Abstract
Interferon γ (IFNG) is a key host response regulator of intracellular pathogen replication, including that of Chlamydia spp The antichlamydial functions of IFNG manifest in a strictly host, cell-type and chlamydial strain dependent manner. It has been recently shown that the IFNG-inducible family of immunity-related GTPases (IRG) proteins plays a key role in the defense against nonhost adapted chlamydia strains in murine epithelial cells. In humans, IFN-inducible guanylate binding proteins (hGBPs) have been shown to potentiate the antichlamydial effect of IFNG; however, how hGBPs regulate this property of IFNG is unknown. In this study, we identified hGBP1/2 as important resistance factors against C. trachomatis infection in IFNG-stimulated human macrophages. Exogenous IFNG reduced chlamydial infectivity by 50 percent in wild-type cells, whereas shRNA hGBP1/2 knockdown macrophages fully supported chlamydial growth in the presence of exogenous IFNG. hGBP1/2 were recruited to bacterial inclusions in human macrophages upon stimulation with IFNG, which triggered rerouting of the typically nonfusogenic bacterial inclusions for lysosomal degradation. Inhibition of lysosomal activity and autophagy impaired the IFNG-mediated elimination of inclusions. Thus, hGBP1/2 are critical effectors of antichlamydial IFNG responses in human macrophages. Through their capacity to remodel classically nonfusogenic chlamydial inclusions and stimulate fusion with autophagosomes, hGBP1/2 disable a major chlamydial virulence mechanism and contribute to IFNG-mediated pathogen clearance.
Introduction
Chlamydia spp are clinically important intracellular bacterial pathogens that are responsible for highly prevalent diseases, such as blinding trachoma and sexually transmitted infections. Chlamydia bacteria complete their infection cycles within an intracellular niche referred to as the inclusion. IFNG mediated host cell immune defenses are important for infection clearance both in vitro and in vivo;Citation1-Citation3 however, many of the IFNG-effector mechanisms involved in the clearance of chlamydia infections are largely unelucidated.
The inhibitory effects of IFNG on chlamydial replication have been extensively studied; the outcome of IFNG responses for Chlamydia spp survival is highly variable in regards to chlamydial strain susceptibilities and antichlamydial effector mechanisms, which vary according to host species (e.g., murine and human) and cell type (e.g., epithelial cell and monocyte/macrophage).Citation4-Citation6 Subinhibitory concentrations of IFNG in vitro can induce chlamydia infections into a reversible state of persistence characterized by aberrant, but viable and metabolically active, inclusions, as assessed by analysis of rRNA transcripts.Citation7
In human epithelial cells, IFNG contributes to the clearance of nonhost adapted chlamydia strains predominantly through the induction of indoleamine 2,3-dioxygenase (IDO), which depletes intracellular tryptophan (Trp) pools.Citation8 Supplementing infected cell culture medium with excess Trp is sufficient to completely reverse IFNG growth inhibitory effects;Citation5,Citation6 C. trachomatis, however, evades IDO-specific IFNG growth inhibitory effects through the production of Trp synthase. Although Trp depletion is a major IFNG antichlamydial effector, other work suggests that additional IFN-effector mechanisms are likely to be important in epithelial cells,Citation9 for example the upregulation of IFN-inducible GTPases.Citation5,Citation10
Chlamydia spp can also infect macrophages.Citation11-Citation16 The mechanisms responsible for the control and regulation of chlamydial intracellular development in macrophages are diverse and include IFNG responses. Supplementing growth medium with excess Trp only partially reverses the growth inhibitory effects of IFNG in macrophages, which suggests that unlike in epithelial cells Trp depletion is not the major IFNG anti-chlamydial effector mechanism in macrophages.Citation6 Similarly, inhibiting production of nitric oxide, an IFNG-inducible effector, in macrophages only partially relieves chlamydia growth arrest.Citation6 Thus, the mechanisms underlying the inhibition of chlamydia growth in human monocytes/macrophages by IFNG have not been fully elucidated.
IFNG induces the expression of large GTPases (including Mx), the very large inducible GTPases, the p47 immunity-related GTPases (IRGs) and guanylate binding proteins (GBPs).Citation17 GBPs are the most abundant class of proteins induced by type II IFN.Citation18 Currently, up to 11 murine GBPs (mGBPs) and 5–7 human GBPs (hGBPs) are known.Citation19,Citation20 Murine GBP1 and 2 possess a terminal CaaX isoprenylation signal, which plays important roles in the membrane association and protein-protein interactions of a number of eukaryotic proteins.Citation21 Isoprenylation causes association of mGBP2 with vesicular cytoplasmic membranes,Citation21 with reported roles in cell growth and protective immunity against bacterial and viral pathogens. IFNG-inducible GTPases are important for resistance to bacterial infections, for example the IRG, Irga6, was shown to have antimicrobial properties against a nonhost-adapted strain, C. trachomatis, in murine embryonic fibroblasts; however, the murine-adapted strain, C. muridarum, evades this IFNG-induced host response.Citation10 A recent report on the essential role of mGBPs for defense against intracellular bacteria shows that mGBP1, 6, 7 and 10 provide broad host protection and immunity against listeria and mycobacterial infections by promoting antimicrobial peptide expression, autophagy and oxidative killing of bacteria.Citation19 In addition, mGBP5 localizes to structures formed by invading Salmonella enterica, which is thought to enhance S. enterica-induced pyroptosis.Citation22 Thus, the role of GBPs in the IFNG response to intracellular bacteria in murine cells has been quite well characterized; by contrast, the roles of hGBPs in immunity have remained elusive for nearly two decades. Recently, some efforts have been made to address the potential role(s) of hGBPs in epithelial cell immunity to chlamydia infections. Carabeo and coworkers suggest a crucial inhibitory role for hGBP1/2 during C. trachomatis infection from their observations that ectopically overexpressed hGBP1/2 delays the growth of C. trachomatis in HeLa cells.Citation9
Here, we demonstrated that the induction of hGBP1/2 is important for the immunoprotective role of exogenous IFNG in C. trachomatis infected macrophages. IFNG-inducible hGBP1/2 mediated fusion of bacterial inclusions with autolysosomes to eliminate C. trachomatis. Thus, we provided new insights into macrophage IFNG effector mechanisms, which, through regulation of autophagy, can contribute to innate immunity during C. trachomatis infection.
Results
IFNG attenuated C. trachomatis infection in human macrophages
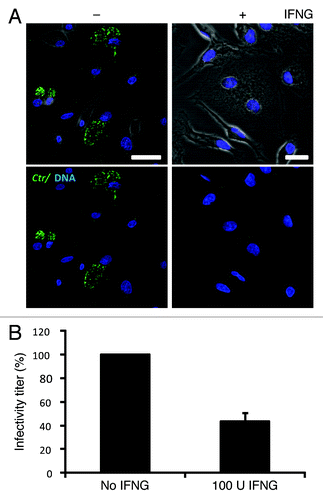
IFNG is a critical mediator of the restriction of chlamydial infection. To assess the effect of IFNG on chlamydial growth in human THP1-derived macrophages, cells were pretreated for 24 h with 100U IFNG. Twenty-four hours post-treatment cells were infected with C. trachomatis at a multiplicity of infection (MOI) of 5 for 2 h. Following 48 h incubation in the presence of IFNG, cells were immunostained and analyzed by microscopy. IFNG treatment resulted in substantially smaller and reduced numbers of C. trachomatis inclusions (). Furthermore, IFNG treatment reduced the production of infectious C. trachomatis elementary bodies (EBs) by more than 50% (). Thus, our observations show that exogenous IFNG treatment attenuates C. trachomatis growth in human macrophages.
Figure 1. IFNG inhibited C. trachomatis growth in human macrophages. THP1 macrophages were treated with 100 U/ml IFNG prior to infection, then infected for 48 h with C. trachomatis (MOI 5) and simultaneously treated with 100 U/ml IFNG or left untreated (control). (A) Immunofluorescence micrographs of macrophages infected with C. trachomatis (green) and DNA (blue). Cytokine treatment resulted in a low number of detectable inclusions in C. trachomatis infected cells. Images are representative of at least three independent experiments (B) Influence of IFNG on development of infectious progeny. The yield of C. trachomatis infectious progeny decreased by 50% upon IFNG stimulation. Infectivity percentages were calculated following infection of HeLa cells: IFU/ml estimated for each treated monolayer / IFU/ml of control cells x 100. Infectivity expressed as a percentage of control cells ± standard deviation (SD) from three independent experiments (n = 3). Ctr, C. trachomatis. Scale bars: 30 μm.
hGBP1/2 shRNA knockdown restored C. trachomatis growth in IFNG-treated macrophages
Because IFNG is considered a potent inducer of hGBP expression,Citation9,Citation19 which can delay the growth of C. trachomatis in human HeLa cells,Citation9 we determined the expression levels of hGBP1/2 in THP1-derived macrophages in response to exogenous IFNG treatment. The addition of exogenous IFNG highly upregulated the expression levels of hGBP1/2 (). To investigate the potential role of hGBP1/2 in IFNG-mediated inhibition of chlamydial growth, IFNG-pretreated (and untreated control) macrophages were infected with C. trachomatis (MOI of 50). Two hours post-infection (h p.i.) cells were processed for indirect immunofluorescence to analyze colocalization of chlamydial inclusions with hGBPs (). Upon IFNG treatment, greater than 60% and 40% of chlamydial inclusions colocalized with hGBP2 and -1, respectively (). To further confirm the involvement of hGBPs in the IFNG-mediated inhibition of chlamydia growth in human macrophages, we conducted short hairpin RNA (shRNA) knockdown of hGBP1/2 in THP1 cells. Knockdown levels of hGBP1/2 were determined and compared with luciferase-specific control knockdown using western blotting and RT-PCR (Fig. S1). Cell viability was unaffected by knockdown with shRNA (data not shown). We infected hGBP1/2-deficient cells with C. trachomatis (MOI of 5) for 48 h in the presence or absence of exogenous IFNG. In contrast with THP1 cells expressing hGBP1/2, IFNG treatment did not inhibit chlamydial growth in hGBP1/2 knockdown cells, as determined by inclusion number (; Fig. S2A). Importantly, the recovery of infectious progeny from hGBP1/2 knockdown cells in the presence of exogenous IFNG was comparable to that from IFNG untreated cells (). Double knockdown of hGBP1/2 provided support to the notion that hGBP1/2 regulated IFNG-induced chlamydial restriction in a redundant manner ().
Figure 2. Early interactions of hGBP1 and GBP2 with chlamydial inclusions mediated IFNG-induced C. trachomatis growth inhibition. (A) Anti-hGBP1 and -GBP2 immunoblot analysis of total lysates from uninfected macrophages, and uninfected macrophages exposed to 100 U/ml IFNG for 24 h. Other monolayers were pretreated with IFNG for 24 h prior to infection and then infected in the presence of IFNG. Host ACTB was used as loading control. (A) hGBP1/2 expression is induced by IFNG, while infection alone had minimal stimulatory effect on hGBP2 expression. Only very low amounts of cellular hGBP2 can be detected in control cells, compared with treated cells. Blot is representative of two independent experiments (B) Double immunofluorescence labeling of hGBP1, hGBP2 and C. trachomatis in macrophages stimulated for 24 h with 100 U/ml IFNG and then infected for 2 h with C. trachomatis (MOI 50). IFNG untreated cells were similarly infected. Upon IFNG induction hGBP1/2 localized to inclusions. For quantification, approximately 100 cells were examined for hGBP1/2-C. trachomatis colocalization in cytokine-treated or untreated cells. Colocalization calculated as a mean percentage: for each treatment, number of hGBP1/2 positive inclusions/total number of inclusions × 100 from two independent experiments (C and D) Deletion of either hGBP1, hGBP2 or both promotes intracellular growth of the bacterial inclusion. Unstimulated and IFNG-stimulated THP1-derived macrophages were infected with C. trachomatis for 48 h (MOI 5). IFNG stimulation did not inhibit chlamydial growth in hGBP1, hGBP2 or double knockdown deficient cells compared with THP1- and control (empty)-derived macrophages. (C) Analysis of inclusion numbers in cells infected (MOI 5) for 48 h was performed by counting inclusions in 100 cells. (D) Infectivity of bacteria in hGBP1 or hGBP2 deficient cells, as well as double knockdown cells is 2-fold higher than in control (empty)- or THP1-derived macrophages. Results depicted as mean percentage normalized to control. Results shown in (C and D) are from three independent experiments. Error bars ± SD. Statistical significance was analyzed by Student’s t-test; *p < 0.01. Scale bars: 10 μm.
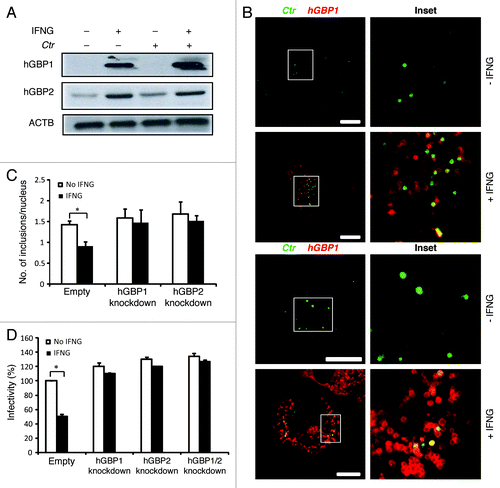
Taken together, growth of C. trachomatis in macrophages was inhibited by exogenous IFNG treatment, whereas chlamydia growth was not affected by IFNG in hGBP1/2 knockdown cells. This suggests that the antichlamydial function of exogenous IFNG is partially dependent on hGBP1/2.
IFNG-induced lysosomal fusion with C. trachomatis inclusions was abrogated in hGBP1/-2 deficient cells
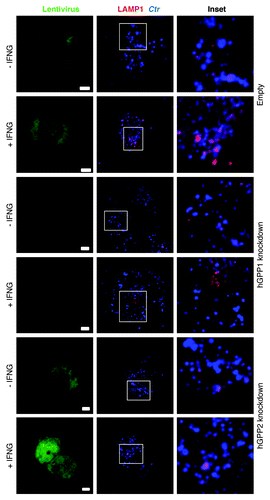
We next investigated whether hGBP1/2-positive C. trachomatis inclusions were directed to lysosomes for degradation by exogenous IFNG treatment. To these ends, we used immunofluorescence microscopy to determine whether C. trachomatis inclusions fuse with lysosomes upon IFNG treatment. First, colocalization of inclusions with the lysosomal marker, lysosomal-associated membrane protein 1 (LAMP1) at 3 h p.i. was monitored (). In control, empty untreated macrophages, 17% of inclusions colocalized with LAMP1. The addition of exogenous IFNG increased colocalization of LAMP1 and inclusions 3-fold (). Importantly, colocalization of LAMP1 and inclusions in hGBP1/2 knockdown THP1 cells was reduced to basal levels and this was refractory to the effects of exogenous IFNG ( and ), which was consistent with our observation that IFNG had no inhibitory effect on chlamydia growth in knockdown cells. These findings clearly indicated that the fusion of bacterial inclusions with lysosomes was induced by IFNG in human macrophages and this process is hGBP1/2 dependent. To confirm that IFNG-induced inclusion-lysosome fusion eliminates C. trachomatis, we studied the effects of bafilomycin A1 (BafA1), a specific vacuolar H+-ATPase inhibitor, which inhibits lysosomal activity. BafA1 treatment rescued chlamydial growth and the production of infectious EBs in IFNG treated macrophages (). These data clearly indicated that the fusion of C. trachomatis inclusions with lysosomes can be elicited by IFNG, which serves to eliminate chlamydia from host macrophages. To further characterize the nature of chlamydial early inclusions after exogenous IFNG stimulation, we examined whether early chlamydial inclusions are acidic using LysoTracker, an acidotropic fluorescent dye that accumulates in acidic cellular compartments (). In wild-type macrophages treated with IFNG, 45% of bacterial phagosomes colocalized with LysoTracker, compared with 20% in control untreated cells. However, in hGBP1/2 knockdown macrophages, colocalization was minimal and comparable to that in untreated wild-type macrophages. This suggested that IFNG treatment in human macrophages rendered chlamydial early inclusions vulnerable to acidification, by a process dependent on hGBP1/2.
Figure 3. IFNG-induced lysosomal interaction with early chlamydial inclusions was abrogated in hGBP1 and hGBP2 knockdown macrophages. (A) Empty (control), hGBP1 and hGBP2-stable knockdown THP1-derived macrophage monolayers were prestimulated for 24 h with 100 U/ml IFNG and then infected with C. trachomatis as described in . IFNG untreated control cells were similarly infected. (A) Double immunofluorescence labeling 3 h p.i. revealed that IFNG stimulated the association of the lysosomal marker LAMP1 (red) with C. trachomatis (blue) inclusions in control cells. While IFNG stimulation demonstrated no recruitment of lysosomes to inclusions in cells deficient of either hGBP1 or hGBP2. Images are representative of two independent experiments. (B) Percentage colocalization Ctr inclusions with LAMP1. Quantification of LAMP1-positive chlamydial inclusions revealed nonsignificant colocalization in cells lacking hGBP1 or hGBP2. Approximately 100 cells per sample were examined. Colocalization expressed as a mean percentage: for each treatment, number of LAMP1 inclusions / total number of inclusions × 100. (C) Inhibition of lysosomal acidification by 100 nM BafA1 abrogated the IFNG-mediated inhibition of C. trachomatis inclusion growth. Infectivity assays were performed as in . BafA1 led to an increase in the yield of infectious progeny in IFNG-treated cells Results shown in (B and C) are from three independent experiments. Error bars ± SD. Statistical significance was analyzed by Student’s t-test; **p < 0.01, *p < 0.05. Scale bars: 5 μm.
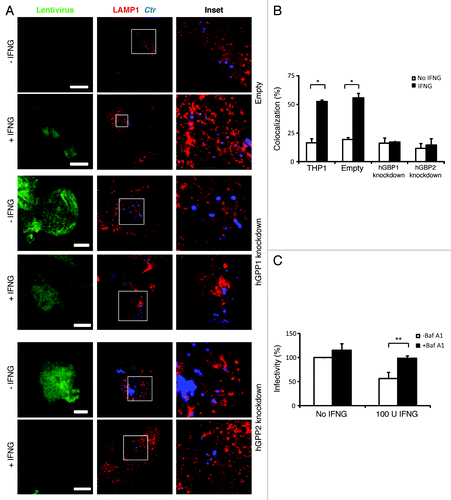
Figure 4. IFNG stimulation increased acidification of chlamydial early inclusions, but not in hGBP1 or -2 knockdown macrophages. Empty (control), hGBP1 and hGBP2-stable knockdown THP1-derived macrophage monolayers were prestimulated for 24 h with 100 U/ml IFNG and then infected with C. trachomatis MOI 100 for 2 h. Confocal microscopy images of acidotropic dye LysoTracker (Red) and C. trachomatis (Blue) revealed that IFNG stimulated the acidification of chlamydial early inclusions in WT macrophages. By contrast, the acidification process is impaired in cells deficient in either hGBP1 or 2, in response to IFNG stimulation. Scale bars: 10 μm.
IFNG-induced autophagy was required for the elimination of C. trachomatis.
IFNG can induce autophagy to eliminate intracellular pathogens.Citation10 We hypothesized that IFNG-induced autophagy reroutes C. trachomatis inclusions to the autophagic compartment for destruction in macrophages. To investigate this further, THP1 cells were infected with C. trachomatis in the presence or absence of exogenous IFNG. Cells were then fixed and stained for endogenous microtubule-associated protein 1 light chain 3 (LC3) using an anti-LC3 antibody. In contrast to IFNG-untreated cells, LC3 exhibited vesicular staining colocalized to C. trachomatis inclusions (53%) in IFNG-treated cells ( and ). A GFP fusion of LC3 and an autophagosomal marker also colocalized with C. trachomatis inclusions in IFNG-treated cells (40%) (Fig. S3). Taken together with our LAMP1 data, these findings clearly suggested that the nature of C. trachomatis inclusions upon IFNG stimulation was autolysosomal. To further support our hypothesis, immunoblotting was used to monitor the formation of early autophagosomal precursors and newly formed autophagosomes by following changes in endogenous LC3-II formation.Citation10,Citation23 LC3-II levels increased in response to IFNG stimulation (). Notably, LC3-II levels were higher in IFNG-treated cells compared with untreated cells (, lane 1 and 2). Further, C. trachomatis infection also increased levels of LC3-II even in the absence of exogenous IFNG (, lane 3). In addition to LC3, SQSTM1/p62 can also be used as a readout of autophagic activity.Citation23,Citation24 SQSTM1 is itself degraded by autophagy and, thus, increased levels of SQSTM1 correlate with inhibition of autophagic activity. Exogenous IFNG reduced levels of SQSTM1 compared with untreated, uninfected cells, demonstrating that IFNG stimulated autophagic function, or flux (). Levels of SQSTM1 in infected cells in the absence of exogenous IFNG were higher compared with IFNG-treated cells. In context with the increased induction of autophagy observed in both IFNG-treated and infected macrophages, IFNG relieved a block to autophagic flux that was evident during infection in the absence of exogenous IFNG (). Interestingly, wild-type and hGBP1/2 knockdown cells treated with rapamycin, a conventional inducer of autophagy, did not restrict chlamydial growth as determined by infectivity titration (Fig. S4). Moreover, the IFNG-induced chlamydial restrictive properties were not potentiated in the presence of rapamycin.
Figure 5. Autophagy-induced chlamydial growth arrest in response to IFNG stimulation was abrogated in cells with impaired autophagy. IFNG induces localization of autophagosomes to inclusions. Macrophages were exposed to 100 U/ml IFNG for 24 h. Cells were then infected with C. trachomatis (MOI 100) for 3 h. IFNG-untreated control cells were similarly infected. LC3 (red) localized to bacterial inclusions (green) in response to IFNG stimulation (A). (B) Quantification of LC3-positive C. trachomatis inclusions in the presence or absence of IFNG. Approximately 30 host cells were examined for LC3- C. trachomatis sequestration per sample. Quantification results shown as mean percentage normalized to control from two independent experiments. Error bars ± SD (C) Anti-LC3 immunoblot analysis of total lysates from uninfected and chlamydia infected macrophages; in the presence or absence of IFNG pre-treatment for 24 h. Host ACTB was used to control equal loading of proteins. Autophagy is induced by IFNG treatment and to a lesser extent after chlamydial infection, as indicated by the amount of LC3-II. IFNG treatment activated autophagic flux as indicated by reduced levels of SQSTM1. Blot is representative of two independent experiments. (D) Normal development of chlamydia in cells with impaired autophagy. ATG5 stable knockdown cells or control (empty) cells were IFNG treated and infected, as in . Infectivity of bacteria from macrophages lacking ATG5 was 2-fold higher than that in control cells. Further, IFNG treatment had minimal activity against bacteria in cells with impaired autophagy. Infectivity expressed as a ratio between infected IFNG-treated and infected-untreated cells from three independent experiments. (E) Influence of ectopic overexpression of hGBP1 on development of infectious progeny in cells lacking ATG5. Infectivity of bacteria in ATG5 deficient cells or ATG5 KD-overexpressing hGBP1 cells is comparable. Results depicted as mean percentage normalized to control. Error bars ± SD. Scale bars: 10 μm.
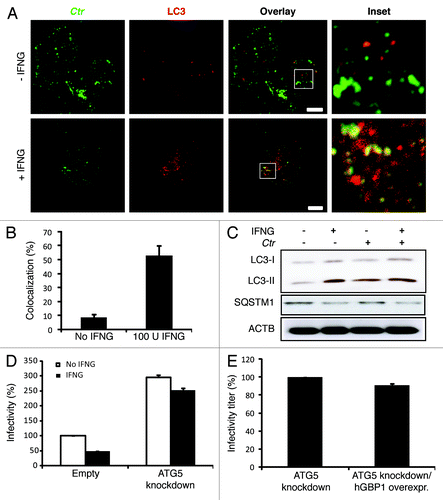
In order to provide further evidence for autophagy-dependent elimination of C. trachomatis in human macrophages, we used shRNA to knock down the key autophagy protein, ATG5. The knockdown level of ATG5 was determined and compared with luciferase-specific control knockdown by western blot (Fig. S5). ATG5 knockdown THP1 cells were infected with C. trachomatis in the presence or absence of exogenous IFNG. Two days p.i., percentage infectivity was assessed using infectivity titration assays. Interestingly, ATG5 knockdown increased infectious progeny production 2.5-fold () compared with wild-type cells and the addition of exogenous IFNG did not inhibit the enhanced infectivity of C. trachomatis in cells depleted of ATG5 (). Further, the ectopic overexpression of hGBP1 was unable to confer a restrictive phenotype on ATG5 knockdown cells, indicating that hGBPs mediate their antichlamydial activity via autophagy (; Fig. S6). Thus, these data, in tandem with our LAMP1 colocalization studies, clearly suggested that IFNG mediates the fusion of C. trachomatis inclusions with lysosomes and the activation of autophagic flux in order to eliminate chlamydial infection from host macrophages.
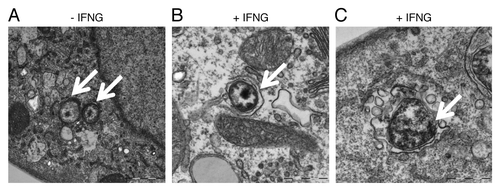
To further characterize the nature of chlamydial early inclusions upon IFNG treatment, we used transmission electron microscopy (TEM) to study the ultrastructural features of chlamydial inclusions. In IFNG-untreated cells () small EBs were present after 6 h of infection (arrows). IFNG treatment induced the formation of autophagosomes around EBs as characterized by the presence of a double membrane around EBs as shown in (). Further, IFNG treatment induced vesicles and vacuoles which accumulated around the morphologically abnormal chlamydial EBs; creating a spacious bacterial vacuole (). By contrast, C. trachomatis early inclusion morphology was normal in control untreated cells, with no double membranes, nonspacious vacuoles, or vesicles surrounding the early inclusion ().
Figure 6. IFNG treatment induced rerouting of chlamydial early inclusions to autophagosomes. Transmission electron microscopy displays ultrastructural features of early C. trachomatis inclusions in infected THP1-derived macrophages. Host cells were exposed to medium containing 100 U/ml IFNG for 24 h or left untreated as a control. (A) Micrograph of infected untreated host cells shows normal chlamydial EBs (arrows) within the early inclusion. (B and C) Micrograph of infected cells exposed to IFNG demonstrates the presence of one EB (early inclusion) surrounded by a double membrane (B) or vesicles surrounding a morphologically abnormal chlamydial early inclusion (arrow) (C).
hGBP1/2 were important for autophagy-mediated C. trachomatis elimination
Next, we determined whether IFNG-inducible hGBP1/2 were required for autophagy induction by following changes in LC3-II formation in cells depleted of hGBP1/2 using immunoblotting. LC3-II levels increased in response to IFNG stimulation in control cells (). Infection with C. trachomatis for 4 to 12 h slightly induced LC3-II expression (); however, exogenous IFNG treatment of infected cells further upregulated LC3-II expression. In general, levels of LC3-II were substantially higher in infected, IFNG-treated cells compared with untreated cells (; Fig S7). Interestingly, in IFNG-treated cells, infection with C. trachomatis stimulated autophagy as the levels of SQSTM1 in these cells were reduced compared with control cells (). In hGBP2-deficient cells, SQSTM1 levels in uninfected and untreated cells were higher compared with control cells (). Moreover, IFNG treatment had a minimal stimulatory effect on SQSTM1 degradation, and therefore on autophagy, in cells depleted of hGBP2 after chlamydia infection (). IFNG treatment also had a minimal stimulatory effect on LC3-II expression, and therefore on autophagy, in comparison to untreated hGBP2-depleted cells (). Similarly, IFNG treatment and C. trachomatis infection had a minimal stimulatory effect on LC3-II expression levels in hGBP1-impaired cells, although to a lesser extent than hGBP2 (). By contrast, rapamycin-induced autophagy had a similar stimulatory effect on SQSTM1 degradation in wild-type and hGBP1/2 knockdown cells after chlamydial infection (Fig. S8). This autophagic activity, induced by rapamycin, did not inhibit chlamydial growth in macrophages (Fig. S4), in contrast to IFNG-induced autophagy. Taken together, these data suggested a role for hGBP2, and possibly hGBP1, in the regulation of the IFNG-induced autophagic pathway.
Figure 7. hGBP1 and hGBP2 played a role in the regulation of the host autophagic machinery. (A–C) Anti-LC3 and SQSTM1 immunoblot analysis of total lysates from uninfected control (empty), hGBP1- and hGBP2-stable knockdown macrophages or from cultures infected for the indicated time points in the presence or absence of 100 U/ml IFNG for 24 h. Other monolayers were pretreated with IFNG for 24 h prior to infection and then infected in the presence of IFNG. Host ACTB was used as loading control. IFNG induced autophagy in control THP1-derived macrophages; similarly treated cells infected with C. trachomatis induced autophagy as indicated by the higher amount of LC3II and reduced amount of SQSTM1. (B and C) hGBP1 or hGBP2 knockdown impaired autophagy induction. Only very low amounts of cellular LC3II, but increasing amounts of SQSTM1 can be detected in hGBP2-knockdown cells, compared with those in control THP1-derived macrophages, while hGBP1 knockdown had a minimal stimulatory effect on autophagy. (D) Anti-ATG5 and ATG12 immunoblots of total lysates from control (empty), hGBP1- and hGBP2-stable knockdown macrophages. Monolayers were pretreated with IFNG for 24 h prior to infection and then infected with C. trachomatis (MOI 10) in the presence of IFNG or left without treatment. Host ACTB was used as loading control. IFNG induced ATG12–ATG5 conjugate formation in all cells. hGBP2, and to a lesser extent hGBP1, knockdown impaired autophagy induction. Monomeric ATG5 and ATG12 were detected in cells lacking hGBP2, and to lesser extents in cells lacking hGBP1, but were absent in control THP1-derived macrophages. Blot shown in (A) is representative of three independent experiments and blots in (B–D) are representatives from two independent experiments.
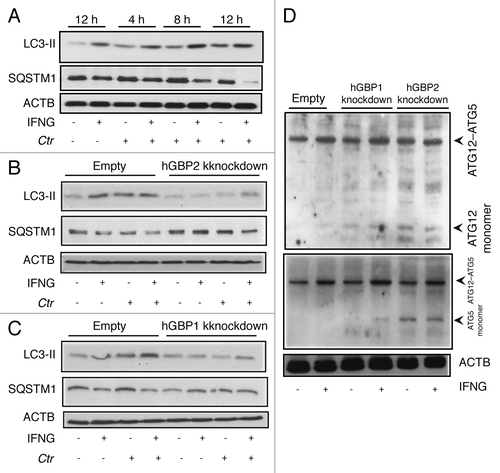
To further confirm the importance of hGBP1/2 in IFNG-induced autophagy, we monitored the expression of ATG5 and ATG12, key regulators of autophagy, by immunoblot. ATG12–ATG5 conjugation has been used in some studies to measure autophagy.Citation25,Citation26 Further, the amount of free ATG5 and ATG12 might be a useful method for following autophagy induction. Here, empty (control), hGBP1/2-depleted cells were pretreated with IFNG for 24 h. Cells were infected with C. trachomatis (MOI of 10) in the presence or absence of IFNG. ATG12–ATG5 conjugate formation was slightly increased in IFNG treated cells (). No monomeric ATG5 or ATG12 was detected in empty control cells (, lanes 1 and 2), suggesting that autophagy is not impaired in these cells. By contrast, monomeric ATG12 and ATG5 were detected in cells depleted of hGBP2 and to lesser extent hGBP1 (, lanes 4 and 6). The data indicated that given similar knockdown levels, hGBP2 exerted a greater influence over autophagy regulation. However, our findings implicated both hGBP1/2 in the regulation of the autophagic process during C. trachomatis infection of macrophages.
Discussion
IFNG is a key immune effector that contributes to the clearance of chlamydia infections in vitro and in vivo.Citation3,Citation27 Here we elucidated the mechanisms underlying IFNG-induced immunity to C. trachomatis in human macrophages. We showed that the human strain C. trachomatis LGV serovar L2 is highly susceptible to the growth inhibitory effects of exogenous IFNG in human macrophages. Inhibition of chlamydia growth was dependent on the IFNG-inducible proteins hGBP1/2. Exogenous IFNG triggered classically nonfusogenic C. trachomatis inclusions to fuse with autolysosomes, leading ultimately to pathogen elimination. Impairment of autophagy and lysosomal function restored chlamydia growth during IFNG treatment. Strikingly, hGBP1/2-deficient macrophages, in which chlamydia growth was enhanced compared with control cells, showed no IFNG-mediated chlamydia elimination. Thus, we have now shown that through the upregulation of its effector proteins, hGBP1/2, IFNG disrupted an important C. trachomatis virulence mechanism in human macrophages, i.e., the maintenance of a nonfusogenic intracellular niche, and targeted inclusions for lysosomal degradation.
There is considerable diversity in the replicative microenvironments established by intracellular pathogens to optimize their survival, i.e., to ensure continuous nutrient and energy supplies and to avoid detection by host immune systems. The chlamydial inclusion is specialized and nonfusogenic in nature;Citation28,Citation29 nonfusogenicity contributes to the survival of Chlamydia spp in host cells as chlamydial inclusions are maintained as distinct entities that do not come into contact with lysosomal degradative enzymes.Citation16,Citation30,Citation31 While it was shown in one study that lysosomal activity promotes chlamydia growth through the provision of essential nutrients,Citation32 we found here that in the context of lysosomal-inclusion fusion, lysosomal activity inhibited chlamydia growth. A block to lysosomal degradation of Chlamydia muridarum inclusions during infection of murine embryonic fibroblasts has previously been suggested to determine Chlamydia spp host tropism and similar to our observations, growth of a nonmurine strain, C. trachomatis, in murine cells was shown elsewhere to be promoted by BafA1.Citation33 However, Yasir et al., report that the inhibition of nonhost adapted strain growth by vATPase-bearing vacuoles occurs independently of colocalization with inclusions, whereas we showed that inclusions colocalized with LAMP1 in IFNG-treated macrophages. The intracellular pathogen, Toxoplasma gondii, is also nonfusogenic with lysosomesCitation34 and IFNG has been found to trigger autophagy-dependent elimination of T. gondii in macrophages.Citation35,Citation36 It has also been suggested that mycobacterial phagosomes are arrested in autolysosomes in activated macrophages.Citation37-Citation39 It is worth noting that despite the propensities of some pathogens to avoid contact with lysosomes, fusion with the lysosomal compartment is not universally detrimental to intracellular pathogen survival. Coxiella burnetti, for example, has evolved mechanisms to survive inside phagolysosomes.Citation40 Coxiella bacteria translocate several proteins from the acidic compartment and delayed phagosome maturation facilitates bacterial survival, a process that is, interestingly, autophagy mediated.Citation41
We report here that autophagy was required for mediating the anti-C. trachomatis effects of exogenous IFNG in human macrophages. Autophagy, originally identified as an intracellular homeostatic mechanism, reroutes cytoplasmic material and damaged organelles to autophagosomes, and subsequently lysosomes, culminating in the formation of autolysosomes for degradation of their cargo. Alongside its important roles in development and programmed cell death, autophagy is being increasingly recognized for its importance in immunity.Citation42 When autophagy was impaired, macrophages supported C. trachomatis infection, even in the presence of exogenous IFNG. IFN-inducible GTPases are, in part, responsible for permissiveness to chlamydia infections in murine epithelial cells.Citation5 Other studies, besides our latest report, have implicated the IFN-inducible GBP protein family, in growth regulation of various intracellular pathogens.Citation9,Citation17,Citation19,Citation43,Citation44 A possible role for mGBP1 and 7 in the induction of LC3-positive structures has also been suggested.Citation19 Here we report that hGBP1/2 remodeled the autophagosomal membrane in human macrophages during C. trachomatis infection, which inhibited chlamydia growth by ~50%. hGBP1/2-depleted cells exhibited reduced levels of LC3II and impairment in autophagy flux; furthermore, monomeric ATG5 and ATG12 species were detected in hGBP1/2 knockdown cells. This suggested a role for hGBPs in regulating autophagy in human macrophages as previously demonstrated,Citation45 although the mechanism underlying this process needs further investigation.
It is likely that effectors such as hGBP1/2, which are induced by IFNG, can stimulate remodeling of the bacterial phagosomal membrane. Similarly, Irga6 enhances resistance to C. trachomatis in an autophagy-dependent manner in murine fibroblasts and IFNG stimulates autophagy and the recruitment of LC3 and LAMP1 to the M. tuberculosis compartment, which inhibits M. tuberculosis growth.Citation10,Citation38 In contrast to IFNG-induced autophagy, rapamycin-induced autophagy did not confer resistance to C. trachomatis in wild-type human macrophages, supporting previous observations in epithelial cells.Citation46 Rapamycin-induced autophagy is responsible for increasing acidification of mycobacterial phagosomes in macrophages, removing the block to phagosome maturation (a prosurvival strategy of this intracellular pathogen).Citation38 Thus, pharmacologically induced autophagy has apparently different antimicrobial capacities for these two intracellular pathogens with differing intracellular niches. In macrophages lacking hGBP1/2, rapamycin-induced autophagy was not impaired, unlike IFNG-induced autophagy. This difference could possibly arise from the different signaling regulation of mammalian autophagy activated by rapamycin compared with IFNG.Citation47,Citation48
We found that both exogenous IFNG and chlamydia infection upregulated autophagy. Crucially, however, IFNG relieved the cellular block to autophagic flux present during infection alone, as determined by decreased levels of SQSTM1; an apparent block to autophagic flux during chlamydia infection has been reported elsewhere.Citation33 We also observed that IFNG stimulated the colocalization of the autophagy protein, LC3, and the lysosomal marker, LAMP1, with C. trachomatis inclusions in human macrophages. Furthermore, we showed how IFNG perturbed ultrastructural features of early chlamydial EBs, e.g., EBs exhibited abnormal morphology, characterized by double-membrane structures, and also recruited vacuoles and vesicles in response to IFNG treatment. This further strengthens the existing observations that Chlamydia bacteria are rerouted to autophagosomes for degradation upon stimulation with exogenous IFNG.
We observed that the inhibition of C. trachomatis growth by IFNG in human macrophages was completely reversed upon GBP1 and GBP2 knockdown. Others have reported that in epithelial cells, hGBP1/2 are not IFNG effectors per se during infection, but rather potentiate the antichlamydial activity of IFNG.Citation9 Tietzel et al., speculate that colocalization of chlamydial inclusions and ectopically-expressed hGBP1/2 can induce interactions between bacterial inclusions and degradative lysosomal compartments of the host cells, although they did not confirm this empirically. Our observations here suggested a role for hGBPs in targeting hGBP1/2-positive cargo to autolysosomes, similar to the role postulated for mGBPs.Citation49 We have previously shown that the IFNG-antichlamydial response to C. trachomatis infection in murine fibroblasts is determined in large part by the murine specific IFNG-inducible protein, Irga6,Citation10 which colocalizes to the inclusion and is essential for IFNG growth arrest through the generation of autolysosomes and lysosomal elimination of bacteria. Irga6 also inhibits the growth of C. trachomatis in murine oviduct epithelial cells;Citation5 however, the authors of that report conclude that an association between Irga6 expression and inhibition of sphingomyelin transport to the chlamydial inclusions is responsible for growth inhibition, without indication of a role for Irga6 in autophagosomal fusion with lysosomes. Similarly, others have suggested that chlamydial growth inhibition by hGBP1/2 could be due to disruption of signaling, or depletion of intracellular guanine nucleotide pools in epithelial cells.Citation9
In summary, hGBP1/2 can contribute to IFNG-mediated protective immunity against chlamydial infection through their potential to mediate fusion of chlamydial inclusions with autolysosomes as a mechanism to eliminate C. trachomatis in human macrophages. Our findings provided new insights into the role of hGBPs for innate immunity to a major clinical pathogen and suggested that manipulation of the host interferon response could be a possible future approach for antichlamydial interventions.
Materials and Methods
Chemicals and antibodies
RPMI-1640 medium (52400) and Dulbecco’s minimal essential medium (DMEM) were purchased from Gibco-Invitrogen (10938) supplemented with 10% fetal bovine serum (FBS; Biochrom AG, S0115). Cycloheximide was obtained from Sigma-Aldrich (01810). IFNG was purchased from Sigma-Aldrich (I3265). BafA1 was purchased from Calbiochem (196000). Rapamycin was obtained from Sirolimus LC Laboratories (SILA 9268A). LysoTracker Red DND-99 was purchased from Molecular Probes (L-7528). Phorbol myristate acetate (PMA) was purchased from Sigma-Aldrich (P1585). Goat antibodies raised against human GBP1 (sc-10579), GBP2 (sc-10581) were purchased from Santa Cruz Biotechnology, Inc. Other serological reagents used were: mouse anti-human CD107a/lysosomal associated membrane protein 1 (LAMP1) (clone H4A3; Southern Biotech, 9835-01); monoclonal antibody to microtubule-associated protein 1 light chain 3 (LC3) (clone 5F10; nanoTools, 0231-100/LC3-5F10); rabbit anti-LC3A/B (Cell Signaling, 4108); mouse monoclonal anti-SQSTM1/p62 (Abcam, ab56416); rabbit anti-ATG5L (Abgent, AP1812b); rabbit polyclonal anti-ATG12 (Zymed Laboratories, 36-6400); mouse monoclonal anti-human ACTB (clone AC-15, Sigma-Aldrich, A5441); rabbit polyclonal anti-Chlamydia genus-specific antibody (Milan Analytica AG, 3-090); and mouse monoclonal anti-C. trachomatis hsp60 (Alexis, ALX-804-072-R100) and LPS (clone CF6J12, Abcam, ab62708-500). The nucleic acid dye, DRAQ5, was obtained from Biostatus Limited (DR50050). Secondary labeled antibodies for immunofluorescence and western blot analyses were purchased from Jackson ImmunoResearch Laboratories (705-165-147, 715-095-151, 715-165-151, 711-165-152, 711-095-152, 703-165-155) and Amersham (NA935).
Cell culture and chlamydial strain
For the generation of macrophages, THP1 cells were seeded in 6-well plates using RPMI supplemented with 10% FBS at a density of 1 × 106 cells/well in the presence of 100 nM PMA. Twenty-four hours post-treatment cells were incubated at 37°C and 5% CO2 in a humidified tissue culture chamber for a further 48 h without PMA, to allow the cells to rest. C. trachomatis serovar L2 was routinely, cultured, prepared, and propagated in HeLa cells (American Type Culture Collection CCL-2.1) as previously described.Citation10
Host infection and infectivity titration assay
Host cells were seeded in 6-well plates, treated as above then infected with C. trachomatis diluted in infectious medium at an MOI 5 and incubated at 35°C and 5% CO2. Two hours post infection (p.i.) cells were washed and loaded with fresh medium containing 100 U/ml IFNG. Control infected cells were not treated with the cytokine. Cell cultures were then incubated as above for 48 h. The formation of infectious C. trachomatis progeny in a secondary infection was assessed by infectivity titration assay as described elsewhere.Citation10 Briefly, infected cells were mechanically destructed using glass beads. Resulting, lysates were serially diluted in infectious medium and then used to inoculate HeLa cells for 2 h. Cells were then washed and further incubated for 24 h in RPMI medium with 5% FBS and 1 µg/ml cycloheximide. One day later, inclusions were manually counted (from each dilution) at 400× magnification in ten randomly selected microscopic fields using phase contrast microscopy. The area of one 400× microscopic field was then estimated using a stage micrometer slide, which could then be used to calculate the total microscopic fields per well by dividing the area of one well by the area of one microscopic field. The infectivity titer is expressed as a number of inclusions (IFU/ml) IFU/ml = no. inclusions in 10 fields/10 × no. fields in a well × dilution factor
Generation of stable knockdown cells
shRNA knockdown cells were generated as follows: shRNA oligonucleotides (Metabion) were annealed and cloned into the lentiviral pLVTHM vector carrying a GFP cassette for monitoring virus production and infection.Citation50 The following sequences were targeted to silence GBP1, GBP2 and ATG5 by shRNA expression: GGA GTA GGA ATT GAG TGA TAA, GCA ACT TCA GAG TGT CAA ACA, and GCT AGC TGG CTG TCC ATA TTG respectively. Viruses carrying GBP1 shRNAs, GBP2 shRNAs, GBP1/2 shRNAs or ATG5 shRNAs were produced by transfecting 293T cells with corresponding pLVTHM constructs together with viral packaging vectors (psPAX2 and pMD2G) by calcium phosphate transfection. GBP1 shRNA, GBP2 shRNAs, GBP1-GBP2 shRNAs- or ATG5 shRNA-containing viruses were harvested from the supernatant 48 h post transfection and applied to THP1 cells for lentiviral infection in the presence of polybrene (5 µg/ml). At 7 d p.i., cells expressing GFP were lysed in sample buffer. The efficiency of gene silencing was determined using western blotting or q-PCR. Empty vector transfected cells were used as a control.
Host cell transfection
Cultured THP1-derived macrophages on coverslips in 12-well-plates were washed once and then transfected with 1 µg/ml plasmid DNA encoding pEGFP-rat LC3 (a kind gift from Tamotsu Yoshimori, Department of Cellular Regulation, Research Institute for Microbial Diseases, Osaka University, Osaka, Japan), using Lipofectamine 2000 (Gibco-Invitrogen, 11668-027) according to manufacturer’s instructions. One day post-transfection, cells were stimulated for 24 h with 100 U/ml IFNG or left without stimulation and incubated as above. Next, transfected cells were infected with C. trachomatis at an MOI of 20 and incubated for the indicated time points at 35°C and 5% CO2, before processing for confocal microscopy.
Confocal microscopy
For fluorescent labeling, cell monolayers were fixed and permeabilized for 5 min with ice-cold 100% methanol at indicated time points p.i. or fixed for 25 min using 4% paraformaldehyde at room temperature (for hGBPs), then permeabilized with Triton X-100 0.3%. For fluorescent labeling for endogenous LC3, cell monolayers were fixed for 25 min using 4% paraformaldehyde followed by methanol fixation for 5 min, then cell monolayers were permeabilized with Triton X-100 (0.3%). Fixed cells were washed three times in PBS and then incubated with 2.5% bovine serum albumin (BSA) in PBS as a blocking reagent. Cells were sequentially incubated for 1 h at room temperature (or overnight at 4°C) with primary antibodies and 1 h at room temperature with secondary antibodies. Then, coverslips were mounted onto glass slides using Mowiol and examined with a Leica TCS-SP laser scanning confocal microscope equipped with a krypton/argon laser (Wetzlar, Germany). Photomicrographs were processed using Adobe Photoshop 6.0 (Adobe Systems) and Microsoft Power-Point.
Immunoblotting
Cell monolayers were washed with cold PBS, and lysed for 30 min on ice in Triton X-100 lysis buffer (20 mM TRIS-HCl; pH 7.6, 150 mM NaCl, 1% Triton X-100), containing 2 mM PMSF and complete protease inhibitor cocktail (Roche, 11836145001). Cell lysates were then centrifuged at 8,000 × g for 10 min. Equal amounts of proteins were subjected to SDS-PAGE and resulting protein bands were transferred electrophoretically onto Immobilon-P polyvinylidene difluoride membranes (Millipore, IPVH00010). The membranes were blocked with 5% fat-skim milk in TBS (Tris-buffered saline; pH 7.5), containing 0.05% Tween-20 for 1 h at room temperature. Next, membranes were incubated with the primary antibodies (overnight, at 4°C) or anti-ACTB (1 h at room temperature) diluted in TBS-0.05% Tween-20. Membranes were washed and then incubated with secondary antibodies conjugated with horseradish peroxidase. Signal detection was performed with the enhanced chemiluminescence system (Amersham, 95038-560).
Staining with acidotropic dye
Labeling of acidic cellular compartments was performed using the pH-sensitive probe LysoTracker Red DND-99. The staining procedure was performed according to the manufacturer’s protocol. Cell monolayers were inoculated with C. trachomatis MOI 100 for 2 h in the presence or absence of IFNG. Cells were then incubated for 30 min with the probe (75 nM), followed by washing with warm PBS. Cell monolayers were then fixed with 4% paraformaldehyde and stained for bacteria as detailed previously.
Inhibition of lysosomal acidification
To examine the effect of the acidification inhibition on chlamydial growth, cell monolayers were inoculated with C. trachomatis (MOI of 5) for 2 h in the presence or absence of IFNG. Cells were then treated with 100 nM BafA1 throughout the experiment period. Control cell monolayers were treated with dimethylsulfoxide, in which BafA1 was dissolved. Two days p.i., cells were assessed for the formation of infectious C. trachomatis by infectivity titration assays on fresh HeLa cell cultures.
Transmission electron microscopy
Control and IFNG-treated cells were seeded into 6-well plates and infected with C. trachomatis for 6 h at MOI 100. Cells were washed with cold PBS and fixed in 2.5% gluteraldehyde. Samples were then incubated with 0.5% osmiumtetroxide and 2% uranylacetate, contrasted with 0.1% tannin acid, dehydrated with graded ethanol and epoxy resin-embedded. Analysis was performed using a Leo 912AB Transmission electron microscope.
Knockdown validation by q-PCR
Total RNA was isolated from host cells using the RNeasy kit (Qiagen, 74104) according to the manufacturer᾽s protocol. The OneStep RT-PCR kit (Qiagen, 210210) was used for cDNA preparation and amplification. Primer sequences were as follow: GBP1 forward 5′-AGG AGT TCC TTC AAA GAT GTG GA-3′, GBP1 reverse 5′-TTC TGA ACA AAG AGA CGA TAG CC-3′, GBP2 forward 5′- CTA TCT GCA ATT ACG CAG CCT-3′, GBP2 reverse 5′-GCC TAG AGA GAA GCC GTT TTT-3′, GAPDH forward 5′-GGT ATC GTG GAA GGA CTC ATG AC-3′, GAPDH reverse 5′-ATG CCA GTG AGC TTC CCG TTC AG-3′.
Plasmid construction
The coding region of human GBP1 was amplified from total cDNA of the THP1 cell line. The construct was constructed by PCR amplification. The PCR product was cloned into the EcoRI sites of the mammalian expression in-house vector pLenti-pEZ149. This lentiviral plasmid was used to generate stable cell line overexpressing GBP1 as mentioned before.
Statistical analysis
For statistical analyses, Student’s t-tests were performed.
| Abbreviations: | ||
| BafA1 | = | bafilomycin A1 |
| DMEM | = | Dulbecco’s minimal essential medium |
| EBs | = | elementary bodies |
| FBS | = | fetal bovine serum |
| GBP1 | = | guanylate-binding protein1, interferon-inducible |
| GFP | = | green fluorescent protein |
| hGBP1 | = | human GBP1 |
| IDO | = | indoleamine 2,3-dioxygenase |
| IFNG | = | interferon-gamma |
| IRG | = | immunity-related GTPases |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| MOI | = | multiplicity of infection |
| mGBP1 | = | murine GBP1 |
| PBS | = | phosphate-buffered saline |
| p.i. | = | post infection |
| PMA | = | phorbol myristate acetate |
| PMSF | = | phenylmethanesulfonylfluoride |
| q-PCR | = | quantitative PCR |
| rRNA | = | ribosomal RNA |
| RT-PCR | = | real-time PCR |
| SDS-PAGE | = | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| shRNA | = | short hairpin RNA |
| TBS | = | tris-buffered saline |
| TEM | = | transmission electron microscopy |
| Trp | = | tryptophan |
| vATPase | = | vacuolar ATPase |
Additional material
Download Zip (3.5 MB)Acknowledgments
We would like to thank Joerg Angermann, Meike Soerensen and Britta Laube for technical support, T. Yoshimori and N. Mizushima for kindly providing us with the rat LC3 construct. The authors also thank Kate Holden-Dye for expert assistance in preparing the manuscript and Markus Kerr for helpful discussions. This work was supported in part by German Academic Exchange Service (DAAD) grants to M.A.A.-Z. and M.A.L. and through a visiting scientist grant from the German Research Foundation to H.M.A.-Y.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/22482
References
- Byrne GI, Krueger DA. Lymphokine-mediated inhibition of Chlamydia replication in mouse fibroblasts is neutralized by anti-gamma interferon immunoglobulin. Infect Immun 1983; 42:1152 - 8; PMID: 6417024
- Rank RG, Ramsey KH, Pack EA, Williams DM. Effect of gamma interferon on resolution of murine chlamydial genital infection. Infect Immun 1992; 60:4427 - 9; PMID: 1398955
- Ito JI, Lyons JM. Role of gamma interferon in controlling murine chlamydial genital tract infection. Infect Immun 1999; 67:5518 - 21; PMID: 10496942
- Morré SA, Lyons JM, Ito JI Jr.. Murine models of Chlamydia trachomatis genital tract infection: use of mouse pneumonitis strain versus human strains. Infect Immun 2000; 68:7209 - 11; http://dx.doi.org/10.1128/IAI.68.12.7209-7211.2000; PMID: 11203323
- Nelson DE, Virok DP, Wood H, Roshick C, Johnson RM, Whitmire WM, et al. Chlamydial IFN-γ immune evasion is linked to host infection tropism. Proc Natl Acad Sci U S A 2005; 102:10658 - 63; http://dx.doi.org/10.1073/pnas.0504198102; PMID: 16020528
- Roshick C, Wood H, Caldwell HD, McClarty G. Comparison of gamma interferon-mediated antichlamydial defense mechanisms in human and mouse cells. Infect Immun 2006; 74:225 - 38; http://dx.doi.org/10.1128/IAI.74.1.225-238.2006; PMID: 16368976
- Koehler L, Nettelnbreker E, Hudson AP, Ott N, Gérard HC, Branigan PJ, et al. Ultrastructural and molecular analyses of the persistence of Chlamydia trachomatis (serovar K) in human monocytes. Microb Pathog 1997; 22:133 - 42; http://dx.doi.org/10.1006/mpat.1996.0103; PMID: 9075216
- Beatty WL, Morrison RP, Byrne GI. Persistent chlamydiae: from cell culture to a paradigm for chlamydial pathogenesis. Microbiol Rev 1994; 58:686 - 99; PMID: 7854252
- Tietzel I, El-Haibi C, Carabeo RA. Human guanylate binding proteins potentiate the anti-chlamydia effects of interferon-gamma. PLoS One 2009; 4:e6499; http://dx.doi.org/10.1371/journal.pone.0006499; PMID: 19652711
- Al-Zeer MA, Al-Younes HM, Braun PR, Zerrahn J, Meyer TF. IFN-gamma-inducible Irga6 mediates host resistance against Chlamydia trachomatis via autophagy. PLoS One 2009; 4:e4588; http://dx.doi.org/10.1371/journal.pone.0004588; PMID: 19242543
- Azenabor AA, Chaudhry AU. Chlamydia pneumoniae survival in macrophages is regulated by free Ca2+ dependent reactive nitrogen and oxygen species. J Infect 2003; 46:120 - 8; http://dx.doi.org/10.1053/jinf.2002.1098; PMID: 12634074
- Beagley KW, Huston WM, Hansbro PM, Timms P. Chlamydial infection of immune cells: altered function and implications for disease. Crit Rev Immunol 2009; 29:275 - 305; http://dx.doi.org/10.1615/CritRevImmunol.v29.i4.10; PMID: 19673684
- Jendro MC, Fingerle F, Deutsch T, Liese A, Köhler L, Kuipers JG, et al. Chlamydia trachomatis-infected macrophages induce apoptosis of activated T cells by secretion of tumor necrosis factor-alpha in vitro. Med Microbiol Immunol 2004; 193:45 - 52; http://dx.doi.org/10.1007/s00430-003-0182-1; PMID: 12750883
- Redecke V, Dalhoff K, Bohnet S, Braun J, Maass M. Interaction of Chlamydia pneumoniae and human alveolar macrophages: infection and inflammatory response. Am J Respir Cell Mol Biol 1998; 19:721 - 7; PMID: 9806736
- Wyrick PB. Intracellular survival by Chlamydia. Cell Microbiol 2000; 2:275 - 82; http://dx.doi.org/10.1046/j.1462-5822.2000.00059.x; PMID: 11207584
- Wyrick PB, Brownridge EA. Growth of Chlamydia psittaci in macrophages. Infect Immun 1978; 19:1054 - 60; PMID: 565338
- MacMicking JD. IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol 2004; 25:601 - 9; http://dx.doi.org/10.1016/j.it.2004.08.010; PMID: 15489189
- Cheng YS, Patterson CE, Staeheli P. Interferon-induced guanylate-binding proteins lack an N(T)KXD consensus motif and bind GMP in addition to GDP and GTP. Mol Cell Biol 1991; 11:4717 - 25; PMID: 1715024
- Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD. A family of IFN-γ-inducible 65-kD GTPases protects against bacterial infection. Science 2011; 332:717 - 21; http://dx.doi.org/10.1126/science.1201711; PMID: 21551061
- Nguyen TT, Hu Y, Widney DP, Mar RA, Smith JB. Murine GBP-5, a new member of the murine guanylate-binding protein family, is coordinately regulated with other GBPs in vivo and in vitro. J Interferon Cytokine Res 2002; 22:899 - 909; http://dx.doi.org/10.1089/107999002760274926; PMID: 12396730
- Vestal DJ, Gorbacheva VY, Sen GC. Different subcellular localizations for the related interferon-induced GTPases, MuGBP-1 and MuGBP-2: implications for different functions?. J Interferon Cytokine Res 2000; 20:991 - 1000; http://dx.doi.org/10.1089/10799900050198435; PMID: 11096456
- Rupper AC, Cardelli JA. Induction of guanylate binding protein 5 by gamma interferon increases susceptibility to Salmonella enterica serovar Typhimurium-induced pyroptosis in RAW 264.7 cells. Infect Immun 2008; 76:2304 - 15; http://dx.doi.org/10.1128/IAI.01437-07; PMID: 18362138
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542 - 5; PMID: 17611390
- Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, et al. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J 2011; 30:395 - 407; http://dx.doi.org/10.1038/emboj.2010.327; PMID: 21139567
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657 - 68; http://dx.doi.org/10.1083/jcb.152.4.657; PMID: 11266458
- Mizushima N, Yoshimori T, Ohsumi Y. Role of the Apg12 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2003; 35:553 - 61; http://dx.doi.org/10.1016/S1357-2725(02)00343-6; PMID: 12672448
- Rottenberg ME, Gigliotti-Rothfuchs A, Wigzell H. The role of IFN-γ in the outcome of chlamydial infection. Curr Opin Immunol 2002; 14:444 - 51; http://dx.doi.org/10.1016/S0952-7915(02)00361-8; PMID: 12088678
- Al-Younes HM, Brinkmann V, Meyer TF. Interaction of Chlamydia trachomatis serovar L2 with the host autophagic pathway. Infect Immun 2004; 72:4751 - 62; http://dx.doi.org/10.1128/IAI.72.8.4751-4762.2004; PMID: 15271937
- van Ooij C, Apodaca G, Engel J. Characterization of the Chlamydia trachomatis vacuole and its interaction with the host endocytic pathway in HeLa cells. Infect Immun 1997; 65:758 - 66; PMID: 9009339
- Al-Younes HM, Rudel T, Meyer TF. Characterization and intracellular trafficking pattern of vacuoles containing Chlamydia pneumoniae in human epithelial cells. Cell Microbiol 1999; 1:237 - 47; http://dx.doi.org/10.1046/j.1462-5822.1999.00024.x; PMID: 11207556
- Friis RR. Interaction of L cells and Chlamydia psittaci: entry of the parasite and host responses to its development. J Bacteriol 1972; 110:706 - 21; PMID: 4336694
- Ouellette SP, Dorsey FC, Moshiach S, Cleveland JL, Carabeo RA. Chlamydia species-dependent differences in the growth requirement for lysosomes. PLoS One 2011; 6:e16783; http://dx.doi.org/10.1371/journal.pone.0016783; PMID: 21408144
- Yasir M, Pachikara ND, Bao X, Pan Z, Fan H. Regulation of chlamydial infection by host autophagy and vacuolar ATPase-bearing organelles. Infect Immun 2011; 79:4019 - 28; http://dx.doi.org/10.1128/IAI.05308-11; PMID: 21807906
- Mordue DG, Sibley LD. Intracellular fate of vacuoles containing Toxoplasma gondii is determined at the time of formation and depends on the mechanism of entry. J Immunol 1997; 159:4452 - 9; PMID: 9379044
- Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, et al. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog 2005; 1:e24; http://dx.doi.org/10.1371/journal.ppat.0010024; PMID: 16304607
- Yap GS, Ling Y, Zhao Y. Autophagic elimination of intracellular parasites: convergent induction by IFN-gamma and CD40 ligation?. Autophagy 2007; 3:163 - 5; PMID: 17204853
- Colombo MI, Gutierrez MG, Romano PS. The two faces of autophagy: Coxiella and Mycobacterium. Autophagy 2006; 2:162 - 4; PMID: 16874070
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119:753 - 66; http://dx.doi.org/10.1016/j.cell.2004.11.038; PMID: 15607973
- Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006; 313:1438 - 41; http://dx.doi.org/10.1126/science.1129577; PMID: 16888103
- Voth DE, Heinzen RA. Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell Microbiol 2007; 9:829 - 40; http://dx.doi.org/10.1111/j.1462-5822.2007.00901.x; PMID: 17381428
- Redd T, Thompson HA. Secretion of proteins by Coxiella burnetii. Microbiology 1995; 141:363 - 9; http://dx.doi.org/10.1099/13500872-141-2-363; PMID: 7704266
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 77; http://dx.doi.org/10.1016/S1534-5807(04)00099-1; PMID: 15068787
- Anderson SL, Carton JM, Lou J, Xing L, Rubin BY. Interferon-induced guanylate binding protein-1 (GBP-1) mediates an antiviral effect against vesicular stomatitis virus and encephalomyocarditis virus. Virology 1999; 256:8 - 14; http://dx.doi.org/10.1006/viro.1999.9614; PMID: 10087221
- Taylor GA, Feng CG, Sher A. Control of IFN-gamma-mediated host resistance to intracellular pathogens by immunity-related GTPases (p47 GTPases). Microbes Infect 2007; 9:1644 - 51; http://dx.doi.org/10.1016/j.micinf.2007.09.004; PMID: 18023232
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8:1124 - 32; http://dx.doi.org/10.1038/ncb1482; PMID: 16998475
- Braun PR, Al-Younes H, Gussmann J, Klein J, Schneider E, Meyer TF. Competitive inhibition of amino acid uptake suppresses chlamydial growth: involvement of the chlamydial amino acid transporter BrnQ. J Bacteriol 2008; 190:1822 - 30; http://dx.doi.org/10.1128/JB.01240-07; PMID: 18024516
- Chang YP, Chen CL, Chen SO, Lin YS, Tsai CC, Huang WC, et al. Autophagy facilitates an IFN-γ response and signal transduction. Microbes Infect 2011; 13:888 - 94; http://dx.doi.org/10.1016/j.micinf.2011.05.008; PMID: 21664983
- Weichhart T. Mammalian target of rapamycin: a signaling kinase for every aspect of cellular life. Methods Mol Biol 2012; 821:1 - 14; http://dx.doi.org/10.1007/978-1-61779-430-8_1; PMID: 22125056
- Kuballa P, Nolte WM, Castoreno AB, Xavier RJ. Autophagy and the immune system. Annu Rev Immunol 2012; 30:611 - 46; http://dx.doi.org/10.1146/annurev-immunol-020711-074948; PMID: 22449030
- Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol 2003; 77:8957 - 61; http://dx.doi.org/10.1128/JVI.77.16.8957-8951.2003; PMID: 12885912