Abstract
The activity of the conserved Atg12–Atg5-Atg16 complex is essential for autophagosome formation. However, little is known about its mechanism of action during this process. In our study we employed in vitro systems consisting of purified proteins and giant unilamellar vesicles (GUVs) or small liposomes to investigate membrane binding by the Atg12–Atg5-Atg16 complex and its interplay with the Atg8 conjugation system. We showed that Atg5 directly binds membranes and that this membrane binding is negatively regulated by Atg12 conjugation but activated by Atg16. Membrane binding by the Atg12–Atg5-Atg16 complex is required for efficient promotion of Atg8 lipidation. Additionally, we found that the Atg12–Atg5-Atg16 complex tethered vesicles in an Atg8-independent manner. In yeast, membrane binding by Atg5 is not required for its recruitment to the phagophore assembly site (PAS) but is essential for efficient promotion of autophagy and the cytoplasm-to-vacuole targeting (Cvt) pathway at a stage preceding Atg8 lipidation and autophagosome closure. Our findings provide new insights into the role of the Atg12–Atg5-Atg16 complex during autophagosome formation.
Autophagy requires the activity of two ubiquitin-like conjugation systems. In the first system, the ubiquitin-like protein Atg8 is conjugated to the head group of the membrane lipid phosphatidylethanolamine (PE). This conjugation is mediated by the conjugating enzymes Atg7 and Atg3. The second system entails the conjugation of the ubiquitin-like protein Atg12 to a lysine residue of Atg5. This conjugation requires the activity of Atg7 and Atg10. The Atg12–Atg5 conjugate in turn associates noncovalently with Atg16. The presence of Atg16 is required for the localization of Atg5 and Atg12 to the PAS. There is an interplay between the Atg8 and Atg12 conjugation systems, as the Atg12–Atg5-Atg16 complex is essential for Atg8–PE formation in vivo, and the Atg12–Atg5 conjugate stimulates Atg8–PE formation in vitro. In our study, we addressed the questions of how the recruitment of the Atg12–Atg5-Atg16 complex to the membrane is regulated, and what the functional consequences of this membrane binding are.
Using purified proteins including Atg5, Atg16, the Atg12–Atg5 conjugate, and the Atg12–Atg5-Atg16 and Atg5-Atg16 complexes in liposome co-sedimentation assays, we analyzed the membrane binding properties of these proteins. While Atg5 efficiently binds to liposomes, membrane binding by the Atg12–Atg5 conjugate is severely reduced compared with Atg5 alone. In contrast, the Atg12–Atg5-Atg16 and Atg5-Atg16 complexes efficiently bind to membranes. These results, supported by fractionation experiments using different yeast deletion strains, suggest that membrane binding is at least in part mediated by Atg5, and inhibited by Atg12, but uncovered upon association of the Atg12–Atg5 conjugate with Atg16. Atg16 may therefore regulate the association of the Atg12–Atg5 conjugate with membranes in vivo.
The Atg12–Atg5-Atg16 complex causes massive aggregation of giant and small unilamellar vesicles in vitro, and this aggregation is Atg16 dependent. Since Atg16 forms a dimer and binds to Atg5, this offers an explanation for its role in vesicle tethering () as Atg16 may dimerize the membrane-binding site located in Atg5. In the absence of Atg12, vesicle tethering is less efficient, suggesting that Atg12 may also be involved in the membrane tethering activity of the Atg12–Atg5-Atg16 complex.
Figure 1. Potential roles of the Atg12–Atg5-Atg16 complex during autophagosome formation. The association of the Atg12–Atg5 conjugate with Atg16 unmasks a membrane-binding site in Atg5 and allows it to bind to phagophore precursors. Due to the dimerization of Atg16, each molecule of the Atg12–Atg5-Atg16 complex contains two molecules of Atg5 enabling Atg12–Atg5-Atg16 to tether vesicles. Atg12 stimulates this tethering activity. In addition, Atg16-dependent membrane localization of Atg12 is required to bring Atg8-bound Atg3 to the membrane. This in turn facilitates the conjugation of Atg8 to PE.
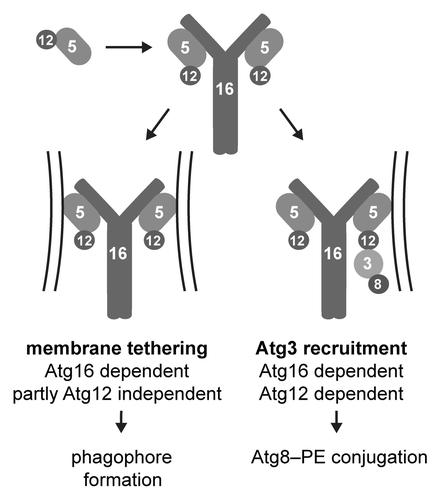
Previous studies reported that Atg16 is required for efficient Atg8 lipidation in vivo but not in a reconstituted in vitro assay employing small unilamellar vesicles (SUVs). We reconstituted Atg8 lipidation using giant unilamellar vesicles (GUVs). In the GUV system membranes are less abundant and less curved than in the SUVs. This assay is therefore more stringent than the SUV assay. Using the GUV system we observed that the Atg12–Atg5-Atg16 complex promotes Atg8 lipidation much more efficiently than Atg12–Atg5 conjugate. Atg12 was shown to interact with Atg3, an enzyme involved in Atg8 lipidation. Due to its ability to bind membranes, the Atg12–Atg5-Atg16 complex is more efficient than the Atg12–Atg5 conjugate in the recruitment of Atg3 to vesicles. Thus, Atg3-bound Atg8 can be more efficiently brought to the membrane by the Atg12–Atg5-Atg16 complex compared with the Atg12–Atg5 conjugate. The Atg12–Atg5-Atg16 complex therefore functions indeed in a manner analogous to E3 enzymes during classical ubiquitination reactions by bringing the Atg8-carrying E2-like enzyme Atg3 in proximity to the substrate (PE).
Knowing that membrane binding by Atg5 requires negatively charged lipids and is inhibited by Atg12 conjugation, we generated an Atg5K160E,R171E mutant. This mutant shows substantially reduced membrane binding in vitro and in vivo. Yeast expressing Atg5K160E,R171E are defective in Atg8 lipidation, prApe1 maturation, and GFP-Atg8 processing. Furthermore, in the protease protection assay, prApe1 is considerably less protected in yeast expressing the Atg5K160E,R171E mutant compared with wild-type Atg5. These data indicate that membrane binding by Atg5 is essential for the correct progression of the Cvt pathway and autophagy at the stage preceding Atg8 lipidation and vesicle closure.
Interestingly, despite the loss of membrane binding, the Atg5K160E,R171E mutant still localizes to the PAS. Atg16 also localizes to the PAS in Atg5K160E,R171E-expressing cells. These results suggest that the recruitment of Atg12–Atg5-Atg16 to the PAS occurs prior to membrane binding and is mediated by another factor, possibly another protein.
In summary, in our study we were able to assign functions to all the components of the Atg12–Atg5-Atg16 complex during autophagosome formation. We found that Atg5 is capable of membrane binding. This membrane-binding activity is required to bring Atg12 to the membrane, which enables it to bring Atg8-laden Atg3 in close proximity to the PE substrate. Atg16 in turn is required to activate membrane binding by Atg5. Additionally, the Atg12–Atg5-Atg16 complex clusters lipid vesicles in vitro in an Atg16-dependent manner. Atg12 has a stimulating effect on this membrane tethering ability. Recent studies reported that small Atg9-containing vesicles coalesce at the PAS. Furthermore, SNARE proteins may then mediate the fusion of these vesicles in order to generate precursors of autophagosomes. The Atg12–Atg5-Atg16 complex may thus contribute to the initial tethering of such precursors.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.