Abstract
Cells require the ability to rapidly detect decreases in concentrations of free amino acids so that homeostatic mechanisms, including autophagy, can be engaged to replenish amino acids. Amino acids are transported into cells where it is generally accepted that they are detected by an intracellular sensor. We now show that the cell surface G protein coupled receptor (GPCR) TAS1R1-TAS1R3 (T1R1-T1R3) can sense extracellular amino acids, activate MTORC1, and inhibit autophagy. This receptor is expressed in most tissues and fasted TAS1R3−/− mice have increased autophagy in the heart, skeletal muscle and liver.
GPCRs as nutrient sensors were first identified in yeast. For example, the GPCR Gpr1 and its regulator Rgs2 sense glucose and sucrose and stimulate signaling pathways that regulate growth. Yeast can sense extracellular amino acids through Ssy1, a cell surface protein that is a member of the amino acid permease family, though it is not able to transport amino acids into the cell. While less is understood about the mechanisms that metazoans use to sense amino acids, it is thought that amino acids have to be transported into cells in order to engage the amino acid sensor, which leads to the activation of MTORC1 and the subsequent increase in translation and inhibition of autophagy.
We began our study by investigating amino acid signaling in pancreatic beta cells. Because amino acids can induce insulin secretion and the biosynthesis of insulin in pancreatic beta cells, we were interested in determining the mechanisms by which amino acids were sensed in this cell type. Our previous work showed that MAPK1-MAPK3 (ERK2-ERK1) are activated by glucose and hormones that induce insulin secretion. Therefore, we hypothesized that amino acids would also be monitored by MAPK1-MAPK3. We determined that amino acids activated MAPK1-MAPK3 with kinetics similar to that observed with carbachol, a muscarinic GPCR agonist. While there are a few amino acid-responsive GPCRs, only the taste receptor TAS1R1-TAS1R3 is significantly activated by most of the 20 amino acids. Therefore, we hypothesized that TAS1R1-TAS1R3 is responsible for activating MAPK1-MAPK3 by amino acids. Knockdown of this receptor in pancreatic beta cells significantly inhibits amino acid-induced MAPK1-MAPK3 activation.
While TAS1R1-TAS1R3 was originally discovered in gustatory neurons, we determined that this receptor is expressed in all of the tissues that we tested, and many cell types, suggesting that TAS1R1-TAS1R3 is more broadly used for amino acid sensing throughout the body. The known amino acid-responsive GPCRs belong to the GPCR family class C, which also includes the sweet taste receptor, metabotropic glutamate receptors, the GABAB receptor, the Ca2+-sensing receptor, and a few orphan receptors. Most members of the class C family function as hetero- or homodimers and contain a large extracellular segment called the Venus Flytrap module that is involved in agonist binding. Interestingly, the Venus Flytrap module shares significant homology with the periplasmic nutrient sensing bacteria proteins. TAS1R3 dimerizes with TAS1R1 to form the receptor responsible for detecting the umami flavor. We hypothesized that TAS1R1-TAS1R3 could send amino acid sufficiency signals into the cell to regulate the activity of the mechanistic target of rapamycin complex 1 (MTORC1). Indeed, when we reduced the expression of TAS1R1-TAS1R3 in heart cells, pancreatic beta cells, and HeLa cells, we observed that amino acids activate MTORC1 much less well than in control cells. We also observed a defect in MTORC1 activity in TAS1R3−/− mice.
Under nutrient-replete conditions, MTORC1 prevents the initiation of autophagy by the phosphorylation-induced inhibition of unc51-like protein kinase ULK1. We found that the knockdown of TAS1R3 in cardiac myoblasts growing in nutrient-replete conditions decreases MTORC1-induced phosphorylation of ULK1 and increases autophagy (). While AMPK activation can increase autophagy by phosphorylating ULK1, we observed a decrease in the phosphorylation of AMPK substrates when TAS1R3 is knocked down. Thus, it appears that TAS1R3 reduction induces autophagy by inhibiting MTORC1 activity and not increasing AMPK activity.
Figure 1. TAS1R1-TAS1R3 directly detects amino acids leading to the activation of MTORC1 and inhibition of autophagy. This receptor activates MTORC1, in part, through the activation of phospholipase C (PLC), the increase in intracellular calcium, and the activation of MAPK1-MAPK3. TAS1R1-TAS1R3 is required for the amino acid-induced MTOR localization to the lysosome, a necessary step in MTORC1 activation.
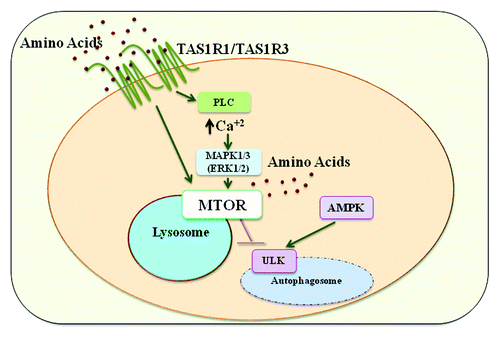
Because it is thought that intracellular amino acids regulate MTORC1 activity and autophagy, we measured intracellular amino acid concentrations. While we observed similar intracellular amino acid levels between the receptor knockdown and control cells growing in nutrient-replete conditions, there was an increase in the expression of several amino acid transporters. There was also a decrease in DDIT4/REDD1 and TSC2 expression, both negative regulators of MTORC1, when TAS1R3 was knocked down. Thus, it appears that these cells are trying to compensate for the perceived deficiency in amino acids.
Excessive lipids are implicated in the development of obesity-related metabolic diseases. Recent evidence suggests that there is also a strong association with elevated branched-chain and aromatic amino acids in these diseases. Thus, research into the mechanisms by which nonobese and obese humans detect and metabolize amino acids will likely lead to a better understanding of how to prevent and treat metabolic diseases.
It will be important to investigate the potential therapeutic usefulness of regulating autophagy and metabolism by targeting TAS1R1-TAS1R3. Diabetes, obesity, cancer and neurodegeneration are all, at least in part, thought of as conditions of altered metabolism. Cancers often take advantage of the PI3K-AKT-MTOR pathway to manipulate nutrient utilization or increase nutrient uptake for production of energy and macromolecules necessary for continual growth and proliferation. In some settings, inhibition of TAS1R1-TAS1R3 might block tumor progression through suppression of MTOR signaling. Alternatively, in diabetes it may be beneficial to activate TAS1R1-TAS1R3 signaling because the reduced expression of this receptor significantly decreases insulin protein content of beta cells. It will be important to determine whether or not increased activation of TAS1R1-TAS1R3 can be used as therapy to combat diabetes, which is associated with a decrease in insulin production in beta cells. Much remains to be determined as to how TAS1R1-TAS1R3 is regulated in cells and the extent to which activation or inhibition of this receptor mechanism may offer therapeutic benefit.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.