Abstract
Crohn disease (CD), one of the major chronic inflammatory bowel diseases, occurs anywhere in the gastrointestinal tract with discontinuous transmural inflammation. A number of studies have now demonstrated that genetic predisposition, environmental influences and a dysregulated immune response to the intestinal microflora are involved. Major CD susceptibility pathways uncovered through genome-wide association studies strongly implicate the innate immune response (NOD2), in addition to the more specific acquired T cell response (IL23R, ICOSLG) and autophagy (ATG16L1, IRGM). Examination of the disease-associated microbiome, although complex, has identified several potentially contributory microorganisms, most notably adherent-invasive E.coli strains (AIEC), which have been isolated by independent investigators in both adult and pediatric CD patients. Here we discuss our recent finding that the type-III intermediate filament (IF) protein VIM/vimentin is a novel NOD2 interacting protein that regulates NOD2 activities including inflammatory NFKB1 signaling, autophagy and bacterial handling.
Keywords: :
Host cell defense against infection requires the coordination of several pathways. Invading pathogens are detected by pattern recognition receptors (PRR), which include toll-like receptors (TLR) and NOD-like receptors (NLR). PRRs detect specific microbe-associated molecular patterns (MAMPs) to trigger the activation of both inflammatory (e.g., through NFKB1) and antimicrobial signaling cascades. Recent studies now support a role for autophagy as an antimicrobial mechanism downstream of TLR and NLR signaling. Stimulation of NOD2 by its ligand muramyl dipeptide (MDP) has been shown to induce autophagy in human monocyte-derived dendritic cells in addition to influencing bacterial handling and antigen presentation; this was dependent on the autophagy proteins ATG5, ATG7 and ATG16L1. The origin of autophagosomes has gained intense interest in the field, and it is now clear that these double-membrane structures can originate from the surface of various cell organelles, including the endoplasmic reticulum, mitochondria and the plasma membrane. One of the major autophagy proteins ATG16L1 interacts with the heavy chain of clathrin, and the plasma membrane has been shown to contribute to the formation of ATG16L1-positive preautophagic structures via endocytosis. Additionally, there is now mounting evidence that NOD2 (previously thought to be a cytosolic protein) also resides and functions at the plasma membrane. This membrane localization has lead to the suggestion that NOD2 functions at sites of infection to directly engage with pathogens and signal an appropriate inflammatory and antimicrobial response. This hypothesis is supported by a recent study demonstrating colocalization of NOD2 and ATG16L1 at the plasma membrane with NOD2 recruiting ATG16L1 to sites of bacterial infection. NOD2 and ATG16L1 localize with invading pathogens at the entry foci, with CD-associated mutant NOD2 or ATG16L1 proteins abrogating this response. Further work has shown that a clathrin- and dynamin-dependent endocytic pathway regulates MDP internalization and NOD2 activation. It has also been proposed that NOD2 can recognize danger-associated molecular patterns (DAMPs) that are exposed when there is disruption of host cell membranes. Pathogens such as Salmonella typhimurium and enteropathogenic E.coli (EPEC) use type-3-secretion systems to inject effector proteins into the cytosol of host cells to mediate adhesion and invasion, and this may act as a DAMP recognized by NOD2 however this awaits experimental verification.
VIM/vimentin is the major IF protein in cells of mesenchymal origin where it plays an important role in the maintenance of cell architecture. VIM is also important for the correct positioning and trafficking of organelles within the cytoplasm and is emerging as an important factor linked to bacterial and viral pathogenicity. Several recent studies have shown that VIM is expressed on the surface of certain cell types, with this surface-expressed VIM possessing lectin-like properties. VIM can bind to the monosaccharide N-acetylglucosamine (GlcNAc), which is cross-linked together with N-acetylmuramic acid to form peptidoglycan, a major constituent in the bacterial cell wall. Furthermore, VIM is involved in the endocytosis of GlcNAc-conjugated liposomes suggesting that VIM is actively involved in the internalization of GlcNAc-bearing ligands on cell surfaces. Cell surface-expressed VIM has in the same way been identified as the binding target for several viruses such as Japanese encephalitis virus with VIM structure and gene expression changing profoundly during infection. Most relevant to CD pathogenesis are recent reports that VIM is expressed on the surface of human brain endothelial cells where it acts as a primary receptor for AIEC strains expressing the virulence factor IbeA, with VIM-mediated signaling required for IbeA+ E.coli invasion of these cells.
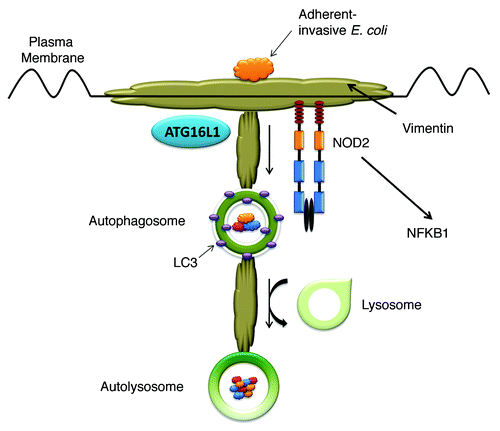
We have now found evidence that VIM is an important host cell receptor for CD-associated AIEC. Following VIM inhibition with the chemical inhibitor withaferin-A (WFA) (or neutralizing antibodies), we find that the ability of NOD2 to signal an appropriate inflammatory response, activate MDP-induced autophagy and limit the invasion and survival of a CD-associated strain of AIEC is impaired. Specifically with regard to autophagy induction, cells stably expressing NOD2 or the CD-associated NOD2 frameshift mutation were transiently transfected with GFP-LC3. MDP efficiently stimulates autophagy in NOD2-expressing cells and is inhibited by treatment with WFA. As expected, MDP has no effect on autophagy induction in cells expressing the frameshift NOD2. We believe this is in part due to our additional observation that VIM is important for the correct localization of NOD2 to the plasma membrane, since CD-associated NOD2 variants are unable to interact with VIM and were mislocalized to the cytosol. Importantly, treatment of cells with WFA disrupts the NOD2-VIM interaction and causes the relocalization of NOD2 from the plasma membrane to the cytosol. These findings led us to further compare the subcellular localization of NOD2 and CD-associated NOD2 variants. Our results reveal that a significant proportion of NOD2 is bound to the cytoskeleton. This is in keeping with previous work showing that NOD2 activity depends on the integrity of the actin cytoskeleton, and drugs that disrupt actin significantly increase the ratio of soluble to insoluble NOD2 as well as reducing AIEC invasion of epithelial cells. An important role for the cytoskeleton in the regulation of autophagy is also emerging. Of particular significance is that the proper formation and distribution of autophagosomes depends on the integrity of IF networks, and that autophagic vacuoles are found to be tightly associated with VIM. We believe that NOD2 may have evolved to use the cytoskeletal network, and in particular VIM, as a means to engage with invading pathogens. NOD2 may then use the intracellular cytoskeleton further for the transport of internalized pathogens via the autophagy pathway to lysosomes for degradation ().
Figure 1. Proposed interaction of VIM, NOD2 and the autophagy pathway during the invasion of host cells by AIEC. VIM expressed on the surface of host cells acts as a receptor for AIEC. The interaction of VIM and NOD2 at the plasma membrane allows NOD2 to engage with pathogens and signal an appropriate inflammatory response (through NFKB1) and antimicrobial response (through recruitment of ATG16L1). Internalized pathogens are trafficked along the cytoskeletal network via the autophagy pathway to lysosomes for degradation.