Abstract
Lysosomal storage diseases are metabolic disorders characterized by the accumulation of acidic vacuoles, and are usually the consequence of the deficiency of an enzyme responsible for the metabolism of vesicular lipids, proteins or carbohydrates. In contrast, mucolipidosis type IV (MLIV), results from the absence of a vesicular Ca2+ release channel called mucolipin 1/transient receptor potential mucolipin 1 (MCOLN1/TRPML1) which is required for the fusion of amphisomes with lysosomes. In Drosophila, ablation of the MCOLN1 homolog (trpml) leads to diminished viability during pupation when the animals rely on autophagy for nutrients. This pupal lethality results from decreased target of rapamycin complex 1 (TORC1) signaling, and is reversed by reactivating TORC1. Our findings indicate that one of the primary causes of toxicity in the absence of TRPML is cellular amino acid starvation, and the resulting decrease in TORC1 activity. Furthermore, our findings raise the intriguing possibility that the neurological dysfunction in MLIV patients may arise from amino acid deprivation in neurons. Therefore, future studies evaluating the levels of amino acids and TORC1 activity in MLIV neurons may aid in the development of novel therapeutic strategies to combat the severe manifestations of MLIV.
Mucolipidosis type IV: Pathophysiology and outstanding questions
Lysosomal storage diseases (LSDs) are a collection of ~50 inherited metabolic diseases, which represent the leading cause of childhood-onset neurodegeneration. The majority of LSDs arise from deficiencies in lysosomal enzymes. However, mucolipidosis type IV, arises from loss-of-function mutations in a gene encoding a lysosomal channel. This channel, called MCOLN1, belongs to the TRP superfamily of cation channels. This channel, as well as its fly homolog, TRPML, is required during autophagy—a lysosomal-dependent process that involves encapsulation and breakdown of cellular macromolecules and organelles (). Despite the demonstration more than a decade ago that MLIV resulted from impairment of MCOLN1, several questions regarding the pathophysiology of MLIV remained unanswered. First, it was not known how the loss of MCOLN1 leads to the accumulation of acidic vesicles, such as late endosomes (LEs) and lysosomes. Second, it was not known whether accumulation of the acidic vesicles was toxic per se, or whether it is a relatively innocent consequence of decreased lysosomal flux. Third, the signaling pathways that modulate autophagy and lysosomal biogenesis, and that are disrupted in MLIV were unresolved. In our recent study we systematically addressed these three questions using a Drosophila model for MLIV.
Figure 1. Autophagy and a model of TRPML function in regulating endosomal trafficking and TORC1 activity.
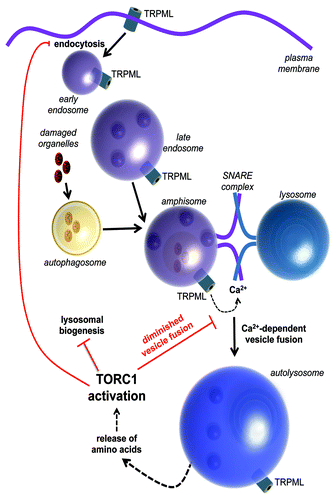
Fusion of amphisomes with lysosomes and activation of TRPML
We evaluated the mechanisms underlying the massive accumulation of acidic vesicles due to loss of the fly TRPML. We found that a subset of vesicles in the mutant cells are multivesicular bodies (MVBs), which accumulate during the cessation of an endocytic signaling pathway (). The cells also amass a large number of amphisomes, many of which are physically coupled to lysosomes. These “fusion-clamped vesicles” indicate that loss of TRPML diminishes vesicle fusion, despite normal docking and tethering. Since SNARE-dependent vesicle fusion is a Ca2+-activated process, it was tempting to speculate that TRPML may be the Ca2+-permeable channel responsible for providing the local Ca2+ elevation necessary to allow the “fusion-clamped” vesicles to complete fusion. Indeed, TRPML-deficient LEs have elevated lumenal Ca2+ supporting the proposal that TRPML is a LE Ca2+ release channel.
Considering that Ca2-dependent vesicle fusion occurs after vesicle tethering, it is intriguing to speculate that vesicle tethering itself leads to the generation of a signal that causes TRPML activation (). If so, possible mechanisms for activation of TRPML include changes in the lipid composition of tethered vesicles, and local membrane deformation. Another possibility is that tethering promotes the apposition of a LE/amphisomal membrane protein with TRPML, which serves as a ligand. We suggest that following vesicle attachment, the putative signal activates TRPML leading to the release of the lumenal Ca2+ required for vesicle fusion. Alternatively, TRPML may be constitutively active, and the appropriate distance between the two fusion vesicles may be sufficient to induce an elevation of Ca2+ necessary for triggering fusion.
Reciprocal control of TRPML and TORC1
TORC1 is a complex that includes the serine/threonine kinase MTOR, and which couples nutrient availability to protein synthesis. The complex is activated by amino acids via RAG small GTPases, which recruit TORC1 to the surface of LEs. TORC1 subsequently interacts with another small GTPase, RHEB, and becomes endowed with the capacity to phosphorylate its targets and promote protein synthesis. The amino acids that initiate this cascade are either taken up from an extracellular source, or are obtained by autophagic degradation of proteins (). Therefore, incomplete amphisome-lysosome fusion in TRPML-deficient cells leads to reduced autophagic flux which, in turn limits the availability of amino acids, and prevents maximal activation of TORC1. As a result, TORC1-mediated suppression of autophagy and lysosomal biogenesis is diminished, and autophagosomes and lysosomes pile up in the mutant cells. Feeding the mutant larvae a protein-rich diet to elevate cellular amino acid levels is sufficient to restore the activity of TORC1 to wild-type levels. Remarkably, restoring TORC1 to wild-type levels reverses the lysosomal storage phenotype despite the continued absence of TRPML.
What may be the other physiological consequence of TORC1 upregulation in TRPML deficient flies? We reported previously that the absence of TRPML leads to lethality of 90% of the animals during the pupal period when autophagy is critical to provide cellular nutrients, since pupae do not feed. In our recent analysis, we found that restoration of TORC1 activity in TRPML-deficient larvae, and the consequent reduction in autophagy, significantly suppresses the pupal lethality. Conversely, inhibiting TORC1 in trpml mutant animals and an increase in autophagy and lysosomal biogenesis exacerbate the lethality. Taken together, our results suggest that conditions that reduce lysosomal accumulation are beneficial to the trpml mutants, and hint at the possibility that the accumulating lysosomes may themselves be harmful to patient cells. Indeed, the probability that these vesicles rupture and induce significant toxicity is a function of the number and size of the vesicles.
It turns out that TORC1 exerts reciprocal control on TRPML. Activating TORC1 in wild-type larvae by a high-protein diet induces a dramatic alteration of TRPML localization from the LE/lysosomal membrane to the plasma membrane. These data indicate that TORC1 activation either prevents internalization of TRPML, or induces exocytosis of TRPML containing vesicles. In either scenario, the end result is that the levels of TRPML in LE/lysosomal membranes is reduced, which diminishes the fusion of amphisomes with lysosomes. Since we find that fusion of these vesicles precedes completion of autophagy and TORC1 activation, we hypothesize that by regulating the subcellular localization of TRPML, TORC1 exerts feedback control on autophagic flux (). These findings indicate that there must be a cost associated with the fusion of amphisomes and lysosomes, which is circumvented under conditions of ample nutrient availability. Another implication of our finding is that TORC1 is able to regulate autophagy at multiple steps—during initiation as described previously, but also during completion (). Since TORC1 has also been shown by others to be required for lysosomal reformation after completion of autophagy, TRPML may also be responsible for lysosomal regenesis following completion of autophagy. Taken together our findings underscore the rich complexity associated with the regulation of autophagy by TORC1.