PM) by molecules that correct ΔF508-CFTR intracellular retention and degradation (correctors).Citation22,Citation23,Citation26-Citation28 However, ΔF508-CFTR that reaches the PM is unstable as result of a [carboxyl-terminus heat shock cognate 70 (HSP70)–interacting protein] (CHIP)-mediated ubiquitination, followed by redirection of the protein from endosomal recycling toward lysosomal delivery and degradation.Citation29,Citation30 Therefore, CF patients carrying the misfolded ΔF508-CFTR are poorly responsive to potentiators of CFTR channel activity that can be used for the treatment of the small subset of CF patients that carry PM-resident CFTR mutants.Citation31,Citation32 A recent clinical trial with the CFTR corrector VX-809 in ΔF508-CFTR homozygous patients demonstrated modest dose-dependent reductions in sweat chloride.Citation33 However, no improvement in lung function or CF complications was reported,Citation33,Citation34 and Phase II clinical studies combining VX-809 and the potentiator VX-770 have to be awaited to evaluate their clinical benefit.Citation34
We have demonstrated that restoring BECN1 or reducing the levels of SQSTM1, a major autophagic substrate,Citation35 can rescue ΔF508-CFTR trafficking to the PM of CF airway epithelial cells.Citation7,Citation8 Here, we explored the possibility that these treatments might conserve functional ΔF508-CFTR at the epithelial surface and enable the beneficial action of potentiators on ΔF508-CFTR homozygous airways. We show that this is the case and outline a novel strategy for improving the function of ΔF508-CFTR.
Results and Discussion
Restoration of autophagy stabilizes functional ΔF508-CFTR expression at the epithelial surface of CF airways
Manipulating proteostasis network has emerged as a novel approach to correct protein misfolding in conformational diseases.Citation23,Citation36 We have described that reducing SQSTM1 expression, restoring autophagy by overexpressing BECN1, as well as addition of proteostasis regulators (PRs), such as cystamine (a transglutaminase-2 inhibitor) or EUK-134 (a superoxide dismutase (SOD)/catalase-mimetic), can improve ΔF508-CFTR trafficking to the plasma membrane (PM) of CF airway epithelial cells.Citation7,Citation8 However, it has been elusive whether manipulating peripheral proteostasis might help improve ΔF508-CFTR PM residence and function.Citation37 Driven by the consideration that CF constitutes the quintessential example of a “conformational disease,”Citation23,Citation36 we wondered whether rescuing autophagy might have positive effects on PM residence and stability of rescued ΔF508-CFTR, thus amplifying the beneficial action of potentiators on ΔF508-CFTR homozygous airways.
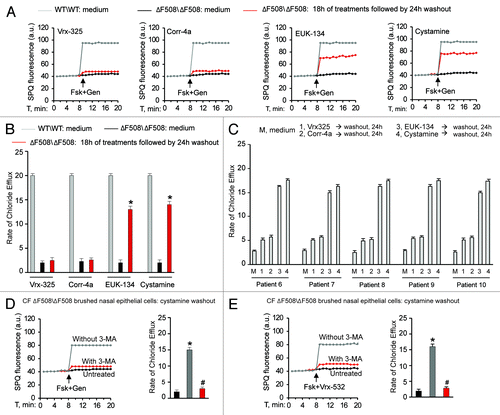
First, we investigated whether PRs, such as cystamine or EUK-134, improve the ΔF508-CFTR PM stability and sustain the residence of functional ΔF508-CFTR at the plasma membrane of CF airway epithelial cells after rescue. For this, we developed an assay in which airway epithelial cells carrying ΔF508/ΔF508 or ΔF508/W1282X CFTR mutations (parental CFBE41o- and IB3-1 cells, respectively),Citation7,Citation29,Citation38 which we refer to as “CF cells,” were first incubated with PRs or the CFTR correctors Corr-4a or Vrx-325Citation26 at 37°C for 18 h and then washed and re-cultured for up to 12 h in the presence of cycloheximide (CHX, 100 μg ml−1, refreshed every 6h) to inhibit protein neosynthesis.Citation29 CHX toxicity in this system was excluded by a 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) cell viability assay.Citation39 The efficacy of protein synthesis inhibition by CHX was validated by the absence of ΔF508-CFTR band B, an immature form of neosynthesized ΔF508-CFTR that is retained in the endoplasmic reticulum and prematurely destroyed, after 12 h of incubation with CHX (Fig. S1A and S1B). Either cystamine or EUK-134, but not correctors, increased the permanence of mature glycosylated ΔF508-CFTR band C at the PM, even in the presence of CHX (; Fig. S2A). Assessment of CFTR channel activity by means of SPQ halide efflux assaysCitation29,Citation40,Citation41 on at least 50 cells per group of treatment revealed that PRs, but not correctors, sustained the ΔF508-CFTR response to forskolin (Fsk) added together with either of two CFTR potentiators genistein or Vrx-532 well beyond drug washout (; Fig. S2B). Similar results were observed in a polarized culture system of CFBE41o- cellsCitation42 (Fig. S3A and S3B).
Figure 1. Cystamine and EUK-134 reduce the turnover of plasma membrane (PM)-located ΔF508 CFTR in human CF airway epithelial cells. (A) CFBE41o- cells were transfected with ΔF508-CFTR at 37°C. Twenty-four hours after transfection the cells were incubated for 18 h with or without Vrx-325, Corr-4a, EUK-134, or cystamine and then with medium for further 12 h in the presence of CHX (100 µg ml−1). Top, surface biotinylation followed by purification of streptavidine-binding PM proteins and immunoblot with anti-CFTR (clone M3A7). E-Cadherin confirmed cell surface protein-specific localization. Bottom, densitometric measurement of residual ΔF508-CFTR band C at PM expressed as percentage of initial amount after rescue normalized to E-cadherin levels; Mean ± SD of triplicates of three independent experiments, *p < 0.05 compared with the corresponding value after rescue (ANOVA). (B) CFBE41o- cells treated as in (A). Fluorescence-based assessment of forskolin plus genistein (Fsk+Gen)-stimulated iodide efflux. SPQ fluorescence intensity (a.u.) (left) and rate of chloride efflux (right) in at least 50 cells per experiment. Mean ± SD of 3 experiments, *p < 0.05 compared with CFBE41o- cells cultured with medium (ANOVA).
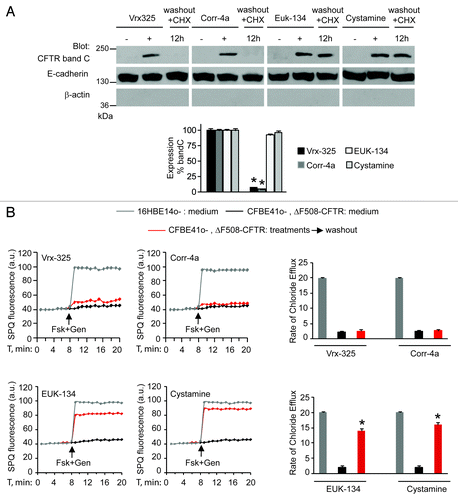
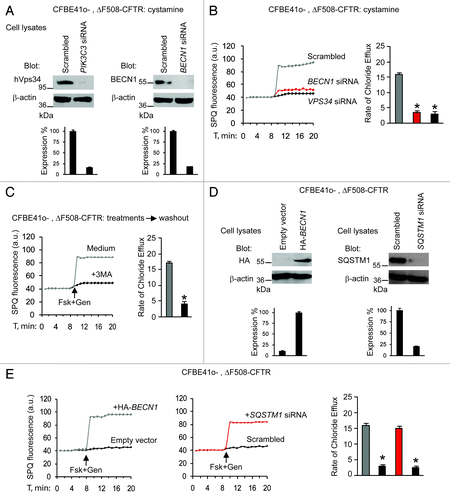
Next, we examined whether the beneficial effects of cystamine on ΔF508-CFTR function would be linked to its capacity to restore the autophagy-stimulatory function of the BECN1/PIK3C3 complexCitation2,Citation3,Citation7 and hence to reduce the abundance of SQSTM1.Citation7,Citation35 siRNA-mediated depletion of BECN1 or PIK3C3, as well as pharmacological inhibition of PIK3C3 activity with 3-methyl-adenine (3-MA),Citation2,Citation3,Citation7 abolished the maintenance of functional ΔF508-CFTR by cystamine in CF cell lines (). Moreover, CF cells were transfected with HA-tagged BECN1 or the empty vector as well as with SQSTM1-specific siRNAs or control siRNAs (). SPQ halide efflux assays on at least 50 cells per group of treatment, revealed that both BECN1 overexpression and SQSTM1 depletion allowed the ΔF508-CFTR response to Fsk added together with either of two CFTR potentiators after 24 h following transfection (; Fig. S4A). Altogether, these results indicate that cystamine and EUK-134 can conserve functional ΔF508-CFTR at the PM of CF epithelial cells by means of their ability to restore autophagy.
Figure 2. Cystamine and EUK-134 increase ΔF508 CFTR function in human CF airway epithelial cells through rescuing BECN1. (A–C) CFBE41o- cells transfected with ΔF508-CFTR and then incubated for 18 h with cystamine followed by 12 h of incubation with medium in the presence of the indicated siRNAs or 3-MA. (A) Validation of siRNAs. Immunoblot after transfection with either scrambled or indicated siRNAs. (B and C) Fluorescence-based assessment of Fsk+Gen-stimulated iodide efflux. SPQ fluorescence intensity (a.u.) (left) and rate of chloride efflux (right) in at least 50 cells per experiment in CFBE41o- cells transfected with siRNAs (B) or incubated with 3-MA (C). Mean ± SD of 3 experiments, *p < 0.05 compared with CFBE41o- cells transfected with scrambled siRNAs or incubated with medium, respectively ANOVA). (D and E) CFBE41o- cells transfected with ΔF508-CFTR in the presence of the enforced expression of HA-tagged BECN1 (HA-BECN1) or SQSTM1 siRNA. (D) Validation of HA-BECN1 transfection (left) or SQSTM1 siRNAs (right). Immunoblot of corresponding proteins. (E) Fluorescence-based assessment of Fsk+Gen-stimulated iodide efflux. SPQ fluorescence intensity (a.u.) (left) and rate of chloride efflux (right) in at least 50 cells per experiment in CFBE41o- cells after transfection.
CF cells cultured at 37°C fail to express ΔF508-CFTR in their PM because at this temperature mutant ΔF508-CFTR is thermolabile.Citation43 However, when CF cells are cultured at a low temperature (26°C), which stabilizes ΔF508-CFTR, they re-express functional CFTR.Citation44,Citation45 Again, this low temperature-rescued ΔF508-CFTR is rapidly dismissed from the PM within a few hours after shifting temperature to 37°C.Citation29 PRs can rescue ΔF508-CFTR, as previously reportedCitation7 and stabilize mutant CFTR at the epithelial surface, as described above, by reducing the abundance of ROS and the activity of TGM2 as they restore autophagy.Citation7 We determined whether treatment with PRs would stabilize functional ΔF508-CFTR in these conditions as well. For this, we developed an assay in which ΔF508-CFTR was first rescued by culture at 26°C for 30 h and the cells were then kept at 37°C for 6 h with cycloheximide alone or in combination with additional pharmacological agents. Cystamine and EUK-134, but neither of the correctors Corr-4a and Vrx-325, stabilized the PM expression of ΔF508-CFTR in these conditions (Fig. S5A and S5B), as they controlled the oxidative stress following temperature shifting (Fig. S6A–C) in a 3-MA inhibitable way (data not shown). Altogether these in vitro results suggest that PRs including cystamine and EUK-134 can restore the environment in which ΔF508-CFTR operates.
Stabilizing ΔF508 at the PM of airway epithelial cells by PRs controls signs of inflammation well beyond drug washout
Defective CFTR mediated oxidative stress, NFκB activation and signs of inflammation characterize CF airway epithelial cells.Citation7,Citation17,Citation18 Restoring CF environment by rescuing autophagy by PRs can interrupt a feed forward loop that sustains inflammation in CF airways.Citation7 Therefore, we examined whether stabilizing a functional CFTR by PRs and rescuing autophagy in CF epithelia could reduce signs of inflammation beyond drug washout. We previously reported that in CF epithelial cells, cystamine induces an increase in peroxisome proliferator activated receptor-γ (PPARγ) levels and a decrease in p42–44 MAPK phosphorylation, TNFA secretion and TGM2 SUMOylation.Citation7,Citation24,Citation25 Here, we report that these effects persisted beyond cystamine washout for 24 h (Fig. S7A), unless either BECN1 (data not shown) or CFTR were depleted by siRNAs during cystamine washout (Fig. S7A).
Beneficial effects of depleting Sqstm1 on CftrF508del mice
The aforementioned results indicate that the pretreatment with PRs improves CFTR function to control signs of inflammation, presumably by rescuing autophagy. Fixing the misfolded ΔF508-CFTR mutant at the PM after rescue is the principal objective of “CFTR-repairing” therapies to enable the beneficial action of CFTR potentiators, thus ensuring a proper control of lung inflammation.Citation34 Therefore, we aimed at translating these findings in vivo, into a mouse model of CF. For this, we took advantage of mice homozygous for the ΔF508-CFTR mutation in the 129/FVB outbred background (Cftrtm1EUR, F508del, FVB/129) (CftrF508del), an established animal model of CF,Citation46-Citation48 that shows constitutive lung inflammation.Citation7,Citation48
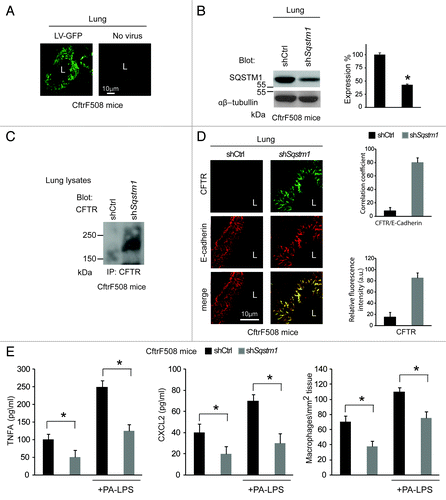
First, CftrF508del mice were transduced intranasally with pLKO.1-CMV-tGFP lentiviral vectors expressing mouse shRNA specific for Sqstm1 or no target sequence (control shRNA) (n = 5 for each group of treatment) (), following a previously described protocol.Citation7 In this setting, Sqstm1 depletion induced the appearance of ΔF508-CFTR Band C () and the relocation of mutant CFTR at the lung epithelial surface lining the airway lumen (). Sqstm1 shRNA significantly decreased the expression of validated markers of lung inflammation,Citation7,Citation25,Citation48 as it reduced the mRNA expression of the inflammatory cytokine Tnfa, the levels of Tnfa and CXCL2 protein, and macrophage infiltration in lungs (p < 0.001), as compared with control shRNA treated CftrF508del mice (; Fig. S8A). Constitutive lung inflammation in CftrF508del mice is enhanced by challenge with PA-LPS.Citation48 Therefore, Sqstm1 shRNA treated mice were challenged with aerosolized PA-LPS (10 μg/20 g body weight) or PBS as a control and sacrificed after 24 h. Sqstm1 shRNA significantly decreased PA-LPS triggered lung inflammation (p < 0.001), as compared with control shRNA treated CftrF508del mice (; Fig. S8A).
Figure 3. Direct Sqstm1 depletion ameliorates either constitutive or LPS-triggered lung inflammation in vivo in CftrF508del homozygous mice. (A) GFP expression in airway tissues from CftrF508del mice after intranasal transduction of a lentiviral vector expressing the GFP (LV-GFP). L, Lumen. Scale bar: 10 μm. (B) Immunoblot and densitometric analysis of SQSTM1 in lungs of CftrF508del mice 4 d after intranasal transduction with a lentiviral vector encoding shSqstm1 or no target sequence (shCtrl) (n = 5 for each group; *p < 0.01; ANOVA). (C) Immunoblot of CFTR (clone CF3) after immunoprecipitation of whole lung lysates with anti-CFTR antibody (clone H182). (D) Left, confocal microscopy images of CFTR (clone H182) and E-cadherin in lung tissues from mice treated as in (B). L, Lumen. Scale bar: 10 μm. Right, relative fluorescence intensity and coefficient of co-localization. (E) ELISA detection of TNFA and CXCL2 protein levels in lung homogenates (left and middle) and number of CD68+ macrophages (per mm2 of lung tissue) counted in 15–20 different random selected fields per lung per mouse for each experimental group (right) in the absence or presence of PA-LPS challenge. Mean ± SD of triplicates for each experiment. *p < 0.01 vs shControl (shCtrl) (ANOVA).
SQSTM1 levels and lung inflammation can be reduced in vivo by the enforced expression of BECN1 through the intranasal transduction of CftrF508del mice with a lentiviral vector encoding Becn1 under the control of the ubiquitous CMV promoter (LV-Becn1).Citation7 We examined whether this treatment would rescue CFTR at the lung epithelial surface in a 3-MA inhibitable way. As compared with untreated or LV-GFP-transduced controls, LV-Becn1 strikingly enhanced the localization of CFTR protein at the respiratory epithelial surface lining the airway lumen (Fig. S9A). Furthermore, another group of CftrF508del mice was intraperitoneally administered with 3-MA (50 mg kg−1 of 3-MA in 100 ml PBS for 5 d),Citation7 in the context of LV-Becn1 or LV-GFP transduction (n = 5 for each group). 3-MA abrogated the effects of LV-Becn1 in reducing SQSTM1 and restoring autophagy (data not shown), as well as in rescuing CFTR localization at the epithelial surface (Fig. S9A). Moreover, the LV-Becn1-mediated reduction in Tnfa production (p < 0.05), CXCL2 protein levels and macrophage counts in lung tissues of CftrF508del mice, were abrogated by 3-MA treatment (Fig. S9B). In contrast, 3-MA had no discernible effects on LV-GFP-transduced control CftrF508del mice (data not shown).
Depleting Sqstm1 enables the action of genistein to control lung inflammation in vivo in CftrF508del mice
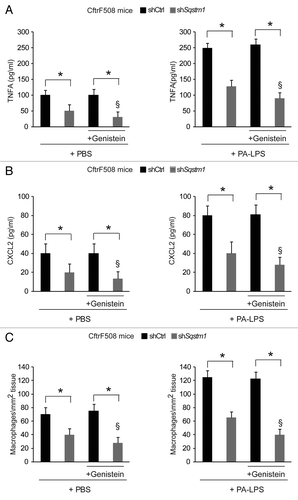
Next, we examined whether the re-expression of ΔF508-CFTR at the epithelial surface induced by Sqstm1 depletion, could enable the action of CFTR potentiator genisteinCitation23,Citation26,Citation27,Citation40,Citation41 in rescuing lung inflammation in vivo. To this aim, Sqstm1-specific shRNA or control shRNAs treated CftrF508del mice were challenged once with genistein or vehicle followed by a final challenge with aerosolized PA-LPS or PBS as a control and sacrificed after 24 h, as described above. While genistein had no effects on its own (), it significantly reduced either the constitutive (, left panels) or PA-LPS-triggered (, right panels) expression of TNFA and CXCL2 protein and decreased macrophage infiltration in lungs from Sqstm1-depleted but not from control shRNAs-treated mice (p < 0.001) (). No effects of sh Sqstm1 or genistein were observed in WT littermates (not shown). This indicates that depletion of Sqstm1 in vivo enables the beneficial action of genistein on ΔF508-CFTR.
Figure 4. Direct Sqstm1 depletion enables the activity of Genistein in controlling either constitutive or LPS-triggered lung inflammation in vivo in CftrF508del homozygous mice. (A–C) Four days after transduction with shControl (shCtrl) or shSqstm1, CftrF508del mice were treated once with intraperitoneal PBS or genistein (Gen) followed by aerosolized PBS (left panels) or PA-LPS (right panels) challenge and sacrificed after 24 h. ELISA detection of TNFA (A) and CXCL2 (B) protein levels in lung homogenates. (C) Number of CD68+ macrophages (per mm2 of lung tissue) counted in 15–20 different random selected fields per lung per mouse for each experimental group. Mean ± SD of triplicates for each experiment. *p < 0.01; §p < 0.05 vs shSQSTM1 in the absence of treatment with Genistein (ANOVA).
Cystamine re-establishes functional ΔF508-CFTR expression in CftrF508del mice well beyond drug washout and enables the beneficial action of genistein to control lung inflammation in vivo
We wondered whether prior pharmacological restoration of autophagy by cystamine would abrogate the reported lack of efficacy of CFTR potentiators in vivo on ΔF508-CFTR homozygous airways.Citation31-Citation34 CftrF508del mice were administered intraperitoneally (i.p.) for 7 d with vehicle alone or cystamine, a regimen that can ameliorate lung inflammation in vivo,Citation7,Citation8,Citation25 or with cystamine (7 d) followed by 10 d of vehicle only. In this experimental model, the effects of cystamine in sustaining the relocation of ΔF508 protein at the respiratory epithelial surface () and in reducing lung inflammation (; Fig. S10A), persisted for 10 d after cystamine withdrawal and were abrogated by intraperitoneal treatment with 3-MA during the phase of withdrawal (not shown).
Figure 5. Prior restoration of autophagy by cystamine enables the activity of Genistein in controlling lung inflammation in CftrF508del homozygous mice. (A and B) CftrF508del mice were treated intraperitoneally (i.p.) for 7 d with PBS or cystamine followed by 10 d of PBS (n = 7 mice per each group of treatment). (A) Left, confocal microscopy images of CFTR (clone H182) and E-cadherin in lung tissues from mice. L, Lumen. Scale bar: 10 μm. Right, relative fluorescence intensity and coefficient of colocalization. (B) ELISA detection of TNFA and CXCL2 protein levels in lung homogenates (left and middle) and number of CD68+ macrophages (per mm2 of lung tissue) counted in 15–20 different random selected fields per lung per mouse for each experimental group (right). Mean ± SD of triplicates for each experiment. *p < 0.01, **p < 0.001 (ANOVA). (C) CftrF508del mice (n = 5 mice per group of treatment) were treated with nebulized PBS or cystamine for 7 d and then were pulsed once with intraperitoneal PBS or genistein (Gen) followed by aerosolized PBS or PA-LPS challenge and sacrificed after 24 h. ELISA detection of TNFA protein levels in lung homogenates (left) and CD68+ macrophages counts (right) measured as in (B). Data represent two pooled experiments (n = 5 mice per group). *p < 0.01, §p < 0.05 vs mice pre-treated with cystamine and pulsed once with intraperitoneal PBS (ANOVA).
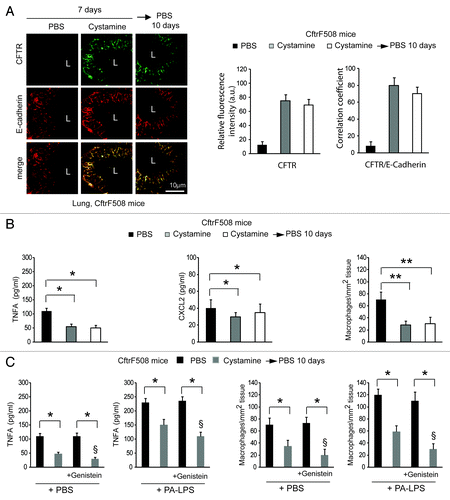
Another group of 5 CftrF508del mice was treated with daily inhalations of aerosolized cystamine (0.2 mg/mouse/day) for one week, a regimen that reduced lung inflammation (data not shown), kept for other 10 d without treatment, and then sequentially pulsed with genistein and either aerosolized PA-LPS or PBS, as above detailed. Genistein further improved the effects of cystamine pretreatment and hence significantly reduced both constitutive (, left panels) and PA-LPS triggered (, right panels; Fig. S10B) signs of lung inflammation, but had no effect on its own, in mice that had not pretreated with cystamine (). Cystamine and genistein were both not effective in wt littermates (data not shown).
Altogether these results indicate that prior restablishment of proteostasis and restoration of autophagy can stabilize functional ΔF508-CFTR at the airway epithelial surface, thus creating the conditions in which CFTR potentiators become capable of alleviating lung inflammation.
Cystamine re-establishes ΔF508-CFTR expression in human nasal polyp mucosae from CF patients
To translate these findings to human CF airways, we used an ex-vivo model of cultured nasal polyp biopsies belonging to CF patients, a useful tool to test potential strategies of modulation of mucosal response to environmental triggers within their natural environment.Citation7,Citation24,Citation25,Citation49
Nasal polyp biopsies from five ΔF508/ΔF508 patients (, Patients #1–5) were treated with cystamine for 18 h, washed and then cultured for further 36 h in the absence of cystamine. The cystamine-mediated rescue of CFTR at the epithelial surface persisted for 36 h after cystamine withdrawal and was abrogated by 3-MA (; Fig. S11A). Moreover, the reduction of signs of mucosal inflammation induced by cystamine, as TGM2 activation and protein tyrosine phosphorylation,Citation7,Citation24,Citation25 persisted after 36 h of washout unless 3-MA was added to the system (; Fig. S11B).
Table 1A. Clinical characteristics of cystic fibrosis patients
Figure 6. Cystamine sustains ΔF508-CFTR at the airway epithelial surface and controls signs of inflammation beyond drug washout in nasal mucosa from ΔF508-CFTR homozygous CF patients. (A and B) Nasal polyp biopsies from ΔF508 homozygous patients cultured for 18 h with medium, or cystamine, followed by 36 h of incubation with medium alone in the presence or absence of 3-MA. (A) Left, confocal microscopy images of CFTR and E-cadherin. Nuclear counterstaining with DAPI in merged images. Scale bar: 10 μm. Data representative of five patients. Right, coefficient of colocalization. (B) Confocal images of in situ detection of TGM2 activity and phospho-tyrosine (anti-PY-99 Ab). Data representative of five patients. Scale bar: 10 μm. (C) Immunoblot of phospho-tyrosine (anti-PY-99 Ab) after immunoprecipitation of whole mucosal lysates with anti-PY99 antibody. (D) Confocal microscopy images of phospho-tyrosine (anti-PY-99 Ab). Left, nasal polyp biopsies from ΔF508/ΔF508 patients (n = 5) pulsed for 30 min with medium or genistein (Gen) followed by 4 h of PA-LPS challenge. Middle, pulses of 30 min with genistein (Gen) followed by 4 h of PA-LPS challenge in nasal polyp biopsies from ΔF508/ΔF508 patients (n = 5) pre-treated with cystamine for 18 h, then washed and kept in medium alone for 24 h without or with 3-MA. Right, relative fluorescence intensity. At least 30 fields were randomly sampled on three slides from each patient. Scale bar: 10 µm.
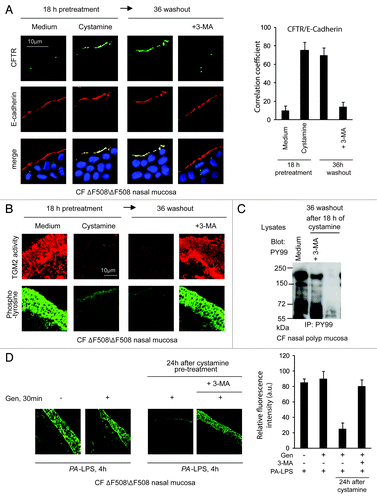
Next, ΔF508/ΔF508 nasal polyp biopsies were pulsed for 30 min with genistein followed by 4 h incubation with PA-LPS. Genistein alone was not effective in reducing epithelial protein phosphorylation (). To examine whether pretreatment with cystamine was effective in improving the action of genistein on human airways, nasal polyp biopsies were incubated with cystamine or medium for 18 h, then washed and kept in medium alone for 24 h and finally pulsed for 30 min with genistein followed by 4 h PA-LPS. In this experimental setting a synergistic (and 3-MA-inhibitable) effect of sequential treatment with cystamine plus genistein was observed ().
Our results indicate that in our system genistein has no effect on its own, but instead potentiates the activity of ΔF508-CFTR, which is still resident at the epithelial surface well beyond washout after cystamine pre-treatment.
Cystamine and antioxidants induce and sustain channel function of rescued ΔF508-CFTR in primary brushed nasal epithelial cells from ΔF508/ΔF508 patients
Targeting TGM2 by cystamine or reducing the levels of ROS, which sustain TGM2 protein expression,Citation7,Citation8,Citation25 can favor ΔF508-CFTR trafficking to the surface of the airway epithelial cells through restoring BECN1 and autophagy and reducing SQSTM1 levels in human and mice CF airway epithelial cells.Citation7,Citation8 Therefore, we examined whether restoring BECN1 and autophagy by means of cystamine or EUK-134Citation7,Citation25 would induce and sustain functional ΔF508-CFTR expression in freshly isolated brushed nasal epithelial cellsCitation51 from ΔF508/ΔF508 patients (, Patients #6–10). We used primary nasal epithelial cells freshly obtained from ΔF508-CFTR homozygous patients to directly test the efficacy of these autophagy-rescuing strategies on CFTR function in human airways. We used SPQ halide efflux assay to test the response to a short pulse of Fsk added together with either of two different CFTR potentiators genistein or Vrx-532, as above described for CF cell lines, and analyzed at least 50 brushed nasal cells for group of treatment in each patient. Freshly isolated brushed nasal epithelial cells were incubated for 18 h with either cystamine or EUK-134 or with the CFTR correctors Corr-4a and Vrx-325. In contrast to Corr-4a and Vrx-325, which only had scarce effects, transient exposure (18 h, followed by wash and reculture for 24 h with medium) to cystamine and EUK-134 rendered the cells capable of conserving ΔF508-CFTR response to Fsk added with either genistein () or Vrx-532 (Fig. S12A–S12C). Finally primary nasal cells from ΔF508-CFTR homozygous patients were exposed to the most popular CFTR potentiator, Vrx-770, after pretreatment with either cystamine or EUK-134. We found that Vrx-770 was effective in stimulating the response to Fsk only when brushed nasal epithelial cells were pretreated with cystamine or EUK-134 (Fig. S13A). The positive effects of such a sequential treatment with PRs and CFTR potentiators, were abolished when 3-MA was added together with the first agent ().
Table 1B. Clinical characteristics of cystic fibrosis patients
Figure 7. Cystamine and EUK-134 sustain ΔF508-CFTR channel function beyond drug washout in freshly isolated brushed nasal epithelial cells from ΔF508-CFTR homozygous CF patients. (A–C) Freshly isolated brushed nasal epithelial cells from 5 ΔF508-CFTR homozygous patients were incubated for 18 h with medium or Vrx-325, Corr-4a, cystamine or EUK-134, washed and then kept with medium alone for 24 h. Brushed nasal epithelial cells from 5 non-CF controls were cultured with medium alone (WT/WT: medium). Assessment of iodide efflux by a fluorescence assay (SPQ) upon stimulation with forskolin (Fsk) plus genistein (Gen). SPQ fluorescence intensity (a.u.) (A) and rate of chloride efflux (B) measured in at least 50 cells per experiment. Mean ± SD of 3 experiments; *p < 0.01 vs ΔF508/ΔF508 cultured with medium (ΔF508/ΔF508: medium); ANOVA. (C) Individual values of the rate of chloride efflux of ΔF508/ΔF508 patients (patients n° 6–10 of ). (D and E) Freshly isolated brushed nasal epithelial cells from 5 ΔF508-CFTR homozygous patients were incubated for 18 h with cystamine, washed and then kept with medium alone for 24 h in presence or absence of 3-MA during cystamine washout. Assessment of iodide efflux by a fluorescence assay (SPQ) upon stimulation with forskolin (Fsk) added with either of two CFTR potentiators genistein (Gen) (D) or Vrx-532 (E). SPQ fluorescence intensity (a.u.) (left) and rate of chloride efflux (right). Mean ± SD of 3 experiments. *p < 0.05 vs untreated cells, #p < 0.01 vs cystamine pretreated cells without 3-MA treatment during washout (ANOVA).
Conclusion
Altogether our results outline a novel approach for the treatment of ΔF508 homozygous CF patients. Our data underscore a functionally important link between BECN1, SQSTM1 and ΔF508-CFTR residence at the surface epithelia of mouse and human ΔF508-CFTR homozygous airways, as they provide pre-clinical evidences that prior restoration of autophagy in CF airways could enable the action of CFTR potentiators in reducing lung inflammation. Restoring ΔF508-CFTR trafficking to the cell surface and preventing its rapid PM disposal can conserve enough mutant protein at the PM to enable the efficacy of CFTR potentiators in ameliorating lung inflammation. It is believed that achieving and maintaining only 10–20% of normal CFTR activity at the PM of epithelial cells would be sufficient to suppress the clinical CF phenotype.Citation50 Our results demonstrate that prior re-establishment of autophagy prolongs the persistence at the epithelial surface of a sufficient amount of ΔF508-CFTR to facilitate the beneficial action of CFTR potentiators. Based on these notions, we are planning to launch a clinical trial in which ΔF508 CF patients will be first treated with cystamine and subsequently pulsed with CFTR potentiators.
Beyond its application to CF, our study suggests a novel approach for the treatment of conformational diseases. Our results indicate that rescuing autophagy may represent a novel approach in human inherited loss-of-function diseases in which protein misfolding compromises the relocation of functional mutants to their site of activity. Manipulating the cellular mechanisms that ultimately link protein misfolding to protein malfunction, instead of specifically targeting misfolded proteins, might constitute a therapeutic strategy for the treatment of a range of conformational diseases.
Materials and Methods
Mice and treatments
CF mice homozygous for the ΔF508-CFTR mutation (abbreviated CftrF508del)Citation7,Citation46-Citation48 in the 129/FVB outbred background (Cftrtm1EUR, F508del, FVB/129) were obtained from Bob Scholte, Erasmus Medical Center Rotterdam, The Netherlands (CF coordinated action program EU FP6 LSHM-CT-2005-018932). These studies and procedures were approved by the local Ethics Committee for Animal Welfare (IACUC N° 382) and conformed to the European Community regulations for animal use in research (CEE no. 86/609). Young adult CF mice and their wild-type (wt) littermates were housed in static isolator cages at the animal care specific pathogen-free facility of Charles River (Calco, Italy). Anaesthetized CftrF508del homozygous mice received a single dose of 4 μl of 1% l-α-lysophosphatidylcholine (LPC; Sigma Aldrich, 62962) in PBS by inhalation-driven instillationCitation7 1 h before intranasal transduction with 1.2 × 107 transducing units (TU) per mouse delivered into the nose (2 10-μl aliquots over several minutes) of pLKO.1-CMV-tGFP validated lentiviral vectors expressing mouse shSqstm1 sequence (CCGGTAGTACAACTGCTAGTTATTTCTCGAGAAATAACTAGCAGTTGTACTATTTTTG) or no target sequence (Sigma Aldrich) (n = 5 mice per group of treatment) . Four days after transduction, animals received intraperitoneal injection of genistein (50 mg/kg, Sigma Aldrich, G6649) 1 h before aerosolized lipopolysaccharide from Pseudomonas aeruginosa (PA-LPS) (10 μg/20 g body weight, Sigma Aldrich, L4005) stimulation. Mice were then killed 24 h after PA-LPS stimulation and lung tissues were collected for analyses. CftrF508del mice were also treated with daily intraperitoneal injections of cystamine (100 µl of 0.01 M in PBS)Citation7 (Sigma Aldrich, 30050) or PBS for 7 d or with cystamine (7 d) followed by 10 d of PBS alone (n = 7 mice per group). Another group of CftrF508del mice received the GFP or Becn1 lentiviral vectors as previously described.Citation7 Mice were killed 5 d after vector administration, and lung tissues were collected for analyses. CftrF508del mice (n = 5) were also administered with 3-mehtyl-adenine (3-MA, Sigma Aldrich, M9281) (intraperitoneal injection of 50 mg kg–1 in 100 μl of PBS) during the Becn1 transduction. Another group of CftrF508del mice was treated by inhalation of nebulized cystamine (0.2 mg/mouse/day) or vehicle for one week and 10 d following cystamine withdrawal were pulsed with a single intraperitoneal administration of genistein (50 mg kg–1) followed by a challenge with aerosolized PA-LPS (10 μg/20 g body weight) (n = 7 mice per group).
Human samples: Ex vivo cultures of nasal polyp mucosal biopsies and brushed nasal epithelial cell
Nasal polyp biopsies from 5 ΔF508 homozygous patients undergoing surgical treatment for nonallergic nasal polyposis (see ) were cultured as previously described,Citation7,Citation24,Citation25,Citation49 for 18 h with medium, cystamine (250 μM) or EUK-134 (50 μg ml−1, Vinci Biochem, 10006329), followed by 36 h of incubation with medium alone in presence or absence of 3-MA, and then pulsed for 30 min with medium or genistein followed by 4 h of PA-LPS challenge.
Nasal epithelial cells freshly isolated by nasal brushing from 5 CF patients carrying ΔF508/ΔF508 CFTR mutations (see ) and 5 non-CF age- and sex-matched controls (3 F, mean age 12.5 y) were immersed in washing solution (PBS, DTT 2 mM, EDTA 10 mM) at 37°C for 1 h on thermal shaker, centrifuged at 2,300 × g for 20 min and washed in PBS. The isolated cells were maintained in 1 ml MEM Earle’s salt L-Glutamine medium supplemented with 10% FBS and the appropriate amount of penicillin/streptomycin.Citation51 Brushed nasal cells were cultured for 18 h with medium, Vrx-325, Corr-4a (10 μM up to 50 μM) (kindly provided by Cystic Fibrosis Foundation), cystamine (250 μM), EUK-134 (50 μg ml−1), followed by 24 h of incubation with medium alone in presence or absence of 3-MA (3 mM).
Informed consent was obtained from all subjects and the ethical committee of the University of Naples Federico II approved the study (N° 290/09).
Cell lines and treatments
CFBE41o-cells (kindly provided by D.C. Gruenert) were cultured in Minimum Essential Medium (MEM) Earle’s salt (200 mM L-Glutamine, 10% FBS and penicillin/streptomycin). IB3-1 human CF bronchial epithelial, S9 or C38 isogenic stably rescued, normal bronchial epithelial 16HBE14o- (LGC Promochem, Milan, Italy) were cultured, as recommended by American Type Culture Collection. Potential problems arising from clonal drift in IB3-1 cells were avoided by using new batches of frozen cells after every 12 passages, as described.Citation42 CFBE41o- and IB3-1 cells were transfected with ΔF508-CFTR at 37°C and incubated for 18 h with cystamine (250 μM),Citation7 EUK-134 (50 μg ml−1), Corr-4a or Vrx-325 (10 μM up to 50 μM) (kindly provided by Cystic Fibrosis Foundation, USA), followed by medium alone for 12 h with or without 3-MA (3 mM), in presence of cycloheximide (CHX, 100 μg ml−1, refreshed every 6 h, Sigma Aldrich, C4859).Citation29 Cells were also transfected with scrambled or SQSTM1 siRNAs or with Hemagglutinin (HA)-BECN1 or empty vector,Citation7 or incubated with cystamine after transfection with scrambled or BECN1 or PIK3C3 or CFTR siRNAs. CFBE41o- cells were also grown in Transwells (Corning, 3470 or 3460) under the normal condition. Briefly, 8 × 104 or 3 × 105 cells were seeded in 6.5-mm diameter or 12-mm diameter collagen-coated Transwells, respectively, and grown until the RT reached 800 to 1,000 Ω · cm2. Transwells with a pore size of 0.4 μm were used. Medium in both the apical and basolateral chambers was changed every other day. Monolayers were immunostained for occludin (Abcam, ab31721) to confirm polarization.Citation52 In another series of experiments, cells were transfected with ΔF508-CFTR, incubated at 26°C for 30 h and then incubated with medium alone, cystamine or EUK-134, at 37°C for further 6 h in the presence of CHX.
Plasmids and lentiviruses
The pcDNA3.1ΔF508-CFTR expression vector (PRIMM), and pcDNA3-HA-BECN1 expression vector (gift from N. Mizushima), were used for transfection experiments, as described.Citation7 pLKO.1-CMV-tGFP validated lentiviral vectors expressing mouse shSqstm1 sequence or no target sequence (Sigma Aldrich) and a lentiviral vectors encoding Becn1 (LV-Becn1) or GFP (LV-GFP) under the control of the ubiquitous CMV promoter were used for in vivo experiments, as described.Citation7
Transfection and RNA interference
Cells were transfected with 50 nM of CFTR (Invitrogen), SQSTM1, BECN1, (PRIMM), PIK3C3 (Sigma Aldrich) siRNAs (see ) or scrambled oligonucleotides by Lipo RnaiMax (Invitrogen, 13778-075) according to the manufacturer’s instructions or transfected with pcDNA3-HA-BECN1 or pcDNA3.1 ΔF508-CFTR or expression vectors, by Lipofectamine 2000 (Invitrogen, 11668–027) according to the manufacturer’s instructions.
Table 2. siRNA sequences
Cell viability assays
Cell viability was measured using the MTT assay.Citation39 Briefly, cells grown in 24- or 96-well plates were treated with CHX (100 μg ml–1) for different times or transfected with ΔF508-CFTR, incubated with cystamine and then treated with CHX. The untreated cells were used as controls for each time point. The culture medium was then removed and replaced with fresh medium (without FBS) containing 0.5 μg ml−1 tetrazolium salt (MTT, 5 mg ml–1, Sigma Alrich, M5665). After 3 h incubation at 37°C in 5% CO2, protected from light, the precipitated formazan was solubilized with DMSO (Sigma Aldrich, D8418) and absorbance was determined at 540 nm using a microplate reader. Four replicates were measured for each sample.
Cell surface biotinylation assay and membrane fractionation
Cell-surface proteins were biotinylated using sulfosuccinimidyl-6-(biotinamido) hexanoate (sulfo-NHS-LC-Biotin, 1 mg ml−1 in PBS, pH 8.2; Pierce, 21335), as described.Citation53 Cells were homogenized in Potter-Elvehjem pestle and glass tube (Sigma Aldrich, P7734) and centrifuged at 2,300 × g for 15 min at 4°C to obtain nuclear pellets. Supernatants that contain the cytoplasmic and plasma membrane fractions were centrifuged 1 h at 16,000 × g at 4°C. Then, the membrane-containing pellet was solubilized in BUFFER A (20 mM TRIS-HCl pH 7.4, 2 mM EDTA, 20 mM 2-ME, 1× PMSF, 1 μg ml−1 inhibitor protease cocktail) +1% Triton X-100 and centrifuged 1 h at 60,000 × g in the ultracentrifuge. The supernatants were collected as plasma membrane fraction. Equivalent amounts of protein (500 μg) were used for streptavidin-agarose pull-down (Pierce, 20349). Biotinylated proteins of plasma membrane fraction were immunoblotted against CFTR (clone M3A7) and E-cadherin or β-actin. Densitometric analyses were performed by means of Image J software, and each data point was expressed as the mean ± SD of triplicate of three independent experiments.
Immunoblot analysis and immunoprecipitation
The protein of cell lines or lung homogenates was obtained from treated and untreated cells or mice, and the amounts of proteins were determined by a Bio-Rad protein assay to ensure equal protein loading before immunoblot analysis. Fifty micrograms of protein were loaded in each lane. Antibodies against PIK3C3 (Sigma Aldrich, 108K4774), 1:1000, SQSTM1, (Sigma Aldrich, 108k4767)1:1000, PPARγ, (Santa Cruz Biotechnology, sc7273) 1:500; BECN1 (Abcam, ab58878), 1:1000, CFTR clone M3A7 (Abcam, ab4067), 1:500, E-Cadherin, (Abcam, ab15148)1:1000; phospho- ERK1/2 (p42/44) (Cell Signaling Technology, 91101) 1:1000, αβ-tubulin, (Cell Signaling Technology, 2148) 1:1000, β-actin, (Cell Signaling Technology, 4970) 1:1000; HA, (BD Bioscience, 631207) 1:1000, were used as primary antibodies.
Immunoprecipitation of CFTR was performed on 500 µg of proteins from whole lung homogenates. The proteins were incubated at 4°C for 8–12 h with CFTR antibody clone H-182 (Santa Cruz Biotechnology, sc10747), followed by the addition of Protein A/G-agarose beads. After thorough washing, bound proteins were analyzed by western blot using CFTR clone CF3 (Abcam, ab2784) 1:1000 antibody. Immunoprecipitation of phospho-tyrosine (PY-99) was performed on 500 µg of proteins from whole lysates of human nasal mucosae and immunoblot with anti PY-99 (Santa Cruz Biotechnoology, sc7020).
Real-time and reverse-transcription PCR analysis
Total RNA was extracted with the RNeasy Mini Kit (Qiagen, 74104). The mRNA was reverse transcribed with a SuperScriptTM III First Strand Synthesis System (Invitrogen, 18080-051). Quantitative RT–PCR was performed with an iCycler iQ Multicolor Real-Time PCR Detector (Bio-Rad) with iQ TM SYBR Green supermix (Bio-Rad, 170-8882). Expression levels of genes were normalized to GAPDH levels in the same sample. The relative amounts of mRNA were calculated by using the comparative Ct method. Real-time RT-PCR analyses were executed for evaluating efficiency of TNFA. The sequence of TNFA primers were: Forward: CCACCACGCTCTTCTGTCTA; Reverse: AGGGTCTGGGCCATAGAACT.
ELISA
Mouse macrophage inflammatory protein 2 (CXCL2) and TNFA were measured in lung homogenates using standard ELISA kits (R&D Systems), as reported.Citation7 Lung tissues were homogenized in lysis buffer, and tissue debris was removed by centrifugation. The supernatants were collected and stored at -80°C until usage. ELISA analysis was performed following the manufacturer’s specification. Samples were read in triplicate at 450 nm in a Microplate Reader (BioRad) using Microplate Manager 5.2.1 software.
Determination of macrophage numbers
We prepared cryostat sections (5 μm) from lung tissues of CftrF508del mice. We fixed section on glass slides with acetone, washed with PBS-Tween (0.2%) and then incubated overnight at 4°C with a 1:50 dilution of monoclonal rat CD68 (Acris, BM4000) in PBS. This was followed by incubation with Alexa-488-conjugated secondary antibodies (1:100, Molecular Probes, A-11006) and DAPI (Invitrogen, D21490) nuclear counterstaining. We then examined them on an LSM 510 confocal microscope (Zeiss). The analysis of macrophage numbers was performed by Image J software and each data point is expressed as the mean ± SD of triplicate of three independent experiments.
Immunofluorescence and confocal microscopy
The procedures were performed as previously described.Citation7,Citation54
Human tissue sections
Five-micrometer frozen human lung tissue sections were fixed in acetone for 10 min. The sections were incubated for 2 h at room temperature with the indicated antibodies (Abs).
Mice lung tissues
Seven-micrometer frozen lung tissue sections from each mice were fixed in acetone for 10 min. The sections were incubated for 2 h at room temperature with the primary Abs. The primary Abs were: CFTR 1:100 (CF3), E-cadherin 1:100 (Abcam), CFTR 1:100 (H-182 (Santa Cruz Biotechnology, sc10747) (used on mouse tissue), phospho-Tyr 1:200 (Santa Cruz Biotechnology), SQSTM1 1:300 (Sigma). These were followed by incubation with Alexa 488 or 546 secondary antibodies (Molecular Probe, Invitrogen). Data were analyzed under fluorescence examination by a LSM510 Zeiss confocal laser-scanning unit (Carl Zeiss).
In situ detection of TGM2 exzyme activity
TGM2 activity in tissue samples was detected by incubating unfixed sections with biotinylated monodansylcadaverine (Molecular Probes, Invitrogen, A1594) for 1 h at 37°C. The incorporation of labeled substrate was visualized by incubation with Alexa 546-conjugated streptavidin (1:100; Molecular Probes, S11224) for 30 min.Citation7,Citation24,Citation25
Iodide efflux
The iodide-sensitive fluorescent indicator, SPQ (Molecular Probes, Invitrogen, M440)Citation29,Citation40,Citation41,Citation55 was introduced into cells in a hypotonic solution of iodide buffer (in mM: 130 NaI, 4 KNO3, 1 Ca(NO3)2, 1 Mg(NO3)2, 10 glucose and 20 HEPES, pH 7.4) diluted 1:1 with water and containing a final concentration of 10 μM SPQ. Cells were loaded for 20 min at 37°C in a humidified chamber with 5% CO2. The SPQ-loaded cells were then mounted on a LSM510 Meta confocal microscope with a 37°C heated stage and perfused with iodide buffer. Changes in CFTR-mediated SPQ fluorescence were monitored at the 445 nm wavelength in response to excitation at 340 nm perfusion at 37°C in nitrate buffer (NaI replaced with 130 mM NaNO3) for 10 min with 20 μM forskolin (Fsk) (Sigma Aldrich, F6886) plus 50 μM genistein, 20 μM Vrx-532 (kindly provided by Cystic Fibrosis Foundation) or 10 µM Vrx-770Citation55 (Selleck, S1144). The peak iodide efflux rate (usually 12 min after Fsk plus genistein or Fsk plus Vrx-532 or Fsk plus Vrx-770) of treated or untreated cells was calculated in accordance with the Stern- Volmer relationship as follows:
(Fo/F) − 1 = KCQ
where F is the observed fluorescence, Fo is the fluorescence in the absence of a quenching anion, CQ is the concentration of the quenching anion, and K is the Stern-Volmer quench constant. The rates were calculated using SigmaPlot Version 7.1 for each mean fluorescence trace generated from the 50 cells examined per population per coverslip.
Statistical analysis
Data are reported as arithmetic mean ± SD. Data distribution was analyzed for normality and statistical analysis performed using the one-way ANOVA. Significant differences are indicated in the figures. All data were obtained from independent measurements. Data were analyzed using SPSS 13 software. Statistical significance was defined as p value of < 0.05.
| Abbreviations: | ||
| CF | = | cystic fibrosis |
| CFTR | = | cystic fibrosis transmembrane conductance regulator |
| TGM2 | = | tissue transglutaminase |
| ROS | = | reactive oxygen species |
| PtdIns3K | = | phosphatidylinositol-3-kinase |
Additional material
Download Zip (1.6 MB)Acknowledgments
We thank Noboru Mizushima (The Tokyo Metropolitan Institute of Medical Sciences, Tokyo, Japan) for the gift of the pcDNA3-HA-BECN1 expression vectors, Dieter C. Gruenert (California Pacific Medical Center Research Institute, San Francisco, CA) for CFBE41o-cell lines; Eliezer Masliah and Brian Spencer (Departments of Neurosciences, University of California, San Diego, La Jolla, CA) for lentiviral vectors encoding Becn1 or GFP. This was supported by the European Institute for Research in Cystic Fibrosis and Italian Cystic Fibrosis Association (L.M.), the Programma di Ricerca Scientifica di Rilevante Interesse Nazionale (2008RMJB3A_004, 2008) of the Ministero dell'Istruzione, dell'Università e della Ricerca (L.M., V.R.), Ligue Nationale contre le Cancer, AXA Chair for Longevity Research, Agence Nationale pour la Recherche, European Commission (Active p53, Apo-Sys, ChemoRes, ApopTrain), Fondation pour la Recherche Médicale, Institut National du Cancer, Cancéropôle Ile-de-France, Fondation Bettencourt-Schueller and the LabEx Onco-Immunology (G.K.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/21483
References
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814 - 22; http://dx.doi.org/10.1038/ncb0910-814; PMID: 20811353
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107 - 32; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005; PMID: 21801009
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280 - 93; http://dx.doi.org/10.1016/j.molcel.2010.09.023; PMID: 20965422
- Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating?. Nat Rev Mol Cell Biol 2012; 13:7 - 12; PMID: 22166994
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323 - 35; http://dx.doi.org/10.1038/nature09782; PMID: 21248839
- Sridhar S, Botbol Y, Macian F, Cuervo AM. Autophagy and disease: always two sides to a problem. J Pathol 2012; 226:255 - 73; http://dx.doi.org/10.1002/path.3025; PMID: 21990109
- Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol 2010; 12:863 - 75; http://dx.doi.org/10.1038/ncb2090; PMID: 20711182
- Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina DL, Settembre C, et al. Cystic fibrosis: a disorder with defective autophagy. Autophagy 2011; 7:104 - 6; http://dx.doi.org/10.4161/auto.7.1.13987; PMID: 21048426
- Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DH, et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy 2011; 7:1359 - 70; http://dx.doi.org/10.4161/auto.7.11.17660; PMID: 21997369
- O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet 2009; 373:1891 - 904; http://dx.doi.org/10.1016/S0140-6736(09)60327-5; PMID: 19403164
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005; 352:1992 - 2001; http://dx.doi.org/10.1056/NEJMra043184; PMID: 15888700
- Accurso FJ. Update in cystic fibrosis 2005. Am J Respir Crit Care Med 2006; 173:944 - 7; http://dx.doi.org/10.1164/rccm.2601006; PMID: 16632633
- Park HW, Nam JH, Kim JY, Namkung W, Yoon JS, Lee JS, et al. Dynamic regulation of CFTR bicarbonate permeability by [Cl-]i and its role in pancreatic bicarbonate secretion. Gastroenterology 2010; 139:620 - 31; http://dx.doi.org/10.1053/j.gastro.2010.04.004; PMID: 20398666
- Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev 1999; 79:Suppl S3 - 22; PMID: 9922374
- Welsh MJ, Ramsey BW, Accurso FJ, Cutting GR. Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill, 2001:5121-88.
- Collins FS. Cystic fibrosis: molecular biology and therapeutic implications. Science 1992; 256:774 - 9; http://dx.doi.org/10.1126/science.1375392; PMID: 1375392
- Vij N, Mazur S, Zeitlin PL. CFTR is a negative regulator of NFkappaB mediated innate immune response. PLoS One 2009; 4:e4664; http://dx.doi.org/10.1371/journal.pone.0004664; PMID: 19247502
- Belcher CN, Vij N. Protein processing and inflammatory signaling in Cystic Fibrosis: challenges and therapeutic strategies. Curr Mol Med 2010; 10:82 - 94; http://dx.doi.org/10.2174/156652410791065408; PMID: 20205681
- Anderson P. Emerging therapies in cystic fibrosis. Ther Adv Respir Dis 2010; 4:177 - 85; http://dx.doi.org/10.1177/1753465810371107; PMID: 20530065
- Ratjen F, Grasemann H. New therapies in cystic fibrosis. Curr Pharm Des 2012; 18:614 - 27; http://dx.doi.org/10.2174/138161212799315984; PMID: 22229570
- Mozzillo E, Franzese A, Valerio G, Sepe A, De Simone I, Mazzarella G, et al. One-year glargine treatment can improve the course of lung disease in children and adolescents with cystic fibrosis and early glucose derangements. Pediatr Diabetes 2009; 10:162 - 7; http://dx.doi.org/10.1111/j.1399-5448.2008.00451.x; PMID: 19207231
- Amaral MD. Targeting CFTR: how to treat cystic fibrosis by CFTR-repairing therapies. Curr Drug Targets 2011; 12:683 - 93; http://dx.doi.org/10.2174/138945011795378586; PMID: 21039334
- Balch WE, Roth DM, Hutt DM. Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harb Perspect Biol 2011; 3:pii:a004499; http://dx.doi.org/10.1101/cshperspect.a004499; PMID: 21421917
- Maiuri L, Luciani A, Giardino I, Raia V, Villella VR, D’Apolito M, et al. Tissue transglutaminase activation modulates inflammation in cystic fibrosis via PPARgamma down-regulation. J Immunol 2008; 180:7697 - 705; PMID: 18490773
- Luciani A, Villella VR, Vasaturo A, Giardino I, Raia V, Pettoello-Mantovani M, et al. SUMOylation of tissue transglutaminase as link between oxidative stress and inflammation. J Immunol 2009; 183:2775 - 84; http://dx.doi.org/10.4049/jimmunol.0900993; PMID: 19625650
- Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest 2005; 115:2564 - 71; http://dx.doi.org/10.1172/JCI24898; PMID: 16127463
- Van Goor F, Straley KS, Cao D, González J, Hadida S, Hazlewood A, et al. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. AJP - Lung Physiol 2006; 260:L1117 - L1130; http://dx.doi.org/10.1152/ajplung.00169.2005
- Kim Chiaw P, Wellhauser L, Huan LJ, Ramjeesingh M, Bear CE. A chemical corrector modifies the channel function of F508del-CFTR. Mol Pharmacol 2010; 78:411 - 8; http://dx.doi.org/10.1124/mol.110.065862; PMID: 20501743
- Okiyoneda T, Barrière H, Bagdány M, Rabeh WM, Du K, Höhfeld J, et al. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010; 329:805 - 10; http://dx.doi.org/10.1126/science.1191542; PMID: 20595578
- Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol Med 2012; 18:81 - 91; http://dx.doi.org/10.1016/j.molmed.2011.10.003; PMID: 22138491
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al, VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365:1663 - 72; http://dx.doi.org/10.1056/NEJMoa1105185; PMID: 22047557
- Davis PB. Therapy for cystic fibrosis--the end of the beginning?. N Engl J Med 2011; 365:1734 - 5; http://dx.doi.org/10.1056/NEJMe1110323; PMID: 22047565
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012; 67:12 - 8; http://dx.doi.org/10.1136/thoraxjnl-2011-200393; PMID: 21825083
- Elborn JS. Fixing cystic fibrosis CFTR with correctors and potentiators. Off to a good start. Thorax 2012; 67:4 - 5; http://dx.doi.org/10.1136/thoraxjnl-2011-201197; PMID: 22058188
- Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009; 137:1001 - 4; http://dx.doi.org/10.1016/j.cell.2009.05.023; PMID: 19524504
- Gidalevitz T, Kikis EA, Morimoto RI. A cellular perspective on conformational disease: the role of genetic background and proteostasis networks. Curr Opin Struct Biol 2010; 20:23 - 32; http://dx.doi.org/10.1016/j.sbi.2009.11.001; PMID: 20053547
- Okiyoneda T, Apaja PM, Lukacs GL. Protein quality control at the plasma membrane. Curr Opin Cell Biol 2011; 23:483 - 91; http://dx.doi.org/10.1016/j.ceb.2011.04.012; PMID: 21190823
- Jurkuvenaite A, Chen L, Bartoszewski R, Goldstein R, Bebok Z, Matalon S, et al. Functional stability of rescued delta F508 cystic fibrosis transmembrane conductance regulator in airway epithelial cells. Am J Respir Cell Mol Biol 2010; 42:363 - 72; http://dx.doi.org/10.1165/rcmb.2008-0434OC; PMID: 19502384
- van Meerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol Biol 2011; 731:237 - 45; http://dx.doi.org/10.1007/978-1-61779-080-5_20; PMID: 21516412
- Silvis MR, Bertrand CA, Ameen N, Golin-Bisello F, Butterworth MB, Frizzell RA, et al. Rab11b regulates the apical recycling of the cystic fibrosis transmembrane conductance regulator in polarized intestinal epithelial cells. Mol Biol Cell 2009; 20:2337 - 50; http://dx.doi.org/10.1091/mbc.E08-01-0084; PMID: 19244346
- Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov 2009; 8:153 - 71; http://dx.doi.org/10.1038/nrd2780; PMID: 19153558
- Singh OV, Pollard HB, Zeitlin PL. Chemical rescue of deltaF508-CFTR mimics genetic repair in cystic fibrosis bronchial epithelial cells. Mol Cell Proteomics 2008; 7:1099 - 110; http://dx.doi.org/10.1074/mcp.M700303-MCP200; PMID: 18285607
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 1995; 83:121 - 7; http://dx.doi.org/10.1016/0092-8674(95)90240-6; PMID: 7553863
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992; 358:761 - 4; http://dx.doi.org/10.1038/358761a0; PMID: 1380673
- Namkung W, Kim KH, Lee MG. Base treatment corrects defects due to misfolding of mutant cystic fibrosis transmembrane conductance regulator. Gastroenterology 2005; 129:1979 - 90; http://dx.doi.org/10.1053/j.gastro.2005.08.049; PMID: 16344066
- Wilke M, Buijs-Offerman RM, Aarbiou J, Colledge WH, Sheppard DN, Touqui L, et al. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst Fibros 2011; 10:Suppl 2 S152 - 71; http://dx.doi.org/10.1016/S1569-1993(11)60020-9; PMID: 21658634
- van Doorninck JH, French PJ, Verbeek E, Peters RH, Morreau H, Bijman J, et al. A mouse model for the cystic fibrosis delta F508 mutation. EMBO J 1995; 14:4403 - 11; PMID: 7556083
- Legssyer R, Huaux F, Lebacq J, Delos M, Marbaix E, Lebecque P, et al. Azithromycin reduces spontaneous and induced inflammation in DeltaF508 cystic fibrosis mice. Respir Res 2006; 7:134; http://dx.doi.org/10.1186/1465-9921-7-134; PMID: 17064416
- Raia V, Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Auricchio S, et al. Inhibition of p38 mitogen activated protein kinase controls airway inflammation in cystic fibrosis. Thorax 2005; 60:773 - 80; http://dx.doi.org/10.1136/thx.2005.042564; PMID: 15994249
- Amaral MD. Processing of CFTR: traversing the cellular maze--how much CFTR needs to go through to avoid cystic fibrosis?. Pediatr Pulmonol 2005; 39:479 - 91; http://dx.doi.org/10.1002/ppul.20168; PMID: 15765539
- Mosler K, Coraux C, Fragaki K, Zahm JM, Bajolet O, Bessaci-Kabouya K, et al. Feasibility of nasal epithelial brushing for the study of airway epithelial functions in CF infants. J Cyst Fibros 2008; 7:44 - 53; http://dx.doi.org/10.1016/j.jcf.2007.04.005; PMID: 17553758
- Sharma M, Pampinella F, Nemes C, Benharouga M, So J, Du K, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol 2004; 164:923 - 33; http://dx.doi.org/10.1083/jcb.200312018; PMID: 15007060
- Caohuy H, Jozwik C, Pollard HB. Rescue of DeltaF508-CFTR by the SGK1/Nedd4-2 signaling pathway. J Biol Chem 2009; 284:25241 - 53; http://dx.doi.org/10.1074/jbc.M109.035345; PMID: 19617352
- Luciani A, Villella VR, Vasaturo A, Giardino I, Pettoello-Mantovani M, Guido S, et al. Lysosomal accumulation of gliadin p31-43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut 2010; 59:311 - 9; http://dx.doi.org/10.1136/gut.2009.183608; PMID: 19951908
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 2011; 108:18843 - 8; http://dx.doi.org/10.1073/pnas.1105787108; PMID: 21976485