Abstract
We have recently shown that arsenic trioxide (As2O3) is a potent inducer of autophagic degradation of the BCR-ABL1 oncoprotein, which is the cause of chronic myeloid leukemia (CML) and Ph+ acute lymphoid leukemia (Ph+ ALL). Our recently published work has shown that pharmacological inhibition of autophagy or molecularly targeting of elements of the autophagic machinery partially reverses the suppressive effects of As2O3 on primitive leukemic precursors from CML patients. Altogether, our studies have provided direct evidence that arsenic-induced, autophagy-mediated, degradation of BCR-ABL1 is an important mechanism for the generation of the effects of As2O3 on BCR-ABL1 transformed leukemic progenitors. These studies raise the potential of future clinical-translational efforts employing combinations of arsenic trioxide with autophagy-modulating agents to promote elimination of early leukemic progenitors and, possibly, leukemia-initiating stem cells.
Despite continuously accumulating knowledge linking autophagy to the pathophysiology of different malignancies, the precise underlying mechanisms that define the ultimate outcome on malignant cell viability still remain unclear, while the existing evidence is in some cases confusing. It has been established that different pharmacological agents with antineoplastic properties can induce autophagic activity, but the upstream cellular mechanisms, the downstream cellular events, and the ultimate phenotypic outcome can be quite variable. The dual role of autophagy as either a cell survival or cell death mechanism has been at the center of this variability and confusion. Autophagy, like apoptosis, is commonly described as a type of programmed cell death, though it is distinct due to a self-catabolic process involving lysosomal proteolytic degradation of cellular components. The nature of the autophagic stimuli, the context and events leading up to autophagy and the cellular targets of that degradation, may be the factors that ultimately define the fate of the cell, leading to either cell survival or death (). An important and clinically relevant question is whether autophagy is dominantly used by cancer cells to gain an advantage during growth and/or to escape death by antineoplastic agents, and whether it can act as a “cellular switch” that can be modified to promote death and elimination of malignant cells.
Figure 1. The dual role of autophagy as a cell survival or cell death mechanism may be defined by the type of the inducing stimuli and/or simultaneously activated pathways and cellular events.
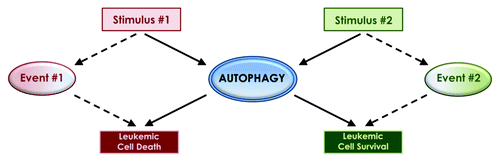
There has been previous evidence that autophagy can be a protective mechanism for the survival of leukemia cells transformed by the BCR-ABL1 oncoprotein, in response to treatment by tyrosine kinase inhibitors (TKIs) or during catalytic dual MTORC1-MTORC2 inhibition. There is also evidence for a similar role for autophagy in the control of cell death by other antineoplastic agents in other malignancies. This has led to the hypothesis that combining autophagy inhibitors with such drugs for the treatment of Ph+ leukemias may result in synergistic responses. This is an important clinical question and should be addressed in future studies. It would be particularly important to do so, as there has been emerging evidence in the recent past that leukemia-initiating stem cells (LICs) are not eliminated by TKIs, even in cases where complete hematological, cytogenetic and/or molecular responses are achieved. Despite the clear direction that has emerged from such previous studies in terms of clinical-translational efforts, the precise role of autophagy in response to other cellular stimuli or other pharmacological agents in Ph+ leukemias remains unclear.
Arsenic trioxide is an agent with well-established antileukemic properties, and its introduction in clinical medicine has had a dramatic impact on the outcome of patients with one form of AML, acute promyelocytic leukemia (APL). Although arsenic is very toxic and poisonous, its successful use in the treatment of APL patients has been possible because of the very high sensitivity of APL cells to its effects that can be achieved at relatively low, clinically tolerant, concentrations of this agent. There is still a hope that this agent will find additional applications in clinical oncology, either by devising ways to sensitize other types of malignant cells to its effects and/or by possibly using it to target cancer stem cells in combination with other drugs. Our recent study demonstrates that degradation of the leukemia-inducing BCR-ABL1 oncoprotein involves induction of autophagy, and that the autophagic process plays a key and essential role for the generation of the antileukemic effects of arsenic trioxide. Crucial to that degradation is the association of the oncoprotein with the polyubiquitin binding protein SQSTM1/p62. Our study also demonstrates that As2O3 treatment promotes formation of SQSTM1/p62-BCR-ABL1 complexes localized to autolysosomes, and that knockdown of key autophagosome-initiating proteins like ATG7 blocks the degradation of BCR-ABL1. These results are intriguing, especially when taken together with work from another group that implicated autophagy as the main mode of degradation of the leukemogenic oncoprotein PML-RARA during arsenic trioxide treatment of APL cells. Additionally, in BCR-ABL1-expressing cells, the lysosome-localized enzymatic protease CTSB seems to play an important role in this process, providing a new insight into the mechanisms by which autophagy-activating drug treatments can target and degrade leukemic oncoproteins.
It remains to be seen whether exploitation of this property of arsenic trioxide will provide an effective approach in the future in efforts to target LICs in Ph+ leukemias and, possibly, other leukemias. What may complicate things in this context is the possible negative effects of autophagy on responses by TKIs that are used routinely in the treatment of CML and Ph+ ALL and on responses by catalytic MTOR inhibitors or other agents that may prove to be useful in cases of TKI-resistance, by targeting essential pathways downstream of BCR-ABL1. For instance, if autophagy plays a critical role in arsenic-induced antileukemic effects, while it impedes antileukemic responses by TKIs in patients with Ph+ leukemias, any potential future LIC-targeting approach using arsenic should avoid its concomitant use with TKIs. However, such potential challenges in the use of agents that modulate autophagy underscores the importance of better understanding the regulatory mechanisms of the autophagic process and the context that defines cell survival or death in response to autophagy.