Abstract
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved catabolic process necessary for normal recycling of cellular constituents and for appropriate response to cellular stress. Although several genes belonging to the core molecular machinery involved in autophagosome formation have been discovered, relatively little is known about the nature of signaling networks controlling autophagy upon intracellular or extracellular stimuli. We discovered ATG8-like proteins (MAP1LC3B, GABARAP and GABARAPL1) as novel interactors of MAPK15/ERK8, a MAP kinase involved in cell proliferation and transformation. Based on the role of these proteins in the autophagic process, we demonstrated that MAPK15 is indeed localized to autophagic compartments and increased, in a kinase-dependent fashion, ATG8-like proteins lipidation, autophagosome formation and SQSTM1 degradation, while decreasing LC3B inhibitory phosphorylation. Interestingly, we also identified a conserved LC3-interacting region (LIR) in MAPK15 responsible for its interaction with ATG8-like proteins, for its localization to autophagic structures and, consequently, for stimulation of the formation of these compartments. Furthermore, we reveal that MAPK15 activity was induced in response to serum and amino-acid starvation and that this stimulus, in turn, required endogenous MAPK15 expression to induce the autophagic process. Altogether, these results suggested a new function for MAPK15 as a regulator of autophagy, acting through interaction with ATG8 family proteins. Also, based on the key role of this process in several human diseases, these results supported the use of this MAP kinase as a potential novel therapeutic target.
Introduction
Autophagy is an intracellular pathway for bulk protein degradation and damaged organelles removal by lysosomes.Citation1 Autophagy is initiated by the formation of a cup-shaped membrane, called phagophore.Citation2 Next, the phagophore enwraps parts of the cytoplasm to form a double-membrane vesicle, the autophagosome, which eventually fuses with lysosomes.Citation1,Citation3 Two evolutionarily conserved ubiquitin-like protein conjugation systems are necessary for the formation of the autophagosome, the ATG12-ATG5 and the ATG8-phosphatidylethanolamine (PE).Citation4 Among them, while ATG8 in yeast is represented by a single gene, its homologs in humans are represented by at least eight members that can be divided into two subgroups based on their amino acid sequence homology: the microtubule-associated protein 1 light chain 3 (LC3) family (LC3A, LC3B, LC3B2 and LC3C) and the GABA(A) receptor-associated protein (GABARAP) family (GABARAP, GABARAPL1, GABARAPL2 and GABARAPL3).Citation5 Importantly, apart from GABARAPL3,Citation6-Citation8 they are all conjugated to PE and, consequently, incorporated into autophagosomal membranes. The two protein subfamilies are essential for autophagosomes biogenesis and probably have distinct roles in the overall autophagic process.Citation8,Citation9 Notably, the functions of ATG8 human orthologs are mostly based on their interaction with a large cohort of proteins, with extensive binding partner overlap between family members.Citation10,Citation11
Autophagy is one of the major responses to stress in eukaryotic cells and is implicated in several pathological conditions such as infections, neurodegenerative diseases and cancer.Citation12-Citation14 Still, as upstream signaling events leading to stimulation of autophagy are still poorly characterized, possibilities to interfere with this process and, eventually, take advantage of its modulation for therapeutic purposes are limited. In this context, mitogen-activated protein (MAP) kinases are a family of proline-directed serine/threonine kinases coordinating the signaling from a variety of extracellular and intracellular stimuli to regulate gene expression, cell metabolism, and cytoskeleton dynamics.Citation15 Importantly, the MAPK1/ERK2, MAPK8/JNK and MAPK14/p38α members of this family have been already involved in the control of the autophagic process.Citation16,Citation17 Mitogen-activated protein kinase 15 (MAPK15/ERK8; ERK7) is the last identified member of the MAP kinase family.Citation18 Although possessing a typical Thr-X-Tyr activation motif (TEY), there is no evidence that MAPK15 is placed in a “classical,” three-tiered, MEKK-MEK-MAPK cascade as the other MAP kinases. Conversely, phosphorylation of TEY motif seems to be catalyzed by MAPK15 itself, therefore suggesting that autophosphorylation has a key role in controlling MAPK15 activity and functions.Citation19 Still, MAPK15 activity can be further modulated by serum, endogenous phosphatases, DNA damage and human oncogenes.Citation18,Citation20,Citation21 In line with the role of all the MAP kinases in controlling gene expression,Citation22 MAPK15 has been also reported to stimulate the activity of the JUN proto-oncogene,Citation23 and to reduce the activity of nuclear receptors such as androgen receptor, glucocorticoid receptors and estrogen-related receptor α (ESRRA/ERRα).Citation24-Citation26 Interestingly, recent data suggest a role for MAPK15 in cell transformationCitation23 and in the protection of genomic integrity, by inhibiting proliferating cell nuclear antigen (PCNA) degradation.Citation27 Ultimately, MAPK15 inhibition strongly affects telomerase activity,Citation28 suggesting this kinase as an important player in the mechanisms contributing to bypass replicative senescence and to immortalize cells.
To better understand the function of MAPK15, we used a yeast two-hybrid assay to screen a mouse cDNA library for potential interacting proteins.Citation25 Through this approach, we identified different members of the mammalian ATG8 family of proteins as binding partners for the C-terminal domain of MAPK15, raising the possibility that this MAP kinase may be involved in the control of autophagy. Here, we provide evidence that, indeed, MAPK15 is localized to autophagic structures and controls, in a kinase-dependent fashion, both basal and starvation-induced autophagy. Since MAPK15 is activated by different stimuli that induce autophagy,Citation19,Citation20 this kinase could therefore provide an unexpected link to connect nutrient- and stress-dependent signaling pathways to the activation of this important cellular process.
Results
MAPK15 interacted with mammalian ATG8-like proteins
In order to identify novel MAPK15 interacting proteins, we performed a yeast two-hybrid screening.Citation25 As MAPK15 has a unique C-terminal portion, which distinguishes this MAP kinase from other members of the family, we decided to use this domain as bait for the screening. Based on the evidence that MAPK15 is highly expressed in the nervous system,Citation18 we chose to screen a mouse brain library of cDNAs. Among the positive clones, we found multiple cDNAs encoding for Gabarap and Gabarapl1, which we confirmed to be devoid of self-activation (, and data not shown), suggesting that MAPK15 was able to interact with these proteins. Also, as they both share high identity and belong to the same protein family of LC3B,Citation29,Citation30 this observation suggested the possibility that MAPK15 also interacts with this protein. Therefore, we performed in vitro affinity precipitation experiments using full-length, HA-tagged, MAPK15 expressed in 293T cells and bacterially expressed, GST-tagged GABARAP, GABARAPL1 and LC3B. As shown in , all GST-tagged ATG8-like proteins, but not GST alone, were able to interact in vitro with MAPK15, albeit with different affinities. To determine whether MAPK15 was able to interact also in vivo with ATG8-like proteins, we performed affinity-precipitation experiments in HeLa cells, co-transfecting 6xHis-tagged MAPK15 with EGFP-tagged, LC3B, GABARAP, and GABARAPL1. As shown, they all coprecipitated in vivo with MAPK15 (), although with different relative affinities, confirming results previously obtained in vitro. Ultimately, we demonstrated that endogenous MAPK15 was also able to interact, in vivo, with endogenous LC3B (), altogether showing that MAPK15 indeed interacts with mammalian ATG8-like proteins.
Figure 1. MAPK15 interacted, in vitro and in vivo, with GABARAP, GABARAPL1 and LC3B. (A) Two positive clones, identified by two-hybrid screening, encoding GABARAP and GABARAPL1 were tested to be devoid of autoactivation. Yeast cells were cotransformed with the pACT2 vector containing the Gabarap (upper row) and Gabarapl1 (lower row) cDNA, respectively, with pGBKT7 alone (left) or with pGBKT7-MAPK15_C-term (right), and streaked on selective medium. (B) Bacterially expressed GST-GABARAP (lanes 3–4), GST-GABARAPL1 (lanes 5–6) and GST-LC3B (lanes 7–8) or GST alone (lanes 1–2), immobilized on glutathione-Sepharose Beads 4B, were incubated, for affinity precipitation (AP), with total lysates (TL) of 293T cells transiently transfected with a control vector or with HA-MAPK15, then analyzed by western blot (WB). (C) HeLa cells were cotransfected with a control vector or with GFP-LC3B, GFP-GABARAPL1, GFP-GABARAP in presence or absence of MAPK15–6XHis. Lysates (1 mg) were incubated with NiNTA-Sepharose Beads 4B, subjected to in vivo affinity precipitation, and then analyzed by WB. The total and MAPK15-bound amounts of GFP-tagged ATG8-like protein were quantified by NIH ImageJ software. (D) HeLa cells lysates (5 mg) were immunoprecipitated with control IgG or anti-MAPK15 antibody and analyzed by WB. LC3B was detected with anti-LC3B antibody (Sigma Aldrich).
MAPK15 localized to autophagic structures
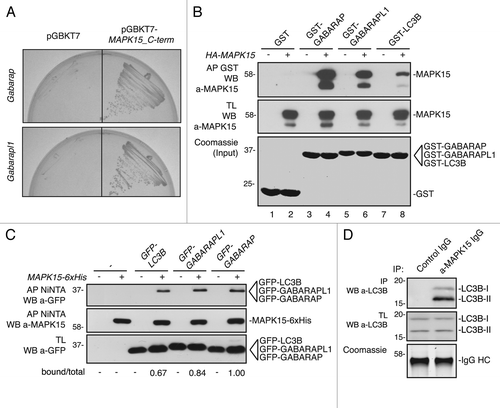
Each member of the LC3/GABARAP family of proteins consists of two forms with different subcellular localizations.Citation5 Among them, form I (e.g., LC3B-I) is localized to the cytoplasm while form II (e.g., LC3B-II), upon addition of a lipidic moiety, is relocalized onto autophagosomal membranes.Citation8,Citation29,Citation31 Hence, we hypothesized that MAPK15 might be recruited to autophagosomal vesicles by interacting with ATG8-like proteins. Still, as no information is currently available regarding MAPK15 localization to intracellular membranes, we first decided to characterize MAPK15 subcellular distribution by confocal microscopy. Immunofluorescence analysis of HeLa cells, transiently expressing MAPK15, revealed that this MAP kinase, besides localizing to the cytoplasm and the nucleus,Citation25 also exhibited a puncta pattern (), suggesting that MAPK15 could have a vesicular membrane localization. To confirm this observation, we also performed cell fractionation experiments. As shown in , MAPK15 was localized to both cytoplasmatic and nuclear fractions, as previously demonstrated.Citation25 Though, the membrane compartment also showed considerable amounts of the MAPK15 protein ().
Figure 2. MAPK15 localized to autophagic structures. (A) HeLa cells were transfected with HA-MAPK15 cDNA. Cells were permeabilized with 0.2% Triton X-100. HA-MAPK15 proteins were immuno-labeled with anti-MAPK15 antibody and revealed with AlexaFluor488-conjugated secondary antibody. Nuclei were stained with DAPI. (B) HeLa cells stably expressing HA-MAPK15 were subjected to fractionation using the Subcellular Protein Fractionation Kit (Thermo Scientific). Lysates were subjected to SDS-PAGE followed by WB with anti-MAPK15 (top), anti-NFKBIA (middle top), anti-Calnexin (middle bottom), and anti-JUN (bottom) antibodies. C, cytoplasmic fraction; M, membrane fraction; N, nuclear fraction. (C) A clone of HeLa cells stably expressing HA-MAPK15 were permeabilized with 100 μg/ml digitonin. Cells were stained with appropriate antibodies (anti-MAPK15, anti-LC3B MBL, anti-GABARAP, anti-SQSTM1 and anti-LAMP1) and revealed with AlexaFluor488- and AlexaFluor555-conjugated secondary antibodies. Nuclei were stained with DAPI. The region enclosed in the white square has been enlarged in the smaller panel for better appreciation of the colocalizations. Arrows indicate colocalization areas. Similar results were obtained in at least 3 independent clones. (D) Colocalization rate of MAPK15 and LC3, GABARAP, SQSTM1 and LAMP1 respectively was obtained by analyzing at least 400 cells/sample from three different experiments (n = 3). (E) HeLa cells stably expressing HA-MAPK15 were transfected with pCEFL-GFP-LC3B. Twenty-four hours later, cells were permeabilized with 100 μg/ml digitonin. Cells were stained with anti-SQSTM1 and anti-MAPK15 antibodies and revealed with AlexaFluor555- and AlexaFluor647-conjugated secondary antibodies. Nuclei were stained with DAPI. The region enclosed in the white square has been enlarged in the lower panels for better appreciation of the colocalizations. Arrows indicate colocalization areas.
Next, we tested the hypothesis that this MAP kinase was also localized to autophagic vesicles. To this aim, we performed immunofluorescence colocalization analysis between MAPK15 and endogenous autophagic vesicle markers, upon permeabilization of cells with digitonin in order to eliminate the background signal due to their cytoplasmatic forms.Citation32 Consistently with its interaction with ATG8-like proteins, MAPK15, indeed, partially colocalized with endogenous LC3B, GABARAP and SQSTM1/p62 (), all classical markers for membranes of autophagosomal origin. However, MAPK15 showed a more limited colocalization rate with LAMP1 (), an established marker for lysosomal and autophagolysosomal vesicles.Citation33 Finally, we also performed triple localization experiments, confirming that this MAP kinase resided on cytoplasmic vesicles of clear autophagosomal origin, being positive for both LC3B and SQSTM1 (). Altogether, our data therefore demonstrated that MAPK15 localizes on autophagic structures and suggested that such localization may be important for this kinase to have full access to pools of specific substrates such as other ATG proteins, to control the autophagic pathway.
MAPK15 induced autophagy
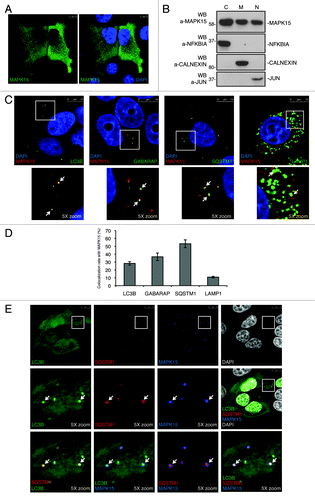
Based on these data, we next decided to investigate the role of MAPK15 in controlling autophagy. As previously described,Citation7,Citation34 induction of autophagy is associated to an increase of the form II of LC3B (and of its related proteins GABARAP and GABARAPL1) that can be monitored by using anti-ATG8 proteins antibodies specifically recognizing their lipidated form II. Indeed, using a well-established stimulus promoting autophagy, the MTOR inhibitor rapamycin,Citation35 form II protein levels increased in our cellular system, reaching a significant enhancement at around 2 h for LC3B-II and 4 h for GABARAP-II (Fig. S1, left panels; refs. Citation36–Citation38). As MAPK15 activity is controlled by autophosphorylation,Citation19,Citation39 we overexpressed it to stimulate its function (Fig. S2; refs. Citation21 and Citation24). As shown in , MAPK15 induced LC3B-II at levels comparable to rapamycin and starvation, suggesting its ability to affect autophagy, as also confirmed by interfering with ATG7 (Fig. S3) and ATG5 (Fig. S4) expression in HeLa cells expressing this MAP kinase. Still, the assay scoring an increase of LC3B-II cannot discriminate between an induction of autophagy or an inhibition in late stage of this cellular process.Citation5,Citation40 To distinguish between these two situations, we therefore used inhibitors of lysosomal acid proteases, according to autophagy methods guidelines.Citation5 In cells overexpressing MAPK15, treatment with protease inhibitors, namely E64d () and leupeptin (Fig. S5), further increased LC3B-II level when compared with MAPK15 overexpression alone. This result indicated that the LC3B-II accumulation, obtained in presence of activated MAPK15, was due to autophagy stimulation and not to an impaired clearance.
Figure 3. MAPK15 induced autophagy. (A) HeLa cells were transfected with control vector or HA-MAPK15. Two hours (h) before harvesting, cells were starved (Starv) or treated with rapamycin (Rap, 200 nM), where indicated. Lysates were analyzed by WB, with indicated antibodies. LC3B was detected with anti-LC3B antibody (Nanotools). The LC3B-II and MAPK1 amounts were quantified by NIH ImageJ software. (B) HeLa cells were transfected with control vector or HA-MAPK15. One hour before harvesting, cells were treated with E64d (10 μg/ml), where indicated. Lysates were analyzed by WB, with indicated antibodies. LC3B was detected with anti-LC3B antibody (Nanotools). The LC3B-II and MAPK1 amounts were quantified by NIH ImageJ software. (C) HeLa T-Rex MAPK15 cells were treated with doxycycline (Doxy, 1 μM) for different periods. Two hrs before harvesting, cells were starved, where indicated. Lysates were analyzed by WB, with indicated antibodies. LC3B was detected with anti-LC3B antibody (Nanotools). The LC3B-II and MAPK1 amounts were quantified by NIH ImageJ software. (D) HeLa T-Rex MAPK15 cells were treated with Doxy for 16 h, where indicated. Cells were fixed and then permeabilized with 100 μg/ml digitonin. Cells were stained with anti-LC3B antibody (MBL) and revealed with AlexaFluor488-conjugated antibody. Nuclei were stained with DAPI. Lower panel of each figure indicates the amount of LC3B dots per cell quantified by Volocity software. Measures were obtained by analyzing at least 400 cells/sample from five different experiments (n = 5). Measures were subjected to one-way ANOVA test. Asterisks were attributed for the following significance value: **p < 0.01. (E) Same as in (D), but, 4 h before fixing, cells were treated with Bafilomycin A1 (Baf, 100 nM). (F) HeLa cells were transfected with control vector or HA-MAPK15. Before harvesting, cells were starved (Starv) for the indicated periods. Lysates were analyzed by WB, with indicated antibodies. (G) HeLa cells were transfected with nonsilencing siRNA or with MAPK15-specific siRNA. After 72 h, cells were fixed and permeabilized with 100 μg/ml digitonin. Cells were stained with anti-LC3B (MBL) antibody and revealed with AlexaFluor488-conjugated secondary antibody. Nuclei were stained with DAPI. Lower panel of each figure indicate the amount of LC3B dots per cell quantified by Volocity software. Measures were obtained by analyzing at least 400 cells/sample from three different experiments (n = 3). Measures were subjected to one-way ANOVA test. Asterisks were attributed for the following significance value: ***p < 0.001. (H) Same as in (G), but, cells were starved for 1 h before fixing.
In order to circumvent potential biases arising from stress induced by transient transfection of the MAPK15 cDNA,Citation41 we next decided to analyze the ability of this kinase to stimulate autophagy by using a MAPK15-inducible cell line, HeLa T-Rex MAPK15. In this cellular system, the expression of MAPK15, induced by doxycycline (; Fig. S6A), also led to the increase of LC3B-II (). Interestingly, the expression in the same inducible system of an exogenous protein, the product of the LacZ gene, did not manifest any effect on LC3B-II (Fig. S6B), indicating the absence of nonspecific effects due to doxycycline or protein overexpression.
Indirect immunofluorescence microscopy, scoring the formation of punctate intracellular vacuoles stained for LC3B, is a key assay to monitor autophagy and to understand its dynamics.Citation5 In order to confirm our previous results and to expand their biological significance, we next performed indirect immunofluorescence analysis in our inducible cell system, aimed at scoring an increase in puncta for endogenous LC3B. As shown in , stimulation of MAPK15 expression in inducible HeLa cells led to a ~6.8-fold increase in the number of autophagic structures, as compared with uninduced cells (quantification performed through unbiased analysis using the Quantitation Module of Volocity software). To better appreciate autophagosomal formation, we also used bafilomycin A1, an inhibitor of lysosomal acidification, to prevent lysosomal degradation of autophagosome-associated LC3B.Citation42 Bafilomycin A1 allows, in fact, the detection of each autophagosome formed in the time-lapse between addition of the drug to cells and harvesting. As shown in , despite the higher background due to the enhancing effect of the inhibitor on the number of autophagic vesicles, MAPK15 expression in bafilomycin A1-treated, HeLa inducible cells also enhanced the amount of autophagic structures (~2.3-fold increase) in comparison to uninduced cells. As a control, MAPK15 protein levels were only slightly affected by this blocker of lysosomal degradation (Fig. S7), supporting the already established preeminent role of the ubiquitin-proteasome pathway in the turnover of the MAPK15 protein.Citation43,Citation44
Similarly to LC3B, also GABARAP and GABARAPL1 are lipidated by the autophagic enzymatic machinery and associated to autophagic vesicles.Citation8,Citation29,Citation37,Citation45 Indeed, in line with results obtained with LC3B, expression of MAPK15 in HeLa cells also led to an increase in the amount of GABARAP-II (Fig. S8A) and to a ~5-fold increase in the number of autophagic structures (Fig. S8B), as compared with uninduced cells, further confirming the ability of MAPK15 to stimulate autophagy.
Zacharogianni et al. recently reported that MAPK15 is stabilized by starvation and mediates its effects on cellular secretion.Citation44 In line with these results, we observed that starvation was also able to induce MAPK15 activity (), with a mechanism possibly involving autophosphorylation,Citation19,Citation39 thereby clearly establishing starvation as a MAPK15-activating stimulus. Still, starvation did not influence the interaction between MAPK15 and LC3B-II (Fig. S9), while it increased localization of this kinase to autophagic structures (Fig. S10), suggesting that this MAP kinase may take advantage of its “constitutive” interaction with ATG8 family members to be recruited to autophagosomal membranes, upon autophagic stimuli. Conversely, rapamycin, a pharmacological stimulus of autophagy acting on the MTOR kinase,Citation35 did not induce MAPK15 activity (Fig. S11), suggesting that MAPK15 may utilize an MTOR-independent mechanism to induce autophagy, a conclusion also supported by the observation that starvation regulates secretion through MAPK15 in an MTOR-independent manner.Citation44
Based on these data, we investigated the role of the endogenous MAPK15 protein in controlling not only basal but also starvation-induced cellular autophagy. To this purpose, MAPK15 expression was silenced using a validated, specific siRNA (Fig. S12; refs. Citation21 and Citation25). By using this tool, we observed ~43% and ~67% reduction in the number of basal (full medium, FM) () and starvation-induced () autophagic structures, respectively, as compared with the corresponding cells transfected with scrambled siRNAs. Altogether, these data support a role for endogenous MAPK15 in controlling the rate of basal cellular autophagy that may be related to its ability to autophosphorylate and, therefore, maintain a constitutive level of activity in the cells even in the absence of upstream stimuli. Still, specific stimuli such as starvation may increase the activity of MAPK15 by possibly enhancing its stabilityCitation44 and the phosphorylation of its TEY activation domain (please, see above), consequently increasing the rate of cellular autophagy.
MAPK15 induced SQSTM1 degradation and reduced LC3B inhibitory phosphorylation
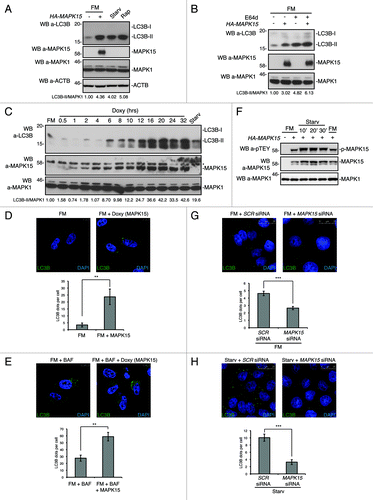
Autophagic flux can be measured by determining the declining abundance of autophagic substrates such as SQSTM1.Citation41,Citation46 Hence, we tested the amount of endogenous SQSTM1 protein upon MAPK15 expression in HeLa cells. As shown in , transiently transfected MAPK15 clearly induced SQSTM1 degradation. Similarly, increasing MAPK15 expression in our HeLa inducible cell line also stimulated SQSTM1 degradation at levels comparable to rapamycin and starvation, our positive controls ().
Figure 4. MAPK15 induced SQSTM1 degradation and reduced LC3B inhibitory phosphorylation. (A) HeLa cells were transfected with control vector or HA-MAPK15. Four hrs before harvesting, cells were treated with Rap or CQ, where indicated. Lysates were analyzed by WB, with indicated antibodies. The SQSTM1 and MAPK1 amounts were quantified by NIH ImageJ software. (B) HeLa T-Rex MAPK15 cells were treated with Doxy for 24 and 32 h, where indicated. Four hrs before harvesting, cells were starved or treated with Rap, where indicated. Lysates were analyzed by WB, with indicated antibodies. The SQSTM1 and MAPK1 amounts were quantified by NIH ImageJ software. (C) HeLa T-Rex MAPK15 cells were treated with Doxy for 16 h or starved as indicated. Cells were fixed and then permeabilized with 100 μg/ml digitonin. Cells were stained with anti-SQSTM1 and anti-MAPK15 antibodies and revealed with AlexaFluor488-conjugated and AlexaFluor555-conjugated secondary antibodies, respectively. Right panels, indicate the amount of SQSTM1 dots per cell quantified by Volocity software. Measures were obtained by analyzing at least 400 cells/sample from four different experiments (n = 4). Measures were subjected to one-way ANOVA test. Asterisks were attributed for the following significance value: ***p < 0.001. (D) HeLa cells were transfected with control vector or HA-MAPK15. One hour before harvesting, cells were treated with Rap, where indicated. Lysates were analyzed by WB, with indicated antibodies. Total LC3B was detected with anti-LC3B antibody (Sigma Aldrich). The phospho-LC3B, LC3B-I and MAPK1 amounts were quantified by NIH ImageJ software.
In HeLa cells, SQSTM1 shows a cytoplasmic distribution in basal conditions while, although degraded at increased rate, it is relocalized to autophagic vesicles upon activation of autophagy.Citation46,Citation47 Indeed, in our inducible HeLa expression system, MAPK15 clearly led to an increase in the number of SQSTM1 positive puncta, similarly to what happens upon starvation, our positive control (). These results therefore indicate that MAPK15 induces both SQSTM1 autophagic relocalization and its autophagic removal.
Recent data have demonstrated that LC3B phosphorylation negatively regulates autophagy.Citation48 We therefore sought to establish whether MAPK15 also was able to impinge on LC3B phosphorylation to control this process. Indeed, MAPK15, while increasing LC3B-II levels, clearly reduced in vivo LC3B phosphorylation both in HeLa () and in 293T cells (Fig. S13). This evidence suggested a mechanism involving MAPK15 in the control of ATG8-like proteins functions through additional proteins (possibly kinases and/or phosphatases).
MAPK15 directly bound mammalian ATG8-like proteins through a specific LIR-containing region in its C-terminal domain
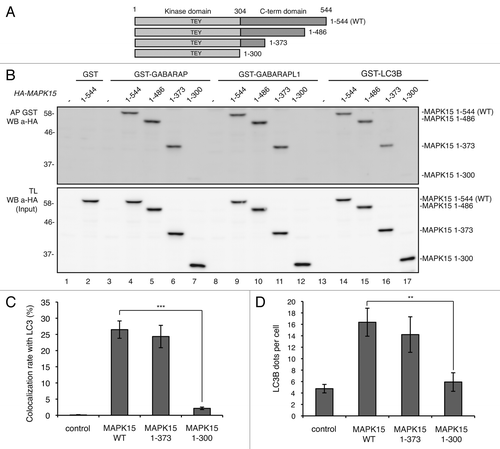
Domains responsible for interaction of MAPK15 with SRC, TGFB1I1, ESRRA and PCNA have already been mapped to its C-terminal region.Citation18,Citation25-Citation27 Here, we sought to identify the MAPK15 C-terminal region allowing interaction with ATG8-like proteins. To this purpose, different MAPK15 deletion mutants were examined (), showing that all mutants containing the region 1–373 efficiently interacted with ATG8-like proteins whereas further deletion up to aa 300 strongly diminished co-precipitation (). Together with two-hybrid screening results, these data suggested that the region of MAPK15 necessary for interaction with LC3B, GABARAP and GABARAPL1 mapped between aa 300 and aa 373. Moreover, the MAPK15 deletion mutant that did not interact with ATG8-like proteins (MAPK15 1–300) was also functionally impaired, being unable to localize to autophagic structures () and to induce the formation of autophagosomal vesicles ().
Figure 5. MAPK15 bound ATG8-like protein through region 300–373 aa. (A) Scheme depicts MAPK15 full length and its deletion mutants. (B) Bacterially expressed GST-GABARAP (lanes 3–7), GST-GABARAPL1 (lanes 8–12), GST-LC3B (lanes 13–17) or GST alone (lanes 1–2) immobilized on glutathione-Sepharose Beads 4B, were incubated with lysates of 293T cells transiently transfected with a control vector or with full length HA-MAPK15_WT or with HA-MAPK15 deletion mutants, subjected to affinity precipitation and then analyzed by WB. (C) Colocalization rate of LC3 with MAPK15 or its deletion mutants in HeLa transfected cells was obtained by analyzing at least 400 cells/sample from three different experiments (n = 3). Measures were subjected to one-way ANOVA test. Asterisks were attributed for the following significance value: ***p < 0.001. (D) The graph indicates the amount of LC3B dots per cell in HeLa cells transfected with MAPK15 or its deletion mutants, quantified by Volocity software. Measures were obtained by analyzing at least 400 cells/sample from three different experiments (n = 3). Measures were subjected to one-way ANOVA test. Asterisks were attributed for the following significance value: **p < 0.01.
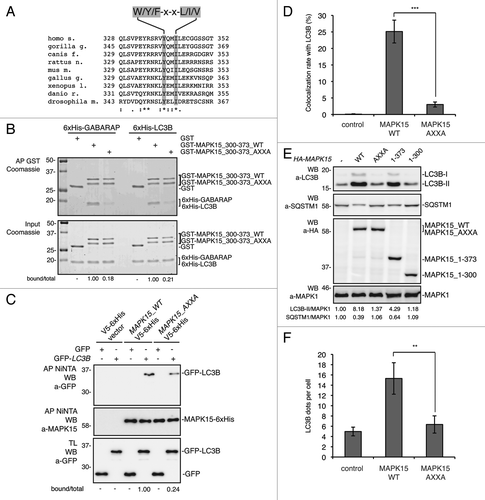
Upon C-terminal lipidation, mammalian ATG8-like proteins are incorporated into the autophagosomal membrane where they promote the recruitment of a large cohort of proteins, with extensive binding partner overlap between family members, and frequent involvement of a conserved surface on ATG8-like proteins known to interact with LC3-interacting regions (LIRs) usually represented by a conserved hydrophobic W/Y/F-X-X-L/I/V motif.Citation10,Citation49,Citation50 Indeed, specific mutations in the LIR often reduce the binding of LIR-containing proteins to ATG8 family members.Citation51-Citation53 Based on the relative low sequence complexity of the LIR consensus, we identified seven potential LIR motifs in full length MAPK15 (Fig S14A). Among them, the most conserved across MAPK15 orthologous genes () was localized within the MAPK15 300–373 region (Fig. S14B). We decided to focus on this LIR, based on the already demonstrated importance of this fragment for the interaction between MAPK15 and ATG8-like proteins. To definitively prove a direct mode of interaction, we performed in vitro affinity precipitation among bacterially purified, His-tagged, LC3B and GABARAP proteins and the GST-tagged 300–373 fragment of MAPK15 (GST-MAPK15_300–373), both in its wild-type (WT) and in its mutated form in the key tyrosine (aa 340) and isoleucine (aa 343) residues of the LIRCitation10,Citation50 (GST-MAPK15_300–373_AXXA). As shown in , both ATG8-like proteins specifically interacted with the purified aa 300–373 of MAPK15 but such interactions were significantly impaired by mutating the conserved LIR motif. Finally, by in vivo affinity-precipitation experiments between the LIR-mutated MAPK15 and LC3B, we confirmed that this specific domain strongly affected the ability of MAPK15 to be recruited by this protein (). Also in this case, we functionally validated these data in HeLa cells, as expression of the MAPK15 protein mutated in the putative LIR motif (HA-MAPK15_AXXA) was unable to localize to autophagic vesicles (), to induce the formation of lipidated LC3B, to stimulate the disposal of SQSTM1 protein () and to induce an increase in the number of autophagosomes (). Altogether, these data further expanded our demonstration of the interaction among these proteins by establishing a direct interaction mechanism mediated by a classical LIR motif contained in the MAPK15 300–373 region, supporting the relevance of this region in the MAPK15-dependent mechanisms controlling autophagy.
Figure 6. MAPK15 bound ATG8-like protein through a specific LIR-containing region. (A) Alignment of a portion of MAPK15 protein sequence across the species showing the position of the conserved LIR motif. (B) GST, GST-MAPK15_300–373_WT or GST-MAPK15_300–373_AXXA were incubated for 1 h at 4°C with 6xHis-GABARAP or 6xHis-LC3B. Proteins were then affinity-purified by glutathione-Sepharose Beads 4B, extensively washed and loaded on SDS-PAGE gels. Total proteins were stained with Coomassie stain. The total and MAPK15-bound amounts of 6xHis-tagged ATG8-like protein were quantified by NIH ImageJ software. (C) HeLa cells were cotransfected with a control vector or with GFP-LC3B, MAPK15_WT-V5–6xHis or MAPK15_AXXA-V5–6xHis. Lysates (1 mg) were incubated with NiNTA-Sepharose Beads 4B, subjected to in vivo affinity precipitation, and then analyzed by WB. The total and MAPK15-bound amounts of GFP-tagged ATG8-like protein were quantified by NIH ImageJ software. (D) Colocalization rate of LC3 with MAPK15_WT or MAPK15_AXXA in HeLa transfected cells was obtained by analyzing at least 400 cells/sample from three different experiments (n = 3). Measures were subjected to one-way ANOVA test. Asterisks were attributed for the following significance value: ***p < 0.001. (E) HeLa cells were transfected with control vector or HA-MAPK15 (WT) or its mutants. Lysates were analyzed by WB, with indicated antibodies. LC3B was detected with anti-LC3B antibody (Nanotools). The LC3B-II, SQSTM1 and MAPK1 amounts were quantified by NIH ImageJ software. (F) The graph indicates the amount of LC3B dots per cell in HeLa cells transfected with MAPK15_WT or MAPK15_AXXA, quantified by Volocity software. Measures were obtained by analyzing at least 400 cells/sample from three different experiments (n = 3). Measures were subjected to one-way ANOVA test. Asterisks were attributed for the following significance value: **p < 0.01.
The catalytic activity of MAPK15 was required for its ability to control autophagy
We next speculated that MAPK15 kinase activity might be involved in the control of autophagy. Therefore, we first used a kinase-dead MAPK15 mutant (MAPK15_KD)Citation19 and established that it was unable to induce an increase of LC3B-II (), suggesting that MAPK15 kinase activity was necessary for this protein to induce autophagy. These data were further confirmed by immunofluorescence microscopy, as MAPK15_KD was also unable to induce the formation of autophagosomal structures (). This effect was not dependent on an impairment of MAPK15 binding to ATG8-like proteins, as MAPK15_KD was able to bind to LC3B () and to colocalize with this protein to autophagosomal structures (; Fig. S15) to an extent comparable to wild-type MAPK15.
Figure 7. The catalytic activity of MAPK15 was required for its ability to control autophagy. (A) HeLa cells were transfected with control vector or HA-MAPK15_WT or HA-MAPK15_KD. Lysates were analyzed by WB, with indicated antibodies. LC3B was detected with anti-LC3B antibody (Nanotools). Right panel, steady-state autophagy. Left panel, autophagic flux. (B) The graph indicates the amount of LC3B dots per cell in HeLa cells transfected with MAPK15_WT or MAPK15_KD, quantified by Volocity software. Measures were obtained by analyzing at least 400 cells/sample from three different experiments (n = 3). Measures were subjected to one-way ANOVA test. Asterisks were attributed for the following significance value: **p < 0.01. (C) For affinity precipitation (AP), bacterially expressed GST-LC3B or GST alone, immobilized on glutathione-Sepharose Beads 4B were incubated with lysates of 293T cells transiently transfected with a control vector or with HA-MAPK15_WT or HA-MAPK15_KD, then analyzed by western blot. (D) Colocalization rate of LC3 and MAPK15 in HeLa cells stably expressing MAPK15_WT or MAPK15_KD was obtained by analyzing at least 400 cells/sample from three different experiments (n = 3). (E) HeLa T-Rex MAPK15 cells were treated with Doxy for 2 h, then Ro-318220 (1 μM) was added to culture medium for the following 6 h, where indicated. Cells were fixed and then permeabilized with 100 μg/ml digitonin. Cells were stained with anti-LC3B (MBL) antibody and revealed with AlexaFluor488-conjugated antibody. Nuclei were stained with DAPI. The LC3B-positive dots per cell were quantified using Volocity software (right panel). Measures were obtained by analyzing at least 400 cells/sample from three different experiments (n = 3). Measures were subjected to one-way ANOVA test. Asterisks were attributed for the following significance values: *p < 0.05, **p < 0.01. (F) HeLa cells were treated with Ro-318220 (1 μM) for 6 h and/or then starved for 1 h, where indicated. Cells were fixed and permeabilized with 100 μg/ml digitonin, then stained with anti-LC3B antibody (MBL) and revealed with AlexaFluor488-conjugated secondary antibody. Nuclei were stained with DAPI. The number of LC3B positive dots per cell was quantified using the Volocity software (right panel). Measures were obtained by analyzing at least 400 cells/sample from three different experiments (n = 3). Measures were subjected to one-way ANOVA test. Asterisks were attributed for the following significance values: *p < 0.05, **p < 0.01. At the concentration used, the Ro-318220 compound did not affect cell viability. (G) HeLa cells were treated as in (E). Lysates were analyzed by WB, with indicated antibodies. LC3B was detected with anti-LC3B antibody (Nanotools). (H) HeLa cells were treated as in (F). Lysates were analyzed by WB, with indicated antibodies. LC3B was detected with anti-LC3B antibody (Nanotools).
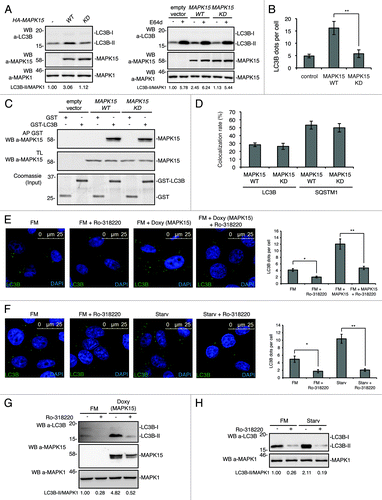
To confirm these data with a different approach, we also set up to investigate the effect of MAPK15 chemical inhibition on autophagy induced by the activated kinase and by starvation. Despite the lack of inhibitors of MAPK15 useful for clinical applications, the Ro-318220 compound has already shown the ability to inhibit this MAP kinase,Citation19,Citation28 as also confirmed in our experimental settings (Fig. S16). In order to demonstrate that, in our MAPK15 inducible system, stimulation of autophagy was dependent on MAPK15 kinase activity, we therefore treated doxycycline-stimulated cells with the Ro-318220 inhibitor and observed a ~61% reduction in the number of punctate intracellular vacuoles stained for LC3B (). In this context, it is important to point out that Ro-318220 is also reported as a PKC inhibitor.Citation54 Still, we feel confident that the inhibitory effect that we observe by using this drug was not attributable to PKC, because, as this kinase inhibits autophagy,Citation55,Citation56 its inhibition should have induced this process. Ultimately, to establish also a role for endogenous MAPK15 kinase activity in controlling starvation-dependent induction of autophagy, we also treated HeLa cells with the MAPK15 inhibitor, observing a ~79% reduction in the number of autophagic structures (), mirroring results observed with MAPK15 siRNA (). Additionally, the immunofluorescence data about the inhibition of autophagy by Ro-318220 were also confirmed by scoring anti-LC3B-II by western blot analysis (). Altogether, these data supported a role for MAPK15 kinase activity in the control of autophagy.
Discussion
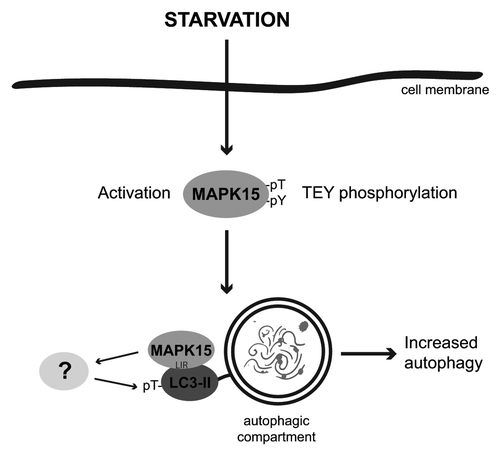
MAPK15 functions are far from being completely understood. The results described here showed that, in basal conditions as well as upon starvation, MAPK15 activity was able to control the rate of the autophagic process by interacting with mammalian ATG8 family proteins (). Based on the kinase activity of MAPK15 and on our results using the kinase-dead mutant and the Ro-318220 inhibitor, phosphorylation of proteins involved in the regulation of autophagy may be the mechanism by which MAPK15 affected this cellular process. However, ATG8 family members do not present conserved minimal MAP kinase phosphorylation motifs (Ser/Thr-Pro) and, therefore, they may not be direct targets of MAPK15 kinase activity. Indeed, recombinant ATG8-like proteins performed very poorly as substrates for MAPK15 in in vitro kinase assays (Fig. S17). Conversely, the interaction between ATG8-like proteins and MAPK15 may be required for the correct localization of the MAP kinase, in order to gain full access to pools of specific substrates, such as other kinases and/or phosphatases, directly impinging on the autophagic process. Ultimately, we demonstrated that MAPK15 indirectly affected LC3B inhibitory phosphorylation in vivo, suggesting that MAPK15 may be involved in complex pathways that integrate different stimuli in a coordinated cellular response. Additional work will be required to address these issues and to specifically dissect all the molecular steps involved in MAPK15-dependent regulation of autophagy.
Figure 8. MAPK15 in the control of the autophagic process. A fraction of the MAPK15 protein is localized to autophagic structures by binding to mammalian ATG8 proteins, through a conserved LIR. When localized to these compartments, in basal conditions as well as upon starvation, MAPK15 enzymatic activity is able to control the rate of autophagy by phosphorylating downstream substrates, possibly additional ATG proteins, ultimately mediating its effect on this process.
Further supporting and expanding our results, MAPK15 has been recently shown to negatively regulate protein secretion.Citation44 More specifically, amino acid starvation, a strong inducer of autophagy, has been demonstrated to cause stabilization of MAPK15 protein and thus to inhibit protein secretion, in an MTOR-independent manner. In this context, our observation that starvation, but not the specific MTOR inhibitor rapamycin,Citation35 stimulated MAPK15 is really intriguing. MAPK15 may therefore coordinate cellular responses to starvation by both turning off the protein secretion pathway and upregulating the autophagic machinery, in order to give the cell more chances to successfully overcome this particularly stressful situation. As such, our data suggested a novel mechanism to understand the role of MAPK15 in cellular physiology and human pathology.
In this work, we also provided several insights into the molecular mechanism of MAPK15-dependent stimulation of autophagy. Specifically, our data underlined the importance of the interaction of MAPK15 with mammalian ATG8-like proteins, through a specific LIR in the kinase C-terminal domain (). Also, they suggested the existence of unknown MAPK15 substrates involved in autophagy, as MAPK15 kinase activity was strictly required to induce autophagy (). In this context, it is worth noting the dual relevance of the 300–373 MAPK15 region for the regulation of autophagy, as it both controlled the binding of the kinase to ATG8-like proteins through the conserved LIR () and was required for kinase activity (Fig. S18; ref. Citation20).
Recent reports have suggested a role for MAPK15 in the response to DNA damage and, possibly, in the maintenance of genomic stability.Citation21,Citation27,Citation28 Interestingly, as previously described, both MAPK15 silencingCitation27 and misregulation of the autophagic processCitation57 are associated with increased DNA damage, gene amplification and aneuploidy, implying a key role for autophagy in limiting chromosomal instability. Our data therefore suggested that induction of autophagy through this MAP kinase may participate, together with PCNA stabilizationCitation27 and induction of telomerase activity,Citation28 to a general MAPK15-dependent program aiming at maintaining genomic integrity.
Mitochondria are the intracellular compartment where oxidative phosphorylation takes place, using glucose and oxygen to produce the ATP required for cell functions.Citation58 This process leads to the formation of reactive oxygen species (ROS), that leak from both normal and, especially, damaged mitochondria, resulting in oxidative damage to proteins, lipids and nucleic acids.Citation59 Autophagy plays a key role in the degradation of damaged mitochondria and, indeed, autophagy-deficient cells exhibit an increase in ROS production.Citation60 Importantly, hydrogen peroxide, a source of ROS, activated MAPK15 (Fig. S11; ref. Citation21). Consequently, our results suggested that MAPK15 may be activated as a consequence of ROS generation to stimulate autophagy, accelerating degradation of damaged mitochondria. Supporting this hypothesis, we recently have demonstrated that MAPK15 also controls the transcriptional activity of ESRRA,Citation25 a key regulator of mitochondrial biogenesis and functions.Citation61 In this context, MAPK15 may be involved in a novel molecular mechanism aimed to mitigate oxidative stress and consequent DNA damage in response to ROS as well as to other stimuli. These and additional hypotheses are currently under investigation.
Autophagy has been recently regarded as an attractive target for successful cancer therapy. However, huge efforts are still needed to address its specific role in cell transformation, mostly concerning its apparently contradictory functions as a proapoptotic or prosurvival mechanism. Indeed, autophagy plays a critical protective role in the maintenance of energy homeostasis and protein and organelle quality control,Citation62 thus protecting cells from damage accumulation under conditions of metabolic stress. Moreover, reduced levels or absence of proteins required for autophagy have been associated to facilitated tumor establishment in mouse models and human tumors.Citation63-Citation65 Based on this evidence, autophagy has been addressed as a tumor suppressor mechanism. Conversely, cells defective for autophagy are more sensitive to metabolic- or chemotherapy-induced stress,Citation14,Citation66 suggesting that autophagy might represent an antiapoptotic mechanism which contributes to resistance to a vast array of antineoplastic drugs.Citation67 Nonetheless, autophagy may also be activated in tumor cells in response to stress and specific human oncogenes,Citation68,Citation69 thus supporting cancer cell survival by eliminating specific apoptosis-inducing proteins.Citation70,Citation71 Consequently, inhibition of autophagy is currently considered a promising approach to sensitize cancer cells to different therapies.Citation72 Based on the already demonstrated ability of different human oncogenes to activate MAPK15,Citation20 we can hypothesize that this kinase, by controlling autophagy, may participate in the tumorigenic process sustained by these oncogenes. This, therefore, allows us to speculate that MAPK15 inhibition may be beneficial for the treatment of these tumors. In this scenario, the precise nature of the stimuli controlling MAPK15 expression and activity and of the mechanisms triggered by MAPK15 to regulate autophagy in physiological as well as in pathological conditions warrant further investigation.
Materials and Methods
Reagents and antibodies
Bafilomycin A1 (Santa Cruz Biotechnology, sc-201550) and Ro-318220 (VWR International, 557521-500) were dissolved in DMSO. Chloroquine (C6628), rapamycin (R0395), doxycycline (D9891) were purchased from Sigma Aldrich. Hank’s medium (H15-010), used as starvation medium, was obtained by PAA. The following primary antibodies were used for western blots, co-immunoprecipitations, and confocal microscopy experiments: anti-MAPK15/ERK8 (custom preparation), anti-HA (Covance, MMS-101R) and anti-GFP (Covance, MMS-118P), anti-LC3B (Nanotools, 0231-1000), anti-LC3B (MBL, M152-3) and anti-GABARAP (MBL, M135-3), anti-LC3B (Sigma Aldrich, L7543), anti-ATG5 (Sigma Aldrich, A0731) and anti-ATG7 (Sigma Aldrich, A2856), anti-phospho-TEY (Cell Signalling, 9101), anti-SQSTM1/p62 (BD Biosciences, 610833), anti-phospho-LC3 (Abgent, AP3301a), anti-MAPK1/ERK2 (Santa Cruz Biotechnology, sc-154), anti-LAMP1 (Santa Cruz Biotechnology, sc-20011), anti-NFKBIA/IκBα (Santa Cruz Biotechnology, sc-371), anti-Calnexin (Santa Cruz Biotechnology, sc-23954) and anti-JUN (Santa Cruz Biotechnology, sc-45). The following secondary antibodies were used for western blot experiments: anti-mouse (Santa Cruz Biotechnology, sc-2004) and anti-rabbit (Santa Cruz Biotechnology, sc-2005) HRP-conjugated IgGs.
Expression vectors
All the vectors used in yeast two-hybrid screening were previously described.Citation25 pCEFL-HA-MAPK15, and all its deletion mutants were already describedCitation20 except for 1–300 MAPK15 deletion mutant which was generated by cloning the cDNA, obtained by PCR amplification, in the pCEFL-HA vector. pcDNA4-Tet-On-MAPK15 was generated by subcloning the MAPK15 cDNA, obtained by restriction enzyme digestion from pCEFL-HA-MAPK15, into the pcDNA4 Tet-inducible vector, kindly provided from Francesca Carlomagno (Università degli Studi di Napoli). pcDNA-Tet-on-LacZ was also kindly provided from F. Carlomagno. A double point mutation of MAPK15 in Y340A and I343A was obtained by QuikChange site-directed mutagenesis kit (Stratagene, 200518) from pCEFL-HA-MAPK15 and generated pCEFL-HA-MAPK15_AXXA. For interaction analysis, MAPK15_WT and MAPK15_AXXA were also subcloned by restriction enzyme digestion on pEF1_V5–6xHis expression vector (Invitrogen, V920-20), generating pEF1_MAPK15_WT-V5-6xHis and pEF1_MAPK15_AXXA-V5-6xHis. MAPK15 300–373 aa (corresponding to 900–1119 bp) cDNA was subcloned by PCR into pGEX4T3 plasmid from pCEFL-HA-MAPK15 and pCEFL-HA-MAPK15_AXXA, generating pGEX4T3-MAPK15_300–373_WT and pGEX4T3-MAPK15_300–373_AXXA. MAPK15 kinase dead mutant D155A (MAPK15 KD) cDNA was a generous gift from Philip Cohen (University of Dundee) and then subcloned into pCEFL-HA plasmid. pCEFL-EGFP-GABARAP, pGEX4T3-GABARAP and pRSET-GABARAP were generated by subcloning in these vectors the GABARAP cDNA obtained by PCR from a pCMVSport6-GABARAP template (Invitrogen, Clone ID 5528016). pCEFL-GFP-GABARAPL1, pGEX4T3-GABARAPL1 were generated by subcloning in these vectors the GABARAPL1 cDNA, obtained by PCR from pCMVSport6-GABARAPL1 template (Invitrogen, Clone ID 6185293). pCEFL-EGFP-LC3B, pGEX4T3-LC3B and pRSET-LC3B were generated by subcloning in these vectors the LC3B cDNA (OpenBiosystem, Clone ID 5276841). pGIPZ plasmids encoding for scrambled shRNA and ATG7 shRNA were kindly provided from Fabrizio Condorelli (Università del Piemonte Orientale).
Cell culture and transfections
293T and HeLa cells were maintained in Dulbecco’s modified Eagle medium (DMEM; PAA, E15-009) supplemented with 10% fetal bovine serum (PAA, A15-151), 2 mM L-glutamine and 100 units/ml penicillin-streptomycin at 37°C in an atmosphere of 5% CO2/air. HeLa HA-MAPK15_WT and HeLa HA-MAPK15_KD were generated by transfecting HeLa cells with pCEFL-HA-MAPK15_WT and pCEFL-HA-MAPK15_KD respectively; stably expressing cells were then plated at limiting diluition in 96-well plates to obtain single cell clones. HeLa T-Rex cells and HeLa T-Rex β-Gal cells, were maintained in DMEM supplemented with 10% fetal bovine serum (Clonetech, 631106), 2 mM L-glutamine, 5 ug/ml blasticidin and 100 units/ml penicillin-streptomycin at 37°C in an atmosphere of 5% CO2/air. Doxycycline-inducible cell lines for MAPK15 were generated by transfecting HeLa T-Rex cells with pcDNA4-Tet-On-MAPK15 plasmid.
For immunofluorescence experiments and western blot analysis, 5 × 104 cells were seeded in 12-well plates (2 × 105 for 6-well plates) and transfected with 200 ng (500 ng in 6-well plates) of each expression vector, using Lipofectamine LTX (Invitrogen, 15338500). All experiments were performed, unless specified, 24 h after transfection. For confocal microscopy experiments, 2.5 × 104 cells were seeded on coverslips placed onto 12-well plates. Each sample was transfected with 200 ng of each plasmid using Lipofectamine LTX.
RNA interference
MAPK15-specific siRNA (target sequence 5′-TTGCTTGGAGGCTACTCCCAA-3′) and nonsilencing siRNA (scrambled, SCR; target sequence 5′-AATTCTCCGAACGTGTCACGT-3′) were obtained from Qiagen. The ATG5-specific siRNA pool (62012000062) was purchased from Riboxx. HeLa cells were transfected with siRNA at a final concentration of 5 nM using HiPerFect (Qiagen, 301707), according to the manufacturer’s instructions. Samples were analyzed, unless specified, 72 h after transfection.
Western blots
For LC3B analysis,Citation73 total lysates were obtained by resuspending washed cellular pellets in 1× Laemmli buffer and incubating for 10 min at 95°C, in continuous agitation. For SQSTM1 analysis,Citation74 total lysates were obtained by resuspending cellular pellets in RIPA buffer (50 mM TRIS-HCl pH 8.0, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% NP-40). For western blot analysis, proteins derived from total lysates, immunoprecipitations or affinity precipitations were loaded on polyacrylamide gels and resolved by SDS-PAGE, transferred to Immobilon-P PVDF membrane (Millipore, IPVH00010), probed with appropriate antibodies and revealed by enhanced chemoluminescence detection (ECL Plus; GE Healthcare, RPN2132). For detection of endogenous MAPK15 protein an amount of 200 μg of total protein extract was loaded for each sample. Densitometric analysis of western blots was performed with NIH Image J 1.43u (National Institutes of Health).
Yeast two-hybrid
Yeast two-hybrid screening was performed as previously described.Citation25 Thanks to the presence of a myc epitope in the resulting fusion protein (myc-GAL4-MAPK15 C-term), we also confirmed the expression of such protein in yeast. We also confirmed that our bait was not able to activate, by itself, the transcription of reporter genes. In addition, we confirmed that our bait could not interact directly with the GAL4 transactivation domain contained in the plasmid employed to engineer the cDNA library used for the screening.
Bacterial expression of GST- and 6xHis-fusion proteins
The BL21(DE3)pLys strain of Escherichia coli was transformed with the pRSET-6xHis alone or encoding for GABARAP and LC3B respectively, and with the pGEX4T3 vector alone or encoding the GABARAP, GABARAPL1, LC3B, MAPK15_300–373_WT and MAPK15_300–373_AXXA fusion proteins respectively. Bacterially expressed 6xHis- and GST-fusion proteins were purified as previously described.Citation75
Coimmunoprecipitations
Whole cell lysates were obtained by resuspending washed pellets in RIPA buffer. Lysates were pre-cleaned with Protein A/G conjugated beads (Santa Cruz Biotechnology, sc-2003), and then incubated with appropriate antibodies for 2 h at 4°C. Immunocomplexes were purified by incubating lysates with Protein A/G-conjugated beads. Immunocomplexes were next resuspended in Laemmli buffer and loaded on gels for SDS-PAGE and western blot analysis.
GST affinity precipitation
Whole cell lysates were obtained by resuspending washed pellets in RIPA buffer. Lysates were pre-cleaned with glutathione conjugated beads (Amersham, 17-5132-03) and then incubated with GST or GST-fusion proteins for 2 h at 4°C. Protein complexes were then purified by glutathione conjugated beads. Protein complexes have been resuspended in Laemmli buffer and loaded on gels for SDS-PAGE and western blot analysis or Coomassie staining.
Ni-NTA affinity precipitation
Whole cell lysates were obtained by resuspending washed pellets in RIPA buffer with 20 mM Imidazole. Protein complexes were then purified by incubating with Ni-NTA-conjugated beads (Qiagen, 30210). Protein complexes were resuspended in Laemmli buffer and loaded on gels for SDS-PAGE and western blot analysis.
GST-pulldown of purified proteins
6xHis-tagged ATG8-like proteins and GST-MAPK15_300–373_WT or GST-MAPK15_300–373_AXXA were incubated in 25 mM TRIS-HCl pH 8.0, 300 mM NaCl, 0.5% Tween 20 for 1 h. Protein complexes were then purified by glutathione conjugated beads. After several washes, protein complexes were resuspended in Laemmli buffer and loaded on gels for SDS-PAGE and western blot analysis.
Coomassie staining
Bacterially expressed proteins were loaded on SDS-PAGE gels, stained with SimplyBlue SafeStain (Invitrogen, LC6060), and revealed using the Odyssey Infrared Imaging System (Li-Cor Biosciences, 9120).
Immunofluorescence (IF)
Cells were washed with PBS, then fixed with 4% paraformaldehyde in PBS for 20 min and permeabilized with 0.2% Triton X-100 solution for 5 min or 100 μg/ml digitonin solution (Invitrogen, BN2006) for 20 min, as indicated. Cells were incubated with appropriate primary antibodies for 1 h, washed three times with PBS, and then incubated with appropriate Alexa Fluor 488-conjugated (Invitrogen, A21202), Alexa Fluor 555-conjugated (Invitrogen, A31272) or Alexa Fluor 647-conjugated (Invitrogen, A21245) secondary antibodies and then washed again three times in PBS. Nuclei were stained with a solution of 1.5 μM of 4′,6-diamidino-2-phenylindole (DAPI; Sigma Aldrich, D9542) in PBS for 5 min. Coverslips were mounted in fluorescence mounting medium (Dako, S3023). Samples were visualized on a TSC SP5 confocal microscope (Leica, 5100000750) installed on an inverted LEICA DMI 6000CS (10741320) microscope and equipped with an oil immersion PlanApo 63× 1.4 NA objective. Images were acquired using the LAS AF acquisition software (Leica, 10210).
Subcellular fractionation
Cytoplasmic, membrane and nuclear fractions were obtained by the Subcellular Protein Fractionation Kit (Thermo Scientific, 78840), according to the manufacturer’s instructions. Briefly, cells were harvested by trypsinization and centrifuged at 500 g for 5 min. Cells were then washed in PBS and resuspended in cytoplasm extraction buffer and centrifuged at 500 g for 5 min. Supernatants were collected as cytoplasm extracts and pellets further suspended in Membrane Extraction Buffer. Following 10 min incubation on ice, resuspended pellets were centrifuged at 3000 g for 5 min. Supernatants were then collected as membrane extracts and pellets further suspended in nuclear extraction buffer. Following 30 min incubation on ice, resuspended pellets were centrifuged at 5000 g for 5 min. Then, supernatants were collected as nuclear extracts. All subcellular fraction extracts were then analyzed by western blot.
Dot counts, colocalization rate and statistical analysis
For the LC3B-, GABARAP- and SQSTM1-positive dots count, we performed intensitometric analysis of fluorescence using the Quantitation Module of Volocity software (PerkinElmer Life Science, I40250). Colocalization rate was also measured by the Quantitation Module of Volocity software. Dot counts and colocalization rate were subjected to statistical analysis. Measures were obtained by analyzing at least 400 cells/sample from three different experiments. Significance (p value) was obtained by one-way ANOVA test. Asterisks were attributed for the following significance values: *p < 0.05, **p < 0.01, ***p < 0.001.
| Abbreviations: | ||
| MAP | = | mitogen-activated protein |
| PE | = | phosphatidylethanolamine |
| aa | = | amino acid |
| FM | = | full medium |
| ROS | = | reactive oxygen species |
| WB | = | western blot |
| ATG | = | autophagy-related |
| EGFP | = | enhanced green fluorescent protein |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| ESRRA | = | estrogen-related receptor alpha |
| Rap | = | rapamycin |
| CQ | = | chloroquine |
| Doxy | = | doxycycline |
| Baf | = | bafilomycin A1 |
| ERK | = | extracellular signal-regulated kinase |
| GABARAP | = | GABA(A) receptor-associated protein |
| LIR | = | LC3-interacting region |
Additional material
Download Zip (3.3 MB)Acknowledgments
We thank Sabrina Di Bartolomeo, Francesca Carlomagno and Laura Salvini for assistance; Federica Sasdelli for critical reading of this manuscript; Philip Cohen for the kind gift of the MAPK15 kinase-dead plasmid; Fabrizio Condorelli for the kind gift of ATG7 shRNA. This work was supported by a start-up grant from Regione Toscana, by the Italian Association for Cancer Research (AIRC) and by a grant to Regione Toscana from Ministero della Salute, in the context of the “Programma di Ricerca Sanitaria Finalizzata 2008.”
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/21857
References
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717 - 21; http://dx.doi.org/10.1126/science.290.5497.1717; PMID: 11099404
- Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol 2010; 12:831 - 5; http://dx.doi.org/10.1038/ncb0910-831; PMID: 20811355
- Seglen PO, Bohley P. Autophagy and other vacuolar protein degradation mechanisms. Experientia 1992; 48:158 - 72; http://dx.doi.org/10.1007/BF01923509; PMID: 1740188
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2:211 - 6; http://dx.doi.org/10.1038/35056522; PMID: 11265251
- Klionsky D, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 18188003
- Chakrama FZ, Seguin-Py S, Le Grand JN, Fraichard A, Delage-Mourroux R, Despouy G, et al. GABARAPL1 (GEC1) associates with autophagic vesicles. Autophagy 2010; 6:495 - 505; http://dx.doi.org/10.4161/auto.6.4.11819; PMID: 20404487
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805 - 12; http://dx.doi.org/10.1242/jcs.01131; PMID: 15169837
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010; 29:1792 - 802; http://dx.doi.org/10.1038/emboj.2010.74; PMID: 20418806
- Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci 2005; 118:7 - 18; http://dx.doi.org/10.1242/jcs.01620; PMID: 15615779
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68 - 76; http://dx.doi.org/10.1038/nature09204; PMID: 20562859
- Noda NN, Ohsumi Y, Inagaki F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett 2010; 584:1379 - 85; http://dx.doi.org/10.1016/j.febslet.2010.01.018; PMID: 20083108
- Cheung ZH, Ip NY. Autophagy deregulation in neurodegenerative diseases - recent advances and future perspectives. J Neurochem 2011; 118:317 - 25; http://dx.doi.org/10.1111/j.1471-4159.2011.07314.x; PMID: 21599666
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323 - 35; http://dx.doi.org/10.1038/nature09782; PMID: 21248839
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21:1367 - 81; http://dx.doi.org/10.1101/gad.1545107; PMID: 17510285
- Gehart H, Kumpf S, Ittner A, Ricci R. MAPK signalling in cellular metabolism: stress or wellness?. EMBO Rep 2010; 11:834 - 40; http://dx.doi.org/10.1038/embor.2010.160; PMID: 20930846
- Corcelle E, Djerbi N, Mari M, Nebout M, Fiorini C, Fénichel P, et al. Control of the autophagy maturation step by the MAPK ERK and p38: lessons from environmental carcinogens. Autophagy 2007; 3:57 - 9; PMID: 17102581
- Lorin S, Pierron G, Ryan KM, Codogno P, Djavaheri-Mergny M. Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy 2010; 6:153 - 4; http://dx.doi.org/10.4161/auto.6.1.10537; PMID: 19949306
- Abe MK, Saelzler MP, Espinosa R 3rd, Kahle KT, Hershenson MB, Le Beau MM, et al. ERK8, a new member of the mitogen-activated protein kinase family. J Biol Chem 2002; 277:16733 - 43; http://dx.doi.org/10.1074/jbc.M112483200; PMID: 11875070
- Klevernic IV, Stafford MJ, Morrice N, Peggie M, Morton S, Cohen P. Characterization of the reversible phosphorylation and activation of ERK8. Biochem J 2006; 394:365 - 73; http://dx.doi.org/10.1042/BJ20051288; PMID: 16336213
- Iavarone C, Acunzo M, Carlomagno F, Catania A, Melillo RM, Carlomagno SM, et al. Activation of the Erk8 mitogen-activated protein (MAP) kinase by RET/PTC3, a constitutively active form of the RET proto-oncogene. J Biol Chem 2006; 281:10567 - 76; http://dx.doi.org/10.1074/jbc.M513397200; PMID: 16484222
- Klevernic IV, Martin NM, Cohen P. Regulation of the activity and expression of ERK8 by DNA damage. FEBS Lett 2009; 583:680 - 4; http://dx.doi.org/10.1016/j.febslet.2009.01.011; PMID: 19166846
- Hazzalin CA, Mahadevan LC. MAPK-regulated transcription: a continuously variable gene switch?. Nat Rev Mol Cell Biol 2002; 3:30 - 40; http://dx.doi.org/10.1038/nrm715; PMID: 11823796
- Xu YM, Zhu F, Cho YY, Carper A, Peng C, Zheng D, et al. Extracellular signal-regulated kinase 8-mediated c-Jun phosphorylation increases tumorigenesis of human colon cancer. Cancer Res 2010; 70:3218 - 27; http://dx.doi.org/10.1158/0008-5472.CAN-09-4306; PMID: 20395206
- Henrich LM, Smith JA, Kitt D, Errington TM, Nguyen B, Traish AM, et al. Extracellular signal-regulated kinase 7, a regulator of hormone-dependent estrogen receptor destruction. Mol Cell Biol 2003; 23:5979 - 88; http://dx.doi.org/10.1128/MCB.23.17.5979-5988.2003; PMID: 12917323
- Rossi M, Colecchia D, Iavarone C, Strambi A, Piccioni F, Verrotti di Pianella A, et al. Extracellular signal-regulated kinase 8 (ERK8) controls estrogen-related receptor α (ERRα) cellular localization and inhibits its transcriptional activity. J Biol Chem 2011; 286:8507 - 22; http://dx.doi.org/10.1074/jbc.M110.179523; PMID: 21190936
- Saelzler MP, Spackman CC, Liu Y, Martinez LC, Harris JP, Abe MK. ERK8 down-regulates transactivation of the glucocorticoid receptor through Hic-5. J Biol Chem 2006; 281:16821 - 32; http://dx.doi.org/10.1074/jbc.M512418200; PMID: 16624805
- Groehler AL, Lannigan DA. A chromatin-bound kinase, ERK8, protects genomic integrity by inhibiting HDM2-mediated degradation of the DNA clamp PCNA. J Cell Biol 2010; 190:575 - 86; http://dx.doi.org/10.1083/jcb.201002124; PMID: 20733054
- Cerone MA, Burgess DJ, Naceur-Lombardelli C, Lord CJ, Ashworth A. High-throughput RNAi screening reveals novel regulators of telomerase. Cancer Res 2011; 71:3328 - 40; http://dx.doi.org/10.1158/0008-5472.CAN-10-2734; PMID: 21531765
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Wang H, Bedford FK, Brandon NJ, Moss SJ, Olsen RW. GABA(A)-receptor-associated protein links GABA(A) receptors and the cytoskeleton. Nature 1999; 397:69 - 72; http://dx.doi.org/10.1038/16264; PMID: 9892355
- Hemelaar J, Lelyveld VS, Kessler BM, Ploegh HL. A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. J Biol Chem 2003; 278:51841 - 50; http://dx.doi.org/10.1074/jbc.M308762200; PMID: 14530254
- Lorenz H, Hailey DW, Lippincott-Schwartz J. Fluorescence protease protection of GFP chimeras to reveal protein topology and subcellular localization. Nat Methods 2006; 3:205 - 10; http://dx.doi.org/10.1038/nmeth857; PMID: 16489338
- Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med 2006; 27:495 - 502; http://dx.doi.org/10.1016/j.mam.2006.08.005; PMID: 16973206
- Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2004; 36:2503 - 18; http://dx.doi.org/10.1016/j.biocel.2004.05.009; PMID: 15325588
- Molnar-Kimber KL. Mechanism of action of rapamycin (Sirolimus, Rapamune). Transplant Proc 1996; 28:964 - 9; PMID: 8623482
- Haspel J, Shaik RS, Ifedigbo E, Nakahira K, Dolinay T, Englert JA, et al. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy 2011; 7:629 - 42; http://dx.doi.org/10.4161/auto.7.6.15100; PMID: 21460622
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 1998; 273:3963 - 6; http://dx.doi.org/10.1074/jbc.273.7.3963; PMID: 9461583
- Tanida I, Tanida-Miyake E, Ueno T, Kominami E. The human homolog of Saccharomyces cerevisiae Apg7p is a Protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J Biol Chem 2001; 276:1701 - 6; PMID: 11096062
- Coulombe P, Meloche S. Atypical mitogen-activated protein kinases: structure, regulation and functions. Biochim Biophys Acta 2007; 1773:1376-87.
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542 - 5; PMID: 17611390
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Shacka JJ, Klocke BJ, Shibata M, Uchiyama Y, Datta G, Schmidt RE, et al. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol Pharmacol 2006; 69:1125 - 36; http://dx.doi.org/10.1124/mol.105.018408; PMID: 16391239
- Kuo WL, Duke CJ, Abe MK, Kaplan EL, Gomes S, Rosner MR. ERK7 expression and kinase activity is regulated by the ubiquitin-proteosome pathway. J Biol Chem 2004; 279:23073 - 81; http://dx.doi.org/10.1074/jbc.M313696200; PMID: 15033983
- Zacharogianni M, Kondylis V, Tang Y, Farhan H, Xanthakis D, Fuchs F, et al. ERK7 is a negative regulator of protein secretion in response to amino-acid starvation by modulating Sec16 membrane association. EMBO J 2011; 30:3684 - 700; http://dx.doi.org/10.1038/emboj.2011.253; PMID: 21847093
- Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 2008; 9:859 - 64; http://dx.doi.org/10.1038/embor.2008.163; PMID: 18704115
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Rusten TE, Stenmark H. p62, an autophagy hero or culprit?. Nat Cell Biol 2010; 12:207 - 9; http://dx.doi.org/10.1038/ncb0310-207; PMID: 20190829
- Cherra SJ 3rd, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, Day BW, et al. Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol 2010; 190:533 - 9; http://dx.doi.org/10.1083/jcb.201002108; PMID: 20713600
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279 - 96; http://dx.doi.org/10.4161/auto.7.3.14487; PMID: 21189453
- Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, et al. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 2008; 13:1211 - 8; http://dx.doi.org/10.1111/j.1365-2443.2008.01238.x; PMID: 19021777
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012; 14:177 - 85; http://dx.doi.org/10.1038/ncb2422; PMID: 22267086
- Popovic D, Akutsu M, Novak I, Harper JW, Behrends C, Dikic I. Rab GTPase-activating proteins in autophagy: regulation of endocytic and autophagy pathways by direct binding to human ATG8 modifiers. Mol Cell Biol 2012; 32:1733 - 44; http://dx.doi.org/10.1128/MCB.06717-11; PMID: 22354992
- Sancho A, Duran J, García-España A, Mauvezin C, Alemu EA, Lamark T, et al. DOR/Tp53inp2 and Tp53inp1 constitute a metazoan gene family encoding dual regulators of autophagy and transcription. PLoS One 2012; 7:e34034; http://dx.doi.org/10.1371/journal.pone.0034034; PMID: 22470510
- Davis PD, Hill CH, Lawton G, Nixon JS, Wilkinson SE, Hurst SA, et al. Inhibitors of protein kinase C. 1. 2,3-Bisarylmaleimides. J Med Chem 1992; 35:177 - 84; http://dx.doi.org/10.1021/jm00079a024; PMID: 1732526
- Coward J, Ambrosini G, Musi E, Truman JP, Haimovitz-Friedman A, Allegood JC, et al. Safingol (L-threo-sphinganine) induces autophagy in solid tumor cells through inhibition of PKC and the PI3-kinase pathway. Autophagy 2009; 5:184 - 93; http://dx.doi.org/10.4161/auto.5.2.7361; PMID: 19098447
- Jiang H, Cheng D, Liu W, Peng J, Feng J. Protein kinase C inhibits autophagy and phosphorylates LC3. Biochem Biophys Res Commun 2010; 395:471 - 6; http://dx.doi.org/10.1016/j.bbrc.2010.04.030; PMID: 20398630
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961 - 7; http://dx.doi.org/10.1038/nrc2254; PMID: 17972889
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol 2006; 16:R551 - 60; http://dx.doi.org/10.1016/j.cub.2006.06.054; PMID: 16860735
- Orrenius S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev 2007; 39:443 - 55; http://dx.doi.org/10.1080/03602530701468516; PMID: 17786631
- Jin S. Autophagy, mitochondrial quality control, and oncogenesis. Autophagy 2006; 2:80 - 4; PMID: 16874075
- Villena J, Henriquez M, Torres V, Moraga F, Díaz-Elizondo J, Arredondo C, et al. Ceramide-induced formation of ROS and ATP depletion trigger necrosis in lymphoid cells. Free Radic Biol Med 2008; 44:1146 - 60; http://dx.doi.org/10.1016/j.freeradbiomed.2007.12.017; PMID: 18191646
- Mathew R, White E. Autophagy, stress, and cancer metabolism: what doesn’t kill you makes you stronger. Cold Spring Harb Symp Quant Biol 2011; 76:389 - 96; http://dx.doi.org/10.1101/sqb.2012.76.011015; PMID: 22442109
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 2008; 10:776 - 87; http://dx.doi.org/10.1038/ncb1740; PMID: 18552835
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 6; http://dx.doi.org/10.1038/45257; PMID: 10604474
- Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Otín C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem 2007; 282:18573 - 83; http://dx.doi.org/10.1074/jbc.M701194200; PMID: 17442669
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51 - 64; http://dx.doi.org/10.1016/j.ccr.2006.06.001; PMID: 16843265
- Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MC, et al. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta 2009; 1793:1524-32.
- Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL, et al. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011; 30:1855 - 67; http://dx.doi.org/10.1038/onc.2010.561; PMID: 21151168
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25:460 - 70; http://dx.doi.org/10.1101/gad.2016311; PMID: 21317241
- Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K, et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol 2012; 14:51 - 60; http://dx.doi.org/10.1038/ncb2386; PMID: 22138575
- Sandilands E, Serrels B, Wilkinson S, Frame MC. Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells. EMBO Rep 2012; 13:733 - 40; http://dx.doi.org/10.1038/embor.2012.92; PMID: 22732841
- Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010; 6:322 - 9; http://dx.doi.org/10.4161/auto.6.3.11625; PMID: 20224296
- Tanida I, Waguri S. Measurement of autophagy in cells and tissues. Methods Mol Biol 2010; 648:193 - 214; http://dx.doi.org/10.1007/978-1-60761-756-3_13; PMID: 20700714
- Lamark T, Perander M, Outzen H, Kristiansen K, Øvervatn A, Michaelsen E, et al. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem 2003; 278:34568 - 81; http://dx.doi.org/10.1074/jbc.M303221200; PMID: 12813044
- Marinissen MJ, Chiariello M, Pallante M, Gutkind JS. A network of mitogen-activated protein kinases links G protein-coupled receptors to the c-jun promoter: a role for c-Jun NH2-terminal kinase, p38s, and extracellular signal-regulated kinase 5. Mol Cell Biol 1999; 19:4289 - 301; PMID: 10330170