Abstract
T cells are essential for defense of the host against invading pathogens. Antigen activation of the T cell receptor (TCR) is required for generation of an adaptive immune response. Several groups have observed that blocking autophagy augments T cell activation, but the molecular basis of this finding has remained elusive. The adaptor protein BCL10 transmits activating signals from the TCR to NFKB1-RELA/NFκB, a transcription factor that is critical for T cell proliferation and function. We recently reported that a TCR-dependent autophagy mechanism selectively targets and degrades BCL10. We found that BCL10 autophagy requires BCL10 K63-polyubiquitination and subsequent binding to the autophagy adaptor SQSTM1/p62. Blocking either one of these processes inhibits BCL10 degradation. Protecting BCL10 from autophagic degradation, either by pharmacological or genetic inhibition of autophagy, results in increased activation of NFKB1-RELA. By demonstrating the mechanism of autophagic uptake and degradation of BCL10, our study has revealed a key mechanism by which selective autophagy controls T cell activation. Here, we discuss the implications of our findings and explore possible directions for future research.
T cell activation is triggered by the recognition of a foreign antigen by the TCR. Antigen engagement of the TCR initiates a complex intracellular signaling cascade, culminating in the activation of key transcription factors, including NFKB1-RELA. The adaptor protein BCL10 is a required intermediate in the pathway that connects the TCR to the inhibitor of kappaB kinase (IKBK/IKK) complex, the kinase holoenzyme that initiates the terminal steps in NFKB1-RELA activation. IKBK activation triggers NFKB1-RELA nuclear translocation, initiating transcription of genes essential for T cell proliferation, differentiation, and function ().
Figure 1. Mechanism of T cell activation and autophagic BCL10 degradation in effector T cells. BCL10 may initiate autophagosome formation by activating the IKBK complex, a known inducer of autophagy. Alternatively, an unknown TCR-dependent autophagy pathway may be responsible for BCL10 degradation.
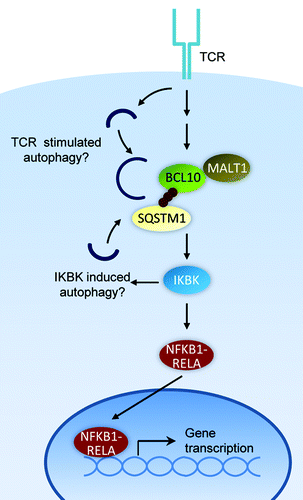
Our findings have revealed new details regarding the mechanism of BCL10 signaling, and regulation of BCL10 activity in this pathway. Specifically, we demonstrated that TCR-dependent BCL10 K63-polyubiquitination triggers interaction with SQSTM1 speckles (cytosolic SQSTM1 clusters). We also showed that the BCL10-SQSTM1 interaction is crucial for TCR signaling to IKBK and consequent activation of NFKB1-RELA. Importantly, we found that SQSTM1 speckles simultaneously interact with BCL10 and LC3+ autophagosomes. The SQSTM1-LC3 interaction results in autophagic uptake and degradation of SQSTM1-bound BCL10, thereby depleting the active polyubiquitinated form of BCL10 and limiting the magnitude of NFKB1-RELA activation. Thus, SQSTM1 is paradoxically required both for BCL10 activation of NFKB1-RELA and for limiting activation of NFKB1-RELA via directing BCL10 autophagy.
Remarkably, this autophagy process is highly selective, resulting in degradation of BCL10, but not its direct binding partner, MALT1. BCL10 is constitutively associated with MALT1, an important signaling partner (). Unexpectedly, we found that LC3 interacts with only BCL10, and not MALT1. Moreover, unlike BCL10, MALT1 is not degraded after T cell activation. Therefore, BCL10 is targeted for autophagy in a highly selective manner. Additionally, our data suggest that an intact autophagy mechanism is required for the physical separation of MALT1 from BCL10. Notably, BCL10 and MALT1 clusters form at locations that cannot be distinguished from sites of autophagic punctum formation by conventional light microscopy. Thus, elucidating the mechanism of selective BCL10 uptake by autophagosomes will require a more detailed understanding of the dynamic molecular interactions occurring at SQSTM1 speckles, following TCR stimulation. Such studies may be aided by electron microscopy or super-resolution microscopy, to assess whether BCL10 and MALT1 become segregated into distinct SQSTM1-proximal subdomains.
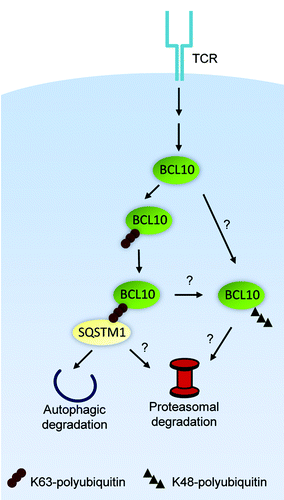
We also found that the specific interaction of BCL10 with LC3+ autophagosomes depends on BCL10 K63-polyubiquitination and BCL10 binding to SQSTM1. This conclusion was supported by the observation that the BCL10 polyubiquitination-deficient mutant, K31,63R (first reported by Jonathan Ashwell’s group at the National Cancer Institute) does not interact with SQSTM1 or LC3, and is not degraded post-TCR stimulation. The SQSTM1-BCL10 interaction is most likely mediated by BCL10 K63-polyubiquitin chains binding to the C-terminal UBA domain of SQSTM1. Recruitment of LC3+ autophagosomes to sites of BCL10-SQSTM1 clusters presumably depends on the SQSTM1 LC3 interacting region (LIR) binding to the N-terminal region of LC3. We observed that BCL10 degradation is profoundly inhibited by SQSTM1 silencing or by blocking BCL10 K63-polyubiquitination. In contrast, pharmacological or genetic inhibition of autophagy only partially inhibits BCL10 degradation. Experiments with the proteasome inhibitor MG132 revealed that proteasomes also play a significant role in BCL10 degradation. As SQSTM1 interacts with proteasomes, the simplest interpretation of these data are that SQSTM1 directs K63-polyubiquitinated BCL10 to autophagosomes and proteasomes. There are also additional possibilities, such as the existence of a parallel pathway of TCR-dependent K48-polyubiquitination of BCL10, targeting some BCL10 for proteasomal degradation (). The precise mechanism of TCR-dependent BCL10 degradation will require further characterization of the requirements for BCL10 proteolysis under conditions of selective inhibition of autophagy or proteasome activity.
Figure 2. Mechanism of degradation of BCL10 by autophagy and the proteasome. K63-polyubiquitinated BCL10 binds to SQSTM1, targeting BCL10 to both autophagosomes and proteasomes. Alternatively, there may be an independent pathway of degradation, whereby K48-polyubiquitinated BCL10 is directed to proteasomes for degradation.
Emerging data suggest that selective autophagy may modulate the activity of diverse signaling pathways. For example, Amy Kiger’s group at the University of California, San Diego demonstrated that selective autophagy of a Rho pathway regulator is essential for cell membrane spreading in macrophages; Takeshi Into’s group at Asahi University reported that a noncanonical autophagy mechanism degrades TICAM1/TRIF and TRAF6 to suppress TLR signaling; Ye-Guang Chen’s group at Tsinghua University observed that autophagy of Dishevelled can limit activation of the Wnt pathway under conditions of nutrient deprivation; and John Kehrl’s group at the National Institutes of Health reported that autophagic degradation of inflammasomes in macrophages decreases IL1B/IL-1β production. The above reports demonstrate a common pattern of target protein ubiquitination and consequent binding to an autophagy adaptor, with subsequent trafficking to autophagosomes for degradation. Importantly, the above studies of signal regulation by autophagy in the TLR and Wnt pathways also identified a role for proteasomes in the destruction of target proteins, analogous to our observations regarding TCR-stimulated proteolysis of BCL10. Collectively, these studies suggest that autophagy adaptors are intimately connected to both the autophagy and proteasomal degradation machinery. We expect that many additional examples of selective autophagy of signaling proteins will soon emerge. Such data would establish selective autophagy as a pervasive regulatory mechanism for diverse cell signaling pathways.
Acknowledgments
Supported by grants from the National Institutes of Health (AI057481 to B.C.S.), the Center for Neuroscience and Regenerative Medicine (CNRM) (to B.C.S.), and a predoctoral fellowship (to S.P.) from the American Heart Association.