Abstract
The autophagy-lysosome system is essential for muscle cell homeostasis and its dysfunction has been linked to muscle disorders that are typically distinguished by massive autophagic buildup. Among them, glycogen storage disease type II (GSDII) is characterized by the presence of large glycogen-filled lysosomes in the skeletal muscle, due to a defect in the lysosomal enzyme acid α-glucosidase (GAA). The accumulation of autophagosomes is believed to be detrimental for myofiber function. However, the role of autophagy in the pathogenesis of GSDII is still unclear. To address this issue we monitored autophagy in muscle biopsies and myotubes of early and late-onset GSDII patients at different time points of disease progression. Moreover we also analyzed muscles from patients treated with enzyme replacement therapy (ERT). Our data suggest that autophagy is a protective mechanism that is required for myofiber survival in late-onset forms of GSDII. Importantly, our findings suggest that a normal autophagy flux is important for a correct maturation of GAA and for the uptake of recombinant human GAA. In conclusion, autophagy failure plays an important role in GSDII disease progression, and the development of new drugs to restore the autophagic flux should be considered to improve ERT efficacy.
The central role of lysosomes in myofiber homeostasis is highlighted by the presence of a group of muscle disorders characterized by lysosomal impairment that ultimately affects the cargo delivery, which includes the autophagy system. Indeed Danon disease is caused by the lack of lysosomal-associated membrane protein 2 (LAMP2), X-linked myopathy with excessive autophagy (XMEA) is triggered by mutations in an essential assembly chaperone of the V-ATPase, the principal mammalian lysosomal proton pump complex, and finally GSDII is due to a defect in lysosomal acid α-glucosidase (GAA), the enzyme responsible for the lysosomal hydrolysis of glycogen to glucose. Furthermore, chloroquine-mediated inhibition of lysosome function induces a well-known myopathy.
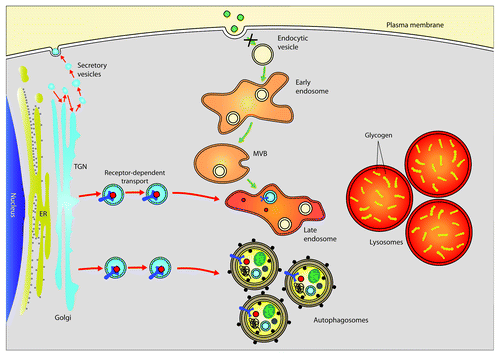
The skeletal muscles of GSDII patients are characterized by a progressive vacuolization. The hallmark of this disorder is the presence of swollen, glycogen-filled lysosomes and the massive accumulation of autophagic debris. Both these characteristics are thought to greatly contribute to the development of muscle weakness. However, the animal models of GSDII disease do not clarify this issue, because inhibition or activation of autophagy neither ameliorates nor worsens the phenotype of GAA-knockout (KO) mice. Surprisingly, glycogen levels also seem to be irrelevant for disease progression. Furthermore, because GAA-KO mice completely lack the GAA enzyme, they are not the best model for understanding the mechanisms of muscle wasting in late-onset GSDII, a condition of partial GAA defect. What animal models underline is a clear correlation between autophagosome accumulation and inefficiency of ERT treatment. This is not surprising because most of the cellular trafficking, including endosomal trafficking, uses proteins that also participate in autophagy. For instance, PIK3C3/VPS34 and PIK3R4/VPS15 are shared factors between endosome trafficking and autophagosome biogenesis. Therefore, autophagosome accumulation can sequester these factors that are also required for endosomal and vesicular trafficking and therefore, for recombinant GAA uptake and delivery to lysosomes ().
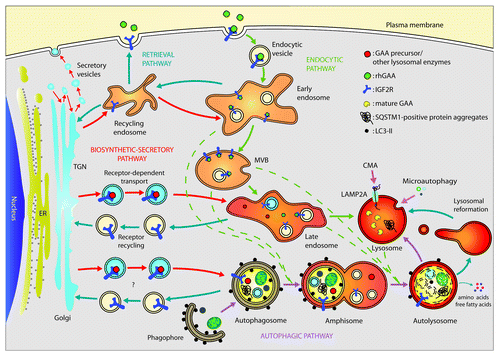
Figure 1. Autophagic, endocytic, biosynthetic-secretory and retrieval pathways merge at lysosomes. Lysosomes are the final destination sites of the autophagic degradation pathway, where it converges with the endocytic pathway, which is important for the uptake and recycling of nutrients and transmembrane receptors. GAA precursor is transported in a complex with IGF2R from the trans-Golgi network (TGN) to late endosomes and, likely, to autophagosomes. There, GAA is released from the IGF2Rs, which are recycled back to the Golgi network by retrieval pathways maintaining the backflow of selected components. In lysosomes, the 110-kDa GAA precursor is cleaved to generate the active forms of 76 and 70 kDa. A small fraction of IGF2R is located on the cell surface, where it mediates the uptake of rhGAA administered by ERT. Once the receptor is internalized in the endocytic pathway in can move to lysosomes via MVBs or recycle through the endocytic recycling compartment (ERC) to the plasma membrane. CMA, chaperone-mediated autophagy.
To clarify the role of autophagy in the pathogenesis of GSDII we have studied 17 muscle biopsies of 12 patients with different phenotypes, ranging from the most severe infantile-onset to the milder childhood, juvenile and adult forms of the disease and at different time points of disease progression. The effects of autophagic impairment on disease progression were monitored using the presence of SQSTM1/p62-containing protein aggregates as a marker of autophagic flux inhibition. In fact we have previously shown that acute inhibition of autophagy in adult muscles leads to formation of SQSTM1 protein aggregates. We then correlated the presence of protein aggregates with vacuolization and myofiber atrophy. Interestingly, the fibers that contain SQSTM1-positive aggregates are significantly smaller than surrounding normal fibers. Moreover, SQSTM1 aggregates do not always correlate with vacuolization, suggesting that the presence of vacuoles does not always mean autophagy impairment. Moreover, SQSTM1 aggregate-positive fibers are more atrophic than vacuolated ones confirming that autophagy impairment is detrimental for muscle mass maintenance. Gene expression analyses strengthened the correlation between autophagy impairment, muscle atrophy and expression of TRIM63/MURF-1, the ubiquitin ligase involved in degradation of contractile proteins. This correlation is particularly evident in adult-onset patients. Interestingly, in this group of patients there was no relationship between myofiber atrophy and expression of FBX032/ATROGIN1, the other atrophy-related muscle-specific ubiquitin ligase.
In order to define a link between disease progression and autophagy impairment we monitored the autophagy system in patients that underwent to a second biopsy and we compared the data with the first biopsy. Moreover, we also checked patients that were treated with ERT. In both cases we can confirm that autophagy impairment correlates with disease progression and that restoration of autophagy flux by ERT treatment increases muscle mass. Moreover, we also confirmed that muscle atrophy was linked to TRIM63/ MURF-1, but not to FBX032/ ATROGIN1, expression.
Importantly, in a few cases, there was no match between accumulation of SQSTM1, revealed by western blotting, with the presence of SQSTM1 aggregates and the increase of LC3-II. This discrepancy was explained by the fact that SQSTM1 is transcriptionally regulated and, therefore, the increase of SQSTM1 protein reflects an upregulation of SQSTM1 transcript. This concept is important and stresses the requirement of monitoring SQSTM1 both at protein and mRNA levels to avoid misinterpretation of autophagy flux based on western blotting analyses.
Impaired autophagy affects vesicle trafficking and inhibits GAA maturation
The comparison of pre- and post-ERT biopsies in one late-treated infantile patient and one early-treated adult patient underlined the concept that functional autophagy is required also for efficient recombinant human GAA (rhGAA) enzyme uptake, maturation and delivery to lysosomes. Indeed, when the autophagic flux is greatly compromised, ERT is inefficient in restoring the flux and in improving muscle morphology and function. The maturation steps of GAA from the synthesis of the immature protein in the endoplasmic reticulum to the final cleaved active protein in lysosomes are complex and require a functional system of vesicle trafficking. GAA precursor and other lysosomal proteins are synthesized and translocated into the ER, transferred to the cis-Golgi area, where GAA interacts with the insulin-like growth factor 2 receptor/cation-independent mannose-6-phosphate receptor (IGF2R), and is finally transported to the secretory vesicles of the trans-Golgi area. Then, the newly produced GAA moves to endosomes where the GAA releases IGF2R and goes to lysosomes. The final step of lysosome delivery may or may not require a fusion process with autophagosomes and other late endosomes to form ‘hybrid’ organelles (). A small fraction of IGF2R is located on the cell surface, where it mediates the uptake of the rhGAA. The delivery of rhGAA to lysosomes occurs via the endocytic-multivesicular body (MVB)-late endosome pathway ().
Since the accumulation of autophagosomes has an important impact on cellular trafficking, we then studied in detail the role of autophagy in GAA maturation by using in vitro cultured myotubes of patients and controls. We induced autophagy in patients’ myotubes by starvation alone or starvation together with rapamycin treatment, an MTOR inhibitor able to induce autophagosome formation. Conversely, we blocked lysosome function by chloroquine treatment in controls, to mimic Pompe disease. Myotubes from patients display vacuolization, while controls do not show any vacuoles unless they are treated with chloroquine. Importantly, autophagy induction reduces the vacuolization observed in patients’ myotubes. We then monitored the maturation of the GAA enzyme under these conditions. Induction of autophagy enhances GAA maturation, whereas chloroquine produces the opposite effect. Quantification of western blots confirmed the positive effect of autophagy on GAA processing, with a reduction in the immature form and enrichment of mature cleaved 76- to 70-kDa forms. Consistently, chloroquine causes an accumulation of the 110-kDa precursor in normal myotubes and disappearance or reduction of the 76- to 70-kDa forms in patient-derived myotubes ().
Figure 2. Abnormalities in lysosomal function have an impact upon vesicle trafficking and therefore on GAA maturation. In GSDII patients, lysosomal dysfunction caused by defective GAA generates a block in the autophagic flux, which also has an impact upon other pathways, impairing cell trafficking and thus inhibiting GAA maturation and rhGAA uptake.
In conclusion, a residual functional autophagic flux counteracts disease and atrophy progression in late-onset GSDII. Moreover, autophagy contributes to GAA maturation, turning out to be an important factor for successful ERT. Such valuable information might help in the near future to develop new drugs targeted to restore the autophagic flux in these patients and to improve the uptake of recombinant GAA.
Acknowledgments
The authors wish to thank Marco Nascimbeni for the artwork on the figures. This work was supported by grants from AFM (14199) to A.N., from Telethon-Italy (GTB07001-DR) and Eurobiobank network (QLRT2001-027769) to C.A., from AFM (15504), ERC (MYOPHAGY) and from Telethon-Italy (TCP04009) to M.S.