Abstract
The multiunit Cullin (CUL)-RING E3 ligase (CRL) controls diverse biological processes by targeting a mass of substrates for ubiquitination and degradation, whereas its dysfunction causes carcinogenesis. Post-translational neddylation of CUL, a process triggered by the NEDD8-activating enzyme E1 subunit 1 (NAE1), is required for CRL activation. Recently, MLN4924 was discovered via a high-throughput screen as a specific NAE1 inhibitor and first-in-class anticancer drug. By blocking CUL neddylation, MLN4924 inactivates CRL and causes the accumulation of CRL substrates that trigger cell cycle arrest, senescence and/or apoptosis to suppress the growth of cancer cells in vitro and in vivo. Recently, we found that MLN4924 also triggers protective autophagy in response to CRL inactivation. MLN4924-induced autophagy is attributed partially to the inhibition of mechanistic target of rapamycin (also known as mammalian target of rapamycin, MTOR) activity by the accumulation of the MTOR inhibitory protein DEPTOR, as well as reactive oxygen species (ROS)-induced stress. Moreover, the blockage of autophagy response enhances apoptosis in MLN4924-treated cells. Together, our findings not only reveal autophagy as a novel cellular response to CRL inactivation by MLN4924, but also provide a piece of proof-of-concept evidence for the combination of MLN4924 with autophagy inhibitors to enhance therapeutic efficacy.
The Cullin-RING E3 ligase (CRL), also known as SKP1-Cullin-F-box (SCF) E3 ligase for its founding member, is the largest multiunit ubiquitin ligase family that controls the ubiquitination and degradation of about 20% of ubiquitin-proteasome system (UPS)-regulated proteins. The core structure of CRL/SCF is a complex of CUL-RBX/ROC by which other essential components of the CRL/SCF are recruited together to form functional E3 ubiquitin ligases (). By targeting various substrates for degradation, CRL/SCF regulates diverse biological processes in physiological conditions, whereas its dysfunction causes carcinogenesis, which renders CRL/SCF a promising anticancer target and stimulates drug discovery aimed at inhibiting CRL/SCF.
Figure 1. NEDD8-activating enzyme E1 subunit 1 (NAE1) inhibitor MLN4924 triggers protective autophagy in cancer cells. CRL/SCF is a multiunit E3 ubiquitin ligase family with a CUL-RBX/ROC complex as its core structure. Post-translational neddylation of CUL is required for the activation of CRL/SCF E3 ligase. During this process, the ubiquitin-like molecule NEDD8 is transferred to CUL by NAE1, NEDD8 conjugating enzyme E2 (UBE2M/Ubc12) and the NEDD8-E3 ligase cascade. The NAE1 inhibitor MLN4924 blocks CUL neddylation and inactivates CRL/SCF to induce protective autophagy, whereas blockage of the autophagy pathway via knockdown of autophagy essential genes (such as ATG5) significantly enhances apoptosis in MLN4924-treated cells. Mechanistically, MLN4924-induced autophagy is attributed to: (a) the inhibition of MTOR activity by the accumulation of the MTOR inhibitory protein DEPTOR, which is a CRL/SCF substrate; (b) reactive oxygen species-induced stress; and (c) dysfunction of other CRL/SCF-regulated substrates and pathways to be identified. Ub, ubiquitin; N8, NEDD8; SR, substrate receptor.
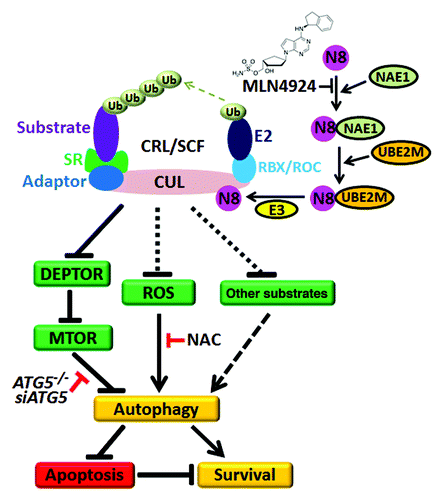
Post-translational neddylation is a process of adding the ubiquitin-like molecule NEDD8 to target proteins, which is triggered by NAE1, NEDD8 conjugating enzyme E2 (UBE2M/Ubc12) and the NEDD8-E3 ligase cascade (). A number of studies demonstrated that CUL neddylation is required for the activation of CRL/SCF E3 ligase. Recently, MLN4924, a specific NAE1 inhibitor, was discovered via a high-throughput screen. By blocking CUL neddylation, MLN4924 inactivates CRL/SCF, causes accumulation of its substrates, and thus triggers a DNA damage response, abnormal cell cycle progression, apoptosis and/or senescence to suppress cancer cell growth. Due to its significant anticancer efficacy and well-tolerated toxicity in preclinical studies, MLN4924 has been advanced into several phase I clinical trials for several solid tumors and hematological malignancies.
While MLN4924 inactivates CRL/SCF and triggers several cell-killing pathways, its potential effect on autophagy is completely unknown. Recently, we demonstrated that MLN4924 also triggers autophagy in response to CRL inactivation in cancer cells based on the following findings that MLN4924 induces: (1) the conversion of LC3-I (microtubule-associated protein 1 light chain 3, soluble form) to lipidated LC3-II; (2) classical punctate distribution of LC3-II; (3) the formation of acidic vesicular organelles (AVOs); (4) the appearance of double-membrane autophagosomes; and (5) the existence of an intact autophagic flux in treated cells. Similarly, we found that genetic inactivation of CRL/SCF via siRNA silencing of its essential component RBX1/ROC1 also triggers autophagy in cancer cells (manuscript in press). These findings indicate that induction of autophagy is a general phenomenon in response to CRL/SCF inactivation by both pharmaceutical and genetic approaches.
Mechanistically, MLN4924-induced autophagy is attributed partially to the inhibition of MTOR activity as well as ROS-induced stress. MTOR is a central regulator of cell survival and metabolism, whereas its inactivation can trigger autophagy. Recently, the MTOR inhibitory protein DEPTOR was identified as a novel substrate of the CRL/SCFβ-TrCP E3 ligase. During the investigation into how MLN4924 triggers autophagy, we found that MLN4924 treatment causes the accumulation of DEPTOR, and thus triggers autophagy by the inhibition of MTOR activity. Similarly, we found that inactivation of CRL/SCF by RBX1/ROC1 knockdown also leads to DEPTOR accumulation and autophagy induction (manuscript in press). Rescue assays by siRNA silencing of DEPTOR indicated that knockdown of this MTOR inhibitory protein significantly inhibits the CRL/SCF inactivation-induced autophagy response, indicating that DEPTOR accumulation and subsequent MTOR inhibition contribute to the autophagy response.
In addition, we found that ROS-induced stress may also contribute to autophagy response upon MLN4924 treatment, since the ROS scavenger NAC partially blocks the conversion of LC3-I to LC3-II. Recently, a mass of potential CRL/SCF substrates have been identified in MLN4924-treated cells via high-throughput proteomic approaches, and some of these potential CRL/SCF substrates may accumulate upon CRL/SCF inactivation induced by MLN4924 and contribute to the autophagy response directly. Thus, MLN4924 may trigger autophagy by integrating several signaling pathways which are regulated by CRL/SCF E3 ligase.
The biological role of autophagy in the regulation of cell survival during cellular stresses is controversial. Some studies indicate that autophagy primarily serves as prosurvival mechanism against unfavorable conditions while others show that autophagy can cause cell death (termed autophagic cell death). Here, we found that MLN4924-induced autophagy serves as a survival signal since blockage of the autophagy pathway enhances apoptosis in ATG5-silenced cancer cells or ATG5-deficient MEF cells. Similarly, we found that inactivation of CRL/SCF by RBX1/ROC1 siRNA silencing also triggers protective autophagy, and blockage of the autophagy pathway via knockdown of the autophagy essential genes ATG5 or BECN1 significantly enhances apoptosis and growth inhibition in cancer cells (manuscript in press). These findings provide a preclinical proof-of-concept for combination therapy with an autophagy inhibitor and CRL/SCF inhibitors (such as MLN4924) to enhance therapeutic efficacy in clinical trials.
Acknowledgments
This work is supported by a National Natural Science Foundation Grant of China (81172092), National Basic Research Program of China (973 program, 2012CB910302) and National Natural Science Foundation Grant of China (31071204) to L.J., and National Natural Science Foundation Grant of China (91129702, 81125001) to J.L.