Abstract
Autophagy is a cellular self-digestion process that mediates protein quality control and serves to protect against neurodegenerative disorders, infections, inflammatory diseases and cancer. Current evidence suggests that autophagy can selectively remove damaged organelles such as the mitochondria. Mitochondria-induced oxidative stress has been shown to play a major role in a wide range of pathologies in several organs, including the heart. Few studies have investigated whether enhanced autophagy can offer protection against mitochondrially-generated oxidative stress. We induced mitochondrial stress in cardiomyocytes using antimycin A (AMA), which increased mitochondrial superoxide generation, decreased mitochondrial membrane potential and depressed cellular respiration. In addition, AMA augmented nuclear DNA oxidation and cell death in cardiomyocytes. Interestingly, although oxidative stress has been proposed to induce autophagy, treatment with AMA did not result in stimulation of autophagy or mitophagy in cardiomyocytes. Our results showed that the MTOR inhibitor rapamycin induced autophagy, promoted mitochondrial clearance and protected cardiomyocytes from the cytotoxic effects of AMA, as assessed by apoptotic marker activation and viability assays in both mouse atrial HL-1 cardiomyocytes and human ventricular AC16 cells. Importantly, rapamycin improved mitochondrial function, as determined by cellular respiration, mitochondrial membrane potential and morphology analysis. Furthermore, autophagy induction by rapamycin suppressed the accumulation of ubiquitinylated proteins induced by AMA. Inhibition of rapamycin-induced autophagy by pharmacological or genetic interventions attenuated the cytoprotective effects of rapamycin against AMA. We propose that rapamycin offers cytoprotection against oxidative stress by a combined approach of removing dysfunctional mitochondria as well as by degrading damaged, ubiquitinated proteins. We conclude that autophagy induction by rapamycin could be utilized as a potential therapeutic strategy against oxidative stress-mediated damage in cardiomyocytes.
Introduction
Progressive accumulation of mitochondrially-generated reactive oxygen species (ROS) has been proposed to be a major player in the pathogenesis of many chronic degenerative diseases, such as cardiovascular disease.Citation1,Citation2 Experimental evidence indicates that in cardiac cells, damaged and dysfunctional mitochondria can produce up to 10-fold more hydrogen peroxide (H2O2) than intact organelles.Citation3 Although ROS is constantly being generated by several enzymatic reactions, such as those catalyzed by the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, cyclooxygenases and xanthine oxidases, the majority of ROS is produced as a byproduct of mitochondrial oxidative phosphorylation.Citation4 It is believed that under physiological conditions, about 0.2 to 2% of the oxygen consumed by the cell is converted to superoxide anions (O2•−) by the mitochondria, with complex I and III of the electron transport chain (ETC) being the major sites of such ROS production.Citation5 Oxidants, in low amounts, can function as essential signaling molecules.Citation6 In contrast, excessive ROS generation is detrimental, negatively affecting the function and integrity of not only the mitochondria, but also of proteins, lipids and other organelles in the cytoplasm and potentially leading to impaired cellular homeostasis and organ functioning.Citation7,Citation8 Importantly, mitochondrial DNA is particularly susceptible to such oxidative damage, primarily due to its close proximity to the site of ROS production, the lack of protective histones, a somewhat less efficient DNA repair system as compared with the nucleus and a lack of introns in the mitochondrial genome.Citation9 Damaged mitochondria in turn generate more ROS, contributing to further oxidative burden and affecting cellular homeostasis.
Enhanced oxidative damage to mitochondrial proteins and lipids has been documented in the myocardium of old rodents.Citation10,Citation11 Mitochondrial dysfunction and oxidative stress are postulated to play a major role in the pathogenesis of cardiac left ventricular hypertrophy as well as in the progression from compensated left ventricular hypertrophy to heart failure.Citation12 Mitochondria-generated oxidative stress also plays a critical role in the pathogenesis of myocardial damage during ischemia/reperfusion, which eventually progresses to cardiac dysfunction.Citation13 Further evidence for the involvement of mitochondrially-generated oxidative stress in mammalian heart senescence is provided by the observation that overexpressing the antioxidant enzyme catalase targeted to the mitochondria (mCAT) delays the development of cardiac pathology in mice.Citation14 The rate of H2O2 generation and mitochondrial oxidative damage are significantly decreased in the heart of old mCAT mice compared with age-matched wild-type controls.Citation14
Apart from the intramitochondrial proteolytic system selectively removing damaged proteins, the only known mechanism by which mitochondria can be cleared from a cell is through autophagy. Autophagy is a self-digestion process whereby the cell degrades long-lived proteins and organelles, generating amino acids and fatty acids that can be reutilized by the cell.Citation15 Autophagy begins with the formation of a double-layered phagophore membrane around the components to be degraded. The phagophore expands, eventually completely engulfing the components and forming the autophagosome. The autophagosome then fuses with the lysosome, forming the autolysosome wherein the cargo is degraded and the digested components transported back to the cytoplasm by vacuolar efflux transporters.Citation16 Growing evidence suggests that autophagy could specifically target damaged and dysfunctional mitochondria for removal, which might otherwise lead to apoptotic induction.Citation17 Additionally, autophagy can also remove oxidatively modified and dysfunctional proteins as a bulk digestion process, thereby maintaining a “cleaner” cell and avoiding potential proteotoxicity arising from their unwanted accumulation.Citation18 It is also worth noting that oxidative stress has been proposed to induce autophagy by specifically regulating autophagy protein ATG4;Citation19 however, recent studies also show that the type of oxidant generated and its site of origin are crucial factors in determining whether it would lead to autophagic stimulation.Citation20
Rapamycin is an immunosuppressant antibiotic widely used as an inducer of autophagy, acting through its inhibitory effect on the mechanistic target of rapamycin (MTOR).Citation21 In the presence of nutrients and growth factors, MTOR is active, hyperphosphorylating and inhibiting the unc-51-like kinase 1 - ATG13 - RB1 inducible coiled-coil protein (ULK1-ATG13-RB1CC1) complex required for the induction of autophagy.Citation22 Rapamycin therefore primarily acts to remove the inhibitory effect of MTOR, thereby inducing autophagy. The cardioprotective properties of rapamycin have been demonstrated in animal models of ischemia/reperfusion as well as in cardiac hypertrophy.Citation23,Citation24 In addition, rapamycin also protects against neurodegeneration by mediating the removal of aggregated proteins through autophagy induction.Citation25
In this study, we investigated whether autophagy induction by rapamycin can be beneficial against mitochondrially-generated oxidative stress in mouse atrial (HL-1 cells) and human ventricular (AC16 cells) cardiomyocytes. Mitochondrial stress has been induced by antimycin A (AMA), a fungicide isolated from Streptomyces kitazawensisCitation26 and known to specifically inhibit complex III of ETC by binding to the Qi site of cytochrome c reductase.Citation27 The binding inhibits the flow of electrons through the ETC, generating O2•− in the mitochondriaCitation28 and inducing apoptosis.Citation29,Citation30 Our study showed that autophagy induction by rapamycin offers cytoprotective effects and improves mitochondrial function in AMA-treated cells and that inhibition of autophagy blocks the beneficial effects of rapamycin. We propose that autophagy enhancement may represent a potential therapeutic strategy against pathological conditions involving mitochondrially-generated oxidative stress.
Results
AMA increases mitochondrial O2•− generation and decreases mitochondrial membrane potential (Δψm)
First, we established the concentration of AMA needed to increase ROS generation in the mitochondria. Cells were pre-labeled with MitoSOX Red, a fluorogenic dye highly selective for the detection of O2•− in the mitochondria,Citation31 followed by treatment with increasing concentrations of AMA or vehicle control. The fluorescence intensity was subsequently analyzed using flow cytometry. In contrast to vehicle-treated cells, which showed minimal MitoSOX Red fluorescence, treatment with AMA resulted in a dose-dependent increase in fluorescence intensity, with 50 µM being the lowest concentration required to reach statistical significance (; p < 0.05). To confirm flow cytometry results, we performed confocal imaging on cells pre-labeled with MitoSOX Red and treated with 50 µM AMA. In contrast to vehicle-treated cells, which showed minimal fluorescence, treatment with 50 µM AMA resulted in a strong MitoSOX Red fluorescence originating from the mitochondria ().
Figure 1. AMA causes cytotoxicity in HL-1 cardiomyocytes. (A) Cells were trypsinized and resuspended in fresh media followed by staining with 3 µM MitoSOX Red. Cells were subsequently incubated with increasing concentrations of AMA or vehicle control for 30 min followed by flow cytometric analysis of MitoSOX Red fluorescence. (B) Cells were incubated with 1 µg/µl Hoechst 33342 and 3 µM MitoSOX Red and subsequently treated with 50 µM AMA or vehicle control for 30 min, followed by confocal imaging. (C) Cells were trypsinized and resuspended in fresh media followed by staining with 50 nM TMRM and were subsequently incubated with increasing concentrations of AMA or vehicle control for 2 h followed by flow cytometric analysis of TMRM fluorescence. (D) Cells were incubated with 1 µg/ul Hoechst 33342 and 50 nM TMRM and subsequently treated with 50 µM AMA or vehicle control for 2 h, followed by confocal imaging. (E) Cells were incubated with increasing concentrations of AMA for 2 h, and were trypsinized and processed for HPLC analysis of DNA (E) and RNA (F) oxidation 24 h later. Data have been normalized to vehicle-treated control. (G) Cells were incubated with increasing concentrations of AMA and cell viability determined using MTT assay after the indicated time points. Data have been normalized to vehicle-treated control. *p < 0.05; **p < 0.01, ***p < 0.001 vs. control. Data are derived from three independent experiments.
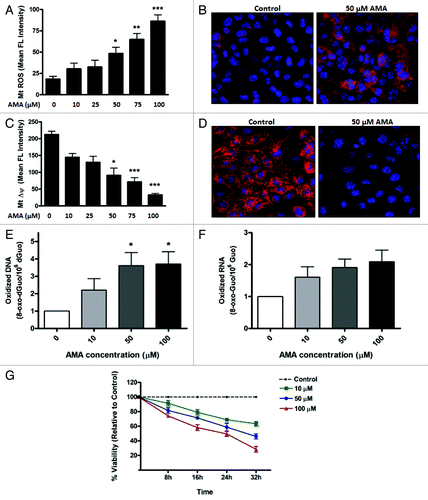
Next, we determined the effects of AMA on Δψm. HL-1 cells were pre-labeled with tetramethyl rhodamine methyl ester (TMRM), a cationic, fluorogenic dye which specifically migrates to bioenergetically active mitochondria and fluoresces red.Citation32 TMRM-loaded cells were treated with increasing concentrations of AMA or vehicle control and the fluorescence intensity of TMRM using flow cytometry. Vehicle-treated cells showed a strong red fluorescence, indicative of normal Δψm, whereas AMA treatment resulted in a dose-dependent decrease in TMRM fluorescence (). We observed that 50 µM is the lowest concentration of AMA required to reach statistically significant decreases in Δψm (p < 0.05). Confocal imaging further confirmed that 50 µM AMA resulted in a drastic decrease in TMRM fluorescence, compared with vehicle-treated cells ().
AMA treatment induces DNA oxidation and decreases cell viability
We next investigated whether AMA-induced ROS can leak out of the mitochondria and have damaging effects on extra-mitochondrial components. To test this, we measured the levels of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dGuo) and 8-oxo-7,8-
dihydroguanosine (8-oxo-Guo) as an indicator of DNA and RNA oxidation, respectively.Citation33 AMA significantly increased the level of DNA oxidation (; p < 0.05) in HL-1 cells at a concentration of 50 µM. RNA oxidation tended to increase with all concentrations, but changes were not statistically significant (; p < 0.10). Finally, the survival of HL-1 cells treated with increasing concentrations of AMA, as determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay at different time points, showed a dose- and time-dependent decrease in viability (). In addition to being used as a viability marker, the MTT assay is also indicative of mitochondrial activity, as most of the dehydrogenases required for the reduction of MTT reside in the mitochondria.
Treatment with AMA does not induce autophagy
Since recent studies have reported a potential role of oxidative stress as an inducer of autophagy,Citation19,Citation34 we investigated whether treatment with AMA can also lead to autophagic induction. All autophagy assessments were conducted in the absence (steady-state) or presence of lysosomal inhibitor bafilomycin A1 (BafA1). To specifically measure autophagic stimulation, we arrested autophagic degradation at the lysosomal-fusion step and determined time-dependent accumulation of autophagic vacuoles. In other words, to determine whether a particular intervention led to autophagic stimulation, we compared LC3-II/LC3-I ratios (or GFP-LC3 puncta) between control and the intervention group under +BafA1 conditions. A statistically significant increase in the LC3-II/LC3-I ratio under +BafA1 conditions in an intervention group compared with vehicle-treated controls was indicative of autophagic stimulation. This method of assessing autophagic stimulation has been used before.Citation35-Citation37 To make sure that the autophagosomes were being degraded in the lysosomes, we also measured autophagic flux by comparing relative increases in LC3-II/LC3-I ratio under +BafA1 conditions in comparison to steady-state conditions. A larger difference in the LC3-II/LC3-I ratios under the two conditions indicated active clearance of autophagosomes by the lysosomes.
To determine whether AMA induces autophagy, HL-1 cells were treated with increasing concentrations of the drug for either 4 h or 24 h in the absence or presence of BafA1 during the final 4 h or 2 h of incubation, respectively. At the 24 h time point, cells were incubated with BafA1 for a shorter period of time to avoid additional toxicity in stressed HL-1 cells. Immunoblot analysis showed no significant autophagic stimulation at either time points with any of the concentrations of AMA used (; Fig. S1A and S1B). Autophagic flux was also not enhanced with AMA treatment (). Similar results were obtained in human ventricular AC16 cells (Fig. S2A–C) and in HL-1 cells treated with lower concentrations of AMA (Fig. S1C and S1D). Immunoblotting results were also confirmed by epifluorescence imaging in GFP-LC3 expressing HL-1 cells, which showed no significant increases in autophagic puncta with AMA treatment (). We also assessed the levels of autophagy proteins BECN1, ATG7 and ATG12–ATG5 conjugate and observed no differences in their expression levels in AMA-treated cells (Fig. S1E).
Figure 2. Treatment with AMA does not induce autophagy in HL-1 cardiomyocytes. (A) Cells were incubated with increasing concentrations of AMA for 4 h, in the presence of DMSO (vehicle) or 75 nM BafA1. Cells were subsequently lysed and immunoblotted for LC3B. GAPDH is used as a loading control. (B) LC3-II/LC3-I ratios were calculated from three independent experiments as in (A). (C) Autophagic flux in HL-1 cells treated with AMA as in (A) was calculated by subtracting LC3-II/LC3-I ratios under steady-state conditions from that obtained under +BafA1 conditions. Data represents three independent experiments. (D) Cells were transfected with GFP-LC3 plasmid and 24 h after transfection, treated with 50 µM AMA for 4 h, in the presence of DMSO (vehicle) or 75 nM BafA1. Cells were subsequently fixed in 4% paraformaldehyde, stained with DAPI and images taken using epifluoresence microscope. Representative images are shown. (E) The number of GFP-LC3 puncta in each cell for (D) was counted to determine the percentage of cells with stimulated autophagy. (F) Cells were treated with vehicle control or 50 µM AMA for 32 h and were subsequently lysed and immunoblotted for VDAC1. GAPDH is used as a loading control.
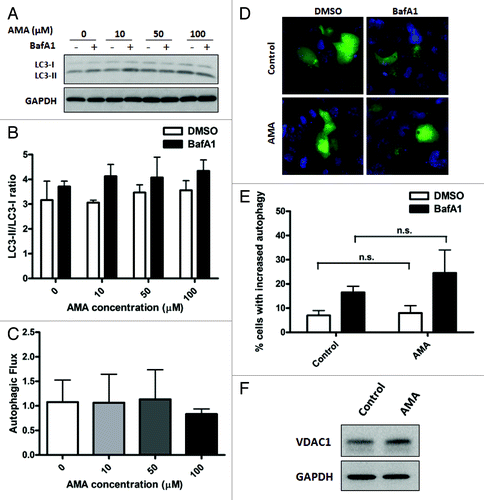
Since AMA treatment led to ROS generation in the mitochondria, we further investigated whether it can lead to the specific degradation of mitochondria by mitophagy. We observed that the abundance of mitochondrial outer membrane protein VDAC1 (voltage-dependent anion channel 1) was unchanged in cells treated with AMA, indicating no significant clearance of mitochondria ().
Treatment with rapamycin induces autophagy in HL-1 cells
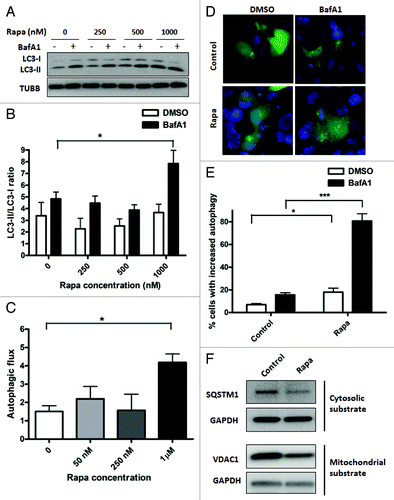
For autophagy induction by rapamycin, cells were incubated with rapamycin or vehicle control for 16 h, in the absence or presence of BafA1 added during the final 4 h of incubation. Incubation of HL-1 cardiomyocytes with rapamycin caused an inhibition of MTOR signaling, as shown by the disappearance of phospho-RPS6, one of the downstream targets of MTOR kinase (Fig. S3A). Rapamycin caused significant autophagic stimulation at a concentration of 1 μM (p < 0.01), with lower concentrations than 1 μM not reaching statistical significance (). Autophagic stimulation by rapamycin was also observed in AC16 cells (Figs. S3C and S3D). Importantly, autophagic flux was also enhanced in rapamycin-treated cells in both HL-1 and AC16 cells (; Fig. S3E). As a further confirmation of our immunoblotting data, we performed epifluorescence imaging on GFP-LC3 expressing HL-1 cells (). Quantification of GFP-LC3 puncta further confirmed induction of autophagy by rapamycin (; p < 0.001). Furthermore, treatment with rapamycin decreased the abundance of both cytosolic and mitochondrial substrates of autophagy, assessed by SQSTM1 and VDAC1 levels, respectively (). To rule out the possibility that the decrease in VDAC1 levels was due to proteasomal degradation of mitochondrial outer membrane proteins, we also assessed mitochondrial inter-membrane space protein, cytochrome c (cyt c). We observed that rapamycin treatment for 48 h decreased cyt c levels in HL-1 cells (Fig. S3B).
Figure 3. Treatment with rapamycin induces autophagy in HL-1 cardiomyocytes. (A) Cells were incubated with increasing concentrations of rapamycin for 16 h, in the presence of DMSO (vehicle) or 75 nM BafA1, added during the final 4 h of incubation. Cells were subsequently lysed and immunoblotted for LC3B. TUBB is used as a loading control. (B) LC3-II/LC3-I ratios were calculated from three independent experiments as in (A). (C) Autophagic flux in rapamycin-treated cells were calculated by subtracting LC3-II/LC3-I ratios under steady-state conditions from that obtained under +BafA1 conditions. (D) Cells were transfected with GFP-LC3 plasmid and 24 h after transfection, treated with 1 µM rapamycin for 16 h, in the presence of DMSO (vehicle) or 75 nM BafA1, added during the final 4 h of incubation. Cells were subsequently fixed in 4% paraformaldehyde, stained with DAPI and images taken using epifluoresence microscope. Representative images are shown. (E) The number of GFP-LC3 puncta in each cell for (D) was counted to determine the percentage of cells with stimulated autophagy. (F) Cells were treated with vehicle control or 1 µM rapamycin for 48 h. Cell lysates were collected and immunoblotted for SQSTM1 or VDAC1. GAPDH is used as a loading control. Rapa, rapamycin. *p < 0.05, ***p < 0.001 vs. control. Data are derived from two to three independent experiments.
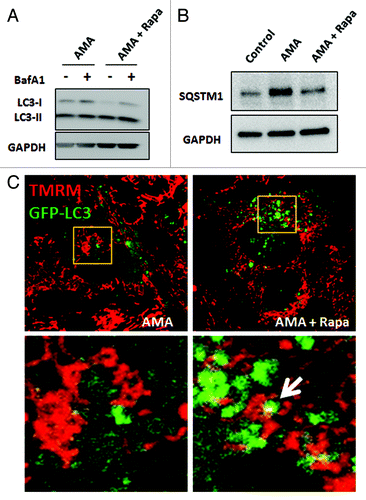
The induction of autophagy by rapamycin was also observed in the presence of AMA as determined by LC3B immunoblotting () in HL-1 cells. As an additional measure of autophagic activity, we investigated SQSTM1/p62 abundance in AMA alone or AMA and rapamycin cotreated cells. SQSTM1 binds to ubiquitinated proteins and also interacts with LC3 through its LC3 binding motif, thereby directing the degradation of ubiquitinated proteins through the autophagic pathway. In the process, SQSTM1 is degraded through autophagy; accordingly, a decrease in SQSTM1 is considered indicative of enhanced autophagic flux.Citation38,Citation39 We observed that treatment with AMA led to an increase in SQSTM1 levels in HL-1 cells and that coincubation with rapamycin decreased its accumulation (). Finally, the results of live cell confocal imaging in HL-1 cardiomyocytes showed colocalization of mitochondria (labeled with TMRM) with autophagosomes (labeled with GFP-LC3) in rapamycin cotreated cells, indicating mitophagy (, right panel). Such colocalization was not observed in cells treated with AMA alone (, left panel).
Figure 4. Rapamycin induces autophagy in the presence of AMA in HL-1 cardiomyocytes. (A) Cells were treated with AMA alone or rapamycin plus AMA for 16 h in the presence of DMSO (vehicle) or 75 nM BafA1, added during the final 4 h of incubation. Cells were subsequently lysed and immunoblotted for LC3B. GAPDH is used as a loading control. (B) Cells were treated with vehicle control, AMA or AMA plus rapamycin for 32 h and were subsequently lysed and immunoblotted for SQSTM1. GAPDH is used as a loading control. (C) Cells were transfected with GFP-LC3 plasmid and 24 h after transfection, treated with 1 µM rapamycin or vehicle control for 16 h. Cells were then incubated with 50 nM TMRM for 30 min, followed by treatment with 50 μM AMA. Cells were subsequently imaged using confocal microscopy. Higher magnification images of the boxed areas are shown in the bottom-panels. Arrow indicates mitochondria surrounded by a growing autophagic vacuole. Rapa, rapamycin.
Rapamycin plays a protective role against AMA-induced toxicity
We next investigated whether rapamycin can protect against AMA-induced toxicity in both HL-1 and AC16 cells. For experiments with HL-1 cells, cells were pretreated with rapamycin for 16 h, followed by addition of AMA in fresh media in the presence of rapamycin for 32 h. Viability analyses by trypan blue exclusion () and MTT assay (Fig. S4A) revealed a drastic decrease (p < 0.01) in cell viability with AMA treatment, which was significantly attenuated by treatment with rapamycin (p < 0.05). MTT assay revealed a slight decrease in cell viability due to rapamycin treatment alone (Fig. S4A), although changes were not statistically significant, and we believe this is due to the antiproliferative effect of the MTOR inhibitor per se, which led to a slightly decreased cell number (as opposed to cell death) in comparison to vehicle-treated controls. This was further supported by the observation that there was no difference in the number of live cells in control and rapamycin-treated cells, when viability was assessed by trypan blue exclusion assay (). For experiments with AC16 cells, cells were either treated with AMA alone or coincubated with rapamycin, and viability was assessed by trypan blue assay at the end of 48 h. Viability assay showed a similar protective effect of rapamycin against AMA-mediated cytotoxicity in AC16 cells ().
Figure 5A–C. Rapamycin protects against the cytotoxic effects of AMA in HL-1 and AC16 cardiomyocytes. (A and B) HL-1 cells were pre-treated with vehicle control or 1 µM rapamycin for 16 h, followed by incubation with 50 µM AMA for an additional 32 h, in the absence or presence of rapamycin. Cell viability was subsequently assessed using trypan blue exclusion assay (A) or cell lysates were collected and immunoblotted for PARP1 (B). GAPDH is used as a loading control. (C). HL-1 cells were pretreated with vehicle control or 1 µM rapamycin for 16 h, followed by incubation with 50 µM AMA for an additional 24 h, in the absence or presence of rapamycin. Phase contrast images of cells were subsequently taken. Representative images are shown.
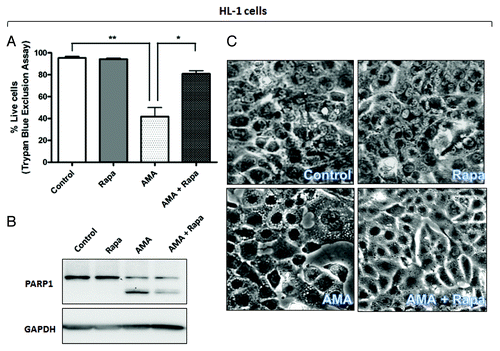
Figure 5D–F. (D and E) AC16 cells were treated with rapamycin or AMA alone or were coincubated with AMA and rapamycin for 48 h. Cell viability was subsequently assessed using trypan blue exclusion assay (D) or cell lysates were collected and immunoblotted for PARP1 (E). (F) AC16 cells were treated with rapamycin or AMA alone or were coincubated with AMA and rapamycin for 36 h. Phase contrast images of cells were subsequently taken. Representative images are shown. Rapa, rapamycin. *p < 0.05; **p < 0.01 vs. control (for A, previous page, and D). Data represent three independent experiments.
AMA-induced cell death in HL-1 cells was characterized by poly (ADP-ribose) polymerase-1 (PARP1) cleavage and CASP3 activation. We observed that rapamycin treatment attenuated such proteolytic processing (; Fig. S4B). Similar results were obtained in AC16 cells (). Additionally, AMA-treated HL-1 cells showed intense vacuolization in the cytoplasm (), a phenomenon often observed with injury, toxicity and cell death.Citation40 Treatment with rapamycin drastically decreased the appearance of such vacuoles (). AMA-treated AC16 cells showed a swollen, rounded morphology and this was prevented by cotreatment with rapamycin ()
Rapamycin prevents AMA-induced accumulation of ubiquitinated proteins
The accumulation of high molecular weight ubiquitinated proteins has been observed in conditions of increased oxidative stress and can originate from an inefficient clearance of damaged proteins through the proteasomal system.Citation41,Citation42 Under these conditions, autophagy induction can mediate the removal of accumulated proteins, averting potential proteotoxicity. We found that AMA induced the accumulation of ubiquitinated proteins in the detergent-insoluble fraction of HL-1 cells and that treatment with rapamycin drastically prevented such an accumulation (Fig. S5). Also, in comparison to vehicle-treated control cells, we observed a decreased accumulation of ubiquitinated proteins in cells treated with rapamycin, which could be attributed to the autophagic clearance of basal levels of uniquitinated proteins by rapamycin.
Rapamycin protects against AMA-induced mitochondrial dysfunction
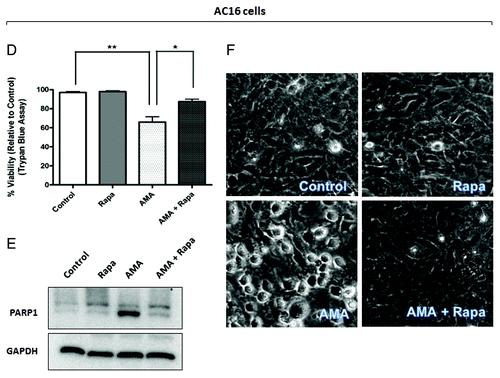
Since mitochondrial clearance was promoted by rapamycin in the presence of AMA, we further investigated whether rapamycin offered protection against mitochondrial dysfunction in AMA-treated cells. A major function of the mitochondria is oxidative phosphorylation, which requires the consumption of O2. Assessing O2 consumption (cellular respiration) in nonpermeabilized cells using polarographic oxygen sensors accurately measures mitochondrial function.Citation43 We found that AMA-treated cells significantly decreased routine respiration even 24 h after treatment (; p < 0.01). This decline in respiration was ameliorated by rapamycin treatment (p < 0.05).
Figure 6. Rapamycin protects against AMA-induced mitochondrial depolarization and respiration dysfunction in HL-1 cardiomyocytes. (A) Cells were pretreated with vehicle control or 1 µM rapamycin for 16 h, followed by treatment with 50 µM AMA for an additional 24 h, in the absence or presence of rapamycin. Cells were subsequently trypsinized, resuspended in Claycomb media and routine respiration analyzed. Data are derived from three independent experiments. *p < 0.05; **p < 0.01. (B) Cells were pretreated with vehicle control or 1 µM rapamycin for 16 h, followed by incubation with 50 µM AMA for an additional 24 h, in the absence or presence of rapamycin. Cells were subsequently stained with 50 nM TMRM and imaged using epifluorescence microscope. Representative images are shown. Arrows represent cells with no Δψm. Arrowheads represent swollen mitochondria. Rapa; rapamycin.
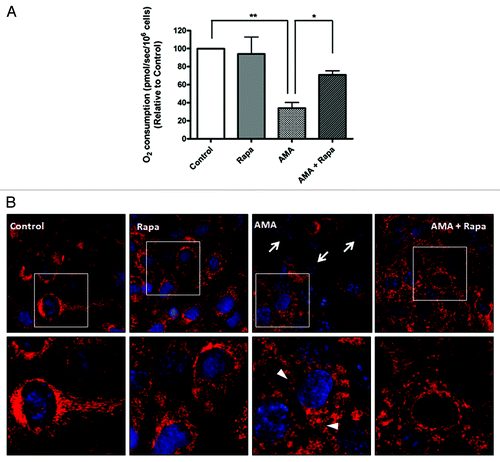
We also measured Δψm as an additional indicator of mitochondrial function. In comparison to control or rapamycin-treated cells, AMA-treated mitochondria had little or no Δψm even 24 h after treatment (, arrows). The few cells that maintained Δψm showed drastically altered mitochondrial morphology, most of them being highly swollen and relatively larger in size (, arrowheads, lower panel). Incubation with rapamycin restored Δψm in AMA-treated cells, as well as maintained normal mitochondrial morphology ().
Inhibition of autophagy blocks the cytoprotective effects of rapamycin against AMA
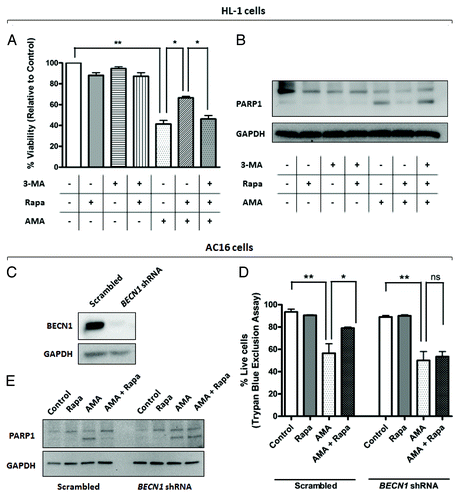
To determine whether the cytoprotective effect of rapamycin against AMA toxicity is mediated through autophagy, we inhibited autophagy by both pharmacological and shRNA approaches. Autophagy was inhibited in HL-1 cells by coincubating cells with the PtdIns3K inhibitor 3-Methyladenine (3-MA), a widely used inhibitor of autophagy.Citation44,Citation45 LC3-II/LC3-I ratios confirmed that 3-MA inhibited rapamycin-induced autophagy (Fig. S6A and S6B). Furthermore, treatment with 3-MA decreased rapamycin-induced colocalization of mitochondrial and autophagic markers in AMA-treated cells (Fig. S6C). We subsequently performed MTT assay to determine the viability of AMA-treated cells coincubated with rapamycin alone or with rapamycin in the presence of 3-MA (). Viability assay revealed that 3-MA attenuated the cytoprotective effects of rapamycin against AMA toxicity (). In addition, 3-MA inhibited rapamycin-induced attenuation of PARP1 and CASP3 cleavage in AMA-treated cells (; Fig. S6D). For inhibition of autophagy in AC16 cells, we employed shRNA approach to knock down the autophagy gene, BECN1. Protein expression analysis confirmed drastic decreases in BECN1 levels in cells treated with BECN1 shRNA in comparison to scrambled-treated control (). Decreased BECN1 levels attenuated rapamycin-induced autophagy (Fig. S7A and S7B) and inhibited rapamycin-mediated protection against AMA toxicity, as assessed by trypan blue exclusion assay () and PARP1 proteolytic processing ().
Figure 7. Inhibition of autophagy attenuates rapamycin-mediated protection against AMA toxicity in HL-1 and AC16 cardiomyocytes. (A) HL-1 cells were pre-treated with vehicle control or 1 µM rapamycin in the absence or presence of 5 mM 3-MA for 16 h, followed by treatment with 50 µM AMA for an additional 32 h, in the presence of rapamycin and 5mM 3-MA. Cell viability was subsequently assessed using MTT assay. *p < 0.05; **p < 0.01. Data represent three independent experiments. (B) HL-1 cells were treated as in (B), cell lysates collected and immunoblotted for PARP1. GAPDH is used as a loading control. (C) AC16 cells were transduced with a lentiviral vector expressing scrambled or BECN1 shRNA. Cells were subsequently lysed and immunoblotted for BECN1 protein expression. GAPDH is used as a loading control. (D) Scrambled or BECN1 shRNA-expressing cells were treated with AMA alone or were coincubated with rapamycin for 48 h. Cell viability was subsequently assessed using trypan blue exclusion assay. *p < 0.05; **p < 0.01.Data represent three independent experiments. (E) AC16 cells were treated as in (D), cell lysates collected and immunoblotted for PARP1. GAPDH is used as a loading control.
Discussion
Pathological conditions can cause the mitochondria to generate elevated amounts of ROS, inflicting macromolecular damage and affecting cellular homeostasis and function in several organs, including the heart.Citation1,Citation46 Mitochondria from heart failure models generate larger amounts of O2•− in the presence of NADH compared with normal mitochondria.Citation47 Oxidative stress can activate signaling pathways involved in myocardial remodeling, affecting cardiomyocyte structure and function.Citation47 Our study showed that the mitochondrial stressor AMA can mimic several features observed under mitochondrial dysfunction-associated pathological conditions. For instance, AMA treatment generated O2•− in the mitochondria and decreased Δψm in HL-1 cardiomyocytes. A decreased Δψm is indicative of defective oxidative phosphorylation and a lower capacity of generating ATP,Citation48 and is used as a marker of cellular and mitochondrial viability.Citation49 It is however worthwhile to note that, to counteract oxidative stress, the mitochondria and the cytoplasm are equipped with detoxifying enzymes and non-enzymatic antioxidants.Citation50 Therefore, although oxidants are constantly being generated by the mitochondria, the antioxidant defense system is capable of detoxifying them, preventing damage to cytoplasmic components.Citation50 Nevertheless, under conditions of stress and disease pathology, damaged mitochondria can generate excessive amounts of ROS, which can overwhelm the defenses and oxidatively modify extramitochondrial components, such as DNA and RNA. We confirmed that ROS generated by AMA can increase DNA oxidation, as determined by the levels of guanine base oxidation product, 8-oxo-dGuo. 8-oxo-dGuo is potentially mutagenicCitation51,Citation52 and is widely used for the estimation of oxidative stress in tissues and urine.Citation53,Citation54 We further observed that most cells treated with AMA had little or no Δψm and the mitochondria were highly swollen. Such aberrant mitochondria, often also characterized by loss of cristae and matrix derangements, has been observed through ultrastructural analysis of the myocardium of aged rodentsCitation55 and in other models of mitochondrial injury,Citation56 and is considered a marker of mitochondrial toxicity.Citation57,Citation58 Furthermore, AMA decreased cell viability and activated apoptotic markers, as well as inhibited cellular respiration in cardiomyocytes, further increasing cytotoxicity ().
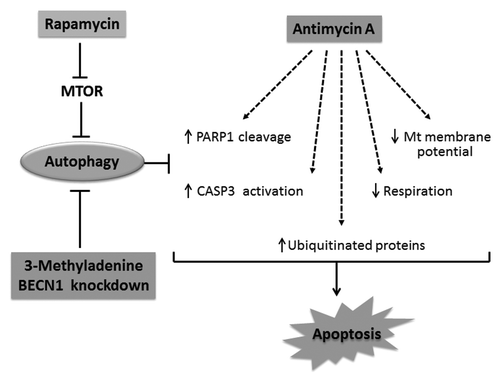
Figure 8. Schematic representation of the protective effects of rapamycin against AMA toxicity. AMA induces PARP1 cleavage and CASP3 activation, decreases respiration, mitochondrial membrane potential as well as mediates the accumulation of ubiquitinated proteins, all of which culminates in apoptotic induction. Rapamycin treatment induces autophagy by removing the inhibitory effect of MTOR on autophagy induction. Stimulation of autophagy suppresses cytotoxic markers induced by AMA. On the other hand, inhibition of rapamycin-induced autophagy by pharmacological (3-MA) or genetic (shRNA) interventions blocks the cytoprotective effects of rapamycin.
Since oxidative stress is postulated to be a major player in the pathogenesis of cardiovascular diseases, one might argue that the administration of antioxidants will offer protection against oxidant-mediated damage. However, clinical studies have been controversial, with limited or no cardiac benefit reported with chronic administration of antioxidants such as vitamin A, vitamin E or β-carotene.Citation59-Citation61 Therefore approaches to remove oxidatively modified macromolecules through autophagy induction represent an appealing alternative. In our study, we observed that rapamycin stimulated autophagy in HL-1 and AC16 cardiomyocytes (; Fig. S3). Importantly, some findings suggest that mitochondrial stress and ROS per se can act as inducers of autophagy.Citation19,Citation34,Citation62 Such autophagic induction could be thought of as an adaptive response of the cell to mediate the clearance of oxidatively damaged components. However, other studies have shown that mitochondrial dysfunction and depolarization might not be sufficient to induce autophagy.Citation63 In our studies, we could not detect any induction of autophagy by treatment with AMA, in both HL-1 and AC16 cardiomyocytes, as assessed by LC3B immunoblotting, GFP-LC3 puncta formation and SQSTM1 protein levels. Rapamycin was able to induce autophagy and mitophagy HL-1 cells () and mediate the clearance of SQSTM1 in AMA-treated cells (), suggesting that the autophagic machinery may not have been impaired by the oxidants generated in AMA-treated cells. It is possible that additional triggers might be necessary for the induction of autophagy, but missing in AMA-treated cells.Citation63 Furthermore, it has been proposed that the inhibition of ETC complex III can actually have an inhibitory effect on autophagy induction, although the molecular mechanisms are as of yet, unknown.Citation64 We also cannot rule out the possibility that the inability to induce autophagy could be cardiomyocyte specific. Nevertheless, stimulating autophagy under oxidative stress conditions could be hypothesized to potentially remove oxidized macromolecules. We tested this hypothesis by additionally incubating AMA-treated cells with the autophagy inducer, rapamycin.
Indeed, we observed that rapamycin-stimulated autophagy had a beneficial effect against AMA-induced toxicity, as assessed by the attenuation of apoptotic signaling molecules, as well as by an improved survival of HL-1 and AC16 cardiomyocytes (). Rapamycin treatment also inhibited the decrease in Δψm as well as the appearance of swollen morphology of the mitochondria in HL-1 cells ( and ). Additionally, respiration analysis in nonpermeabilized HL-1 cells showed that autophagy induction by rapamycin attenuated the decline in cellular respiration induced by AMA ( and ). These protective effects of rapamycin could be partly due to the result of autophagy-mediated removal of dysfunctional mitochondria, which if not removed, can produce greater amounts of ROS and/or induce apoptosis through the release of proapoptotic factors usually sequestered in the intermembrane space.Citation17 The encapsulation and degradation of dysfunctional mitochondria by rapamycin-induced autophagy has been previously reported in numerous studies.Citation65,Citation66 Consistent with these studies, we have observed colocalization of mitochondrial and autophagic markers in rapamycin-treated cells (). The occurrence of mitophagy in rapamycin-treated cells (in the absence or presence of AMA) was however modest, with colocalization being detected at a rate of mostly 2 to 4 per cell. The apparently low level of mitophagy could be attributed to the fact that such experiments were conducted under steady-state conditions, rather than under conditions of inhibited downstream degradation. However, in comparison to the mitophagy observed in cells treated with AMA and rapamycin, treatment with AMA alone showed no colocalization of mitochondrial and autophagosomal markers, confirming that rapamycin enhanced mitophagy. In addition to colocalization experiments, mitochondrial clearance in rapamycin-treated cells was also confirmed by assessing VDAC1 (mitochondrial outer membrane protein) and cyt c (mitochondrial inter membrane space protein) levels (; Fig. S3C). However, it is worth noting that although colocalization of mitochondrial and autophagosomal markers was observed in rapamycin-treated cells, we cannot conclude that the autophagic cargo was mitochondria-specific. Given the fact that rapamycin is a general inducer of autophagy, it is possible that the mitochondria were cleared along with other autophagic cargo, such as aggregated ubiquitinated proteins, in the same autophagic vesicle. Nevertheless, the clearance of damaged and dysfunctional mitochondria was supported by respiration analysis (). We observed that respiration was inhibited to the same extent in AMA and AMA plus rapamycin treated cells 2 h after treatment (data not shown). However, after 24 h, respiration was significantly increased in AMA-treated cells additionally exposed to rapamycin, suggesting that some of the dysfunctional mitochondria might have been removed by autophagy during this time period. Furthermore, Δψm imaging analysis showed that the accumulation of dysfunctional (lacking Δψm) and swollen mitochondria was seen only in AMA-treated cells; such damaged mitochondria could not be detected in cells additionally treated with rapamycin. Finally, in order to establish that the protective effects of rapamycin against AMA toxicity are autophagy specific, we used both pharmacological (3-MA) and genetic (shRNA) approaches to inhibit autophagy. Treatment of HL-1 cells with 3-MA, known to inhibit class III PtdIns3K,Citation44,Citation45 not only inhibited rapamycin-induced autophagy and mitophagy (Fig. S6A–C), but also attenuated the beneficial effects of rapamycin against AMA toxicity (; Fig. S6D). Knockdown of autophagy protein BECN1 using shRNA approach in AC16 cells also attenuated rapamycin-induced autophagy (Fig. S7A and S7B) and ameliorated the protective effects of rapamycin against AMA toxicity ().
Similar to the mitochondria, an increase in the ‘dwell time’ of oxidatively modified cytoplasmic proteins enhances their probability of being post-translationally modified, which could further increase cellular toxicity.Citation67,Citation68 Damaged cellular components are marked by ubiquitination, to be subsequently removed by the ubiquitin-proteasome system (UPS).Citation41 Growing evidence suggests that under conditions of increased oxidative burden, accumulation of high molecular weight ubiquitinated proteins is frequently observed, owing to their inefficient removal through the UPS system.Citation41,Citation42,Citation69 Autophagy induction under these circumstances has been shown to mediate the removal of such accumulated proteins. In our studies, rapamycin-induced autophagy attenuated the accumulation of ubiquitinated proteins that resulted from AMA-induced exposure. Such an accumulation was specifically detected in the insoluble fraction, suggesting that they might be aggregated in nature. Notably, our observation is consistent with studies showing that autophagy induction alleviates toxicity in cells expressing mutant aggregate-prone proteins such as SNCACitation21 and HTT.Citation21,Citation70
Our data using BECN1 knockdown in AC16 cells suggested that rapamycin-induced protection from AMA was mediated by autophagy. However, it is possible that prolonged periods of incubation with rapamycin activated the phosphatidylinositol 3-kinase (PtdIns3K) pathway, resulting in an antiapoptotic effect against AMA, independent of autophagy. Inhibiting autophagic degradation by siRNA-mediated knockdown of ATG5 or ATG7 (PtdIns3K-independent) in AMA-treated cells would best address this possibility. However, our observations of enhanced mitochondrial clearance, improved Δψm and respiration in rapamycin-treated cells suggested that the beneficial effects were likely mediated through autophagy. Consistent with our observations on the beneficial role of autophagy, other studies have shown that autophagy plays a protective role against anoxia-regeneration in isolated cardiomyocytes,Citation71 as well as required for the preconditioning effect of adenosine.Citation72 On the other hand, inhibition of autophagy through cardiac specific deletion of the gene encoding ATG5 results in mitochondrial misalignment and cardiac hypertrophy in response to pressure overload induced by transverse aortic constriction. This eventually progresses into left ventricular hypertrophy, contractile dysfunction and cardiac dilatation.Citation73 Inhibition of the autophagic clearance pathway has also been implicated in the pathogenesis of Danon disease, which is characterized by cardiomyopathy and heart failure.Citation74
In summary, it is worthwhile to note that cardiomyocytes are terminally differentiated; therefore any damages accumulated during its life span of several decades cannot be diluted by cell proliferation. Furthermore, the heart is highly metabolic and hence challenged with a considerable amount of oxidative stress during its lifetime. Given the host of cardiac pathologies that can result from the accumulation of damaged mitochondria (or from the oxidants generated thereof), the maintenance of a functional pool of mitochondria, coupled with the removal of potentially toxic, oxidatively modified cellular components, are vital for the preservation of cardiomyocyte homeostasis. It therefore stands reasonable to assume that the housekeeping property of autophagy will play a protective role. However, although oxidative stress and damaged mitochondria have been proposed as inducers of autophagy and mitophagy, it is possible that the disease pathology prevents sufficient induction of the autophagic process. We showed that under such circumstances, inducing autophagy by rapamycin can mediate the removal of dysfunctional mitochondria as well as aggregated, ubiquitinated proteins in cardiomyocytes, leading to an overall protective effect of autophagy induction against oxidative stress (). We therefore propose rapamycin as a potential therapeutic strategy against cardiovascular disorders associated with chronic generation of oxidation stress.
Materials and Methods
Cell culture conditions
HL-1 mouse atrial cardiomyocytes were a kind gift of Dr. W. Claycomb (Louisiana State University Health Science Center) and were cultured in Claycomb Media (JRH Biosciences, 51800C) supplemented with 10% fetal bovine serum (Sigma, F2442), 2 mM L-glutamine (Life Technologies, 25030-081), 0.1 mM norepinephrine (Sigma, A0937), 100 U/ml penicillin and 100 mg/ml streptomycin (Life Technologies, 15140-122), on fibronectin-gelatin coated plates. Human ventricular AC16 cells were obtained from ATCC (Designation No. PTA-1500) and were cultured in DMEM media (Mediatech Cellgro, 10-090-CV) with 12.5% FBS (Hyclone, SH30910-03), 100 U/ml penicillin and 100 mg/ml streptomycin (Life Technologies, 15140-122). Both cell lines were cultured at 37°C in a humidified atmosphere, with 5% CO2.
Chemical reagents
AMA (Sigma, A8674) was dissolved in ethanol at a concentration of 15 mM; Rapamycin (Calbiochem, 553211) was made up in ethanol at 5 mM; BafA1 (Sigma, B1793) was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 25 µM and 3-MA (Sigma, M9281) was dissolved in deionized water at 180 mM.
Flow cytometric determination of mitochondrial O2•− generation
O2•− generation in the mitochondria was analyzed using the fluorescence dye MitoSOX Red (Invitrogen, M36008). It is specifically targeted to bioenergetically active mitochondria where it is rapidly and selectively oxidized by O2•− molecules. The oxidized product binds to mitochondrial DNA and fluoresces red.Citation31 HL-1 cells were trypsinized and resuspended in fresh media at a density of 2 × 106 cells/ml and MitoSOX Red was added to a final concentration of 3 μM. Cells were allowed to load MitoSOX Red for a period of 30 min at 37°C, washed twice with Dulbecco’s Phosphate Buffered Saline (DPBS) and the media replaced with fresh media. Cells were subsequently incubated with increasing concentrations of AMA or vehicle control for 30 min at 37°C, followed by flow cytometric analysis of MitoSOX Red fluorescence using FACSCalibur flow cytometer (BD Biosciences). MitoSOX Red was excited by laser at 488 nm and the data collected using forward scatter (FSC) and side scatter (SSC) and FL2 (575 ± 12.5 nm) channels. Cell debris is represented by distinct low FSC and SSC and was gated out for analysis. The data shown represents mean MitoSOX Red fluorescence intensity of 10,000 HL-1 cells treated with the indicated concentrations of AMA.
Confocal imaging of mitochondrial O2•− generation
HL-1 cells were plated on No. 1.5 coverslip-glass bottom dishes (MatTek Corporation) and were incubated with 3 μM MitoSOX Red and 1 µg/ml Hoechst 33342 (Invitrogen, H3570) for a period of 30 min at 37°C, washed twice with DPBS and the media replaced with fresh media. Cells were subsequently treated with 50 µM AMA or vehicle control for 30 min and imaged for MitoSOX Red fluorescence using Leica TCS SP2 Laser Scanning Confocal Microscope (Leica Microsystems GmbH) using an excitation of 514 nm and emission of 560 to 600 nm. Hoechst 33342 was visualized using 405 nm excitation and 440 to 470 nm emission. The pictures were merged using Leica LCS software (Leica Microsystems GmbH).
Flow cytometric determination of mitochondrial membrane potential (Δψm)
Δψm was analyzed using the cationic fluorescent dye, TMRM (Invitrogen, T668), which is specifically targeted to and accumulates in bioenergetically active mitochondria.Citation32 HL-1 cells were trypsinized and resuspended in fresh media at a density of 2 × 106 cells/ml, after which TMRM was added to a final concentration of 50 nM. Cells were allowed to load TMRM for a period of 30 min at 37°C, washed twice with DPBS and the media replaced with fresh media. Cells were subsequently incubated with increasing concentrations of AMA or vehicle control for 2 h at 37°C, followed by flow cytometric analysis of TMRM fluorescence using the same excitation-emission and gating protocol as that of MitoSOX Red.
Confocal imaging of Δψm
HL-1 cells were incubated with 50 nM TMRM and 1 µg/ml Hoechst 33342 at 37°C for a period of 30 min, washed twice with DPBS and the media replaced with fresh media. Cells were subsequently treated with 50 µM AMA or vehicle control for 2 h and imaged for TMRM fluorescence using Leica TCS SP2 Laser Scanning Confocal Microscope with an excitation of 543 nm and emission of 560 to 600 nm. The pictures were merged using Leica LCS software.
Determination of cell viability
Cell viability was determined by MTT (Sigma, M5655) and trypan blue dye exclusion assay (Sigma, T8154). To determine cell viability by MTT assay, cells in 24-well plates were incubated with 0.5 mg/ml MTT for a period of 30 min at 37°C. The purple formazan crystals formed were dissolved in DMSO at 37°C for 10 min and the absorbance of the resulting solution measured at 540 nm using Biotek Synergy plate reader (Biotek). The background absorbance was subtracted at 630 nm. The values represented are normalized to control vehicle-treated group. To determine cell viability by trypan blue assay, cells were trypsinized and resuspended in fresh media containing equal volume of 0.4% trypan blue for 5 min and the number of live (bright) or dead (dark) cells counted using a hemocytometer.
Determination of DNA/RNA oxidation
8-oxo-dGuo (DNA oxidation) and 8-oxo-Guo (RNA oxidation) concentrations were measured in cells using HPLC coupled to electrochemical detection (HPLC-ECD) analysis (ESA Coulochem III electrochemical detector) according to protocol described before.Citation33 Briefly, HL-1 cells were treated with AMA or vehicle control for 2 h, and were trypsinized 24 h later. Cells were subsequently homogenized in 3 M guanidium thiocyanate lysis buffer (GTC, 0.2% w/v N-lauroylsarcosinate, 20 mM Tris, pH 7.5) in the presence of the metal chelator deferoxamine mesylate (DFOM; Sigma, D9533) to prevent environmental oxidation during processing. Cellular DNA/RNA were isolated using phenol-chloroform method and precipitated in isopropanol. The nucleic acid pellet was subsequently washed with ethanol, air-dried and dissolved in 10 mM DFOM followed by hydrolysis in 10 ul Nuclease P1 (stock of 0.4 U/µl in 300 mM sodium acetate, 0.2 mM ZnCl2, pH 5.3; MP Biomedicals, N8630) and 5 ul alkaline phosphatase (1 U/µl; Sigma, P6774-10KU) for a period of 60 min at 50°C. The hydrolysate was filtered to remove enzymes and subsequently loaded for HPLC-ECD analysis. For quantification, HPLC peaks were compared with calibration curves of 8-oxo-dGuo and dGuo standards (Sigma, D7154). The amount of 8-oxo-Guo and 8-oxo-dGuo present was normalized to the total amount of non-oxidized DNA and RNA, respectively.
Induction and assessment of autophagy by immunoblotting
Autophagic stimulation converts the cytosolic and unconjugated form of LC3 (LC3-I) to the carboxy terminal-cleaved, phosphatidylethanolamine (PE) conjugated form (LC3-II) and this causes a change in the molecular weight of the protein, allowing both forms to be resolved in an SDS-PAGE gel. The LC3-II/LC3-I ratio is therefore indicative of the amount of ongoing autophagy. However, an increase in the LC3-II/LC3-I ratio could be due to increased autophagic stimulation or due to a decreased clearance of autophagic vacuoles in the lysosomes. Hence all autophagic responses were monitored in the absence or presence of the lysosomal inhibitor BafA1. We measured the rate of autophagosome formation (autophagic stimulation) by comparing LC3-II/LC3-I ratios under +BafA1 conditions. This method has been extensively used previously for measuring autophagic stimulation.Citation35-Citation37 In addition, we measured autophagic flux by subtracting LC3-II/LC3-I ratio under –BafA1 conditions from than of +BafA1 conditions.
We used rapamycin to induce autophagy in HL-1 and AC16 cells. To determine the optimal concentration of rapamycin for inducing autophagy in HL-1 cardiomyocytes, cells were treated with increasing concentrations of the drug for a period of 16 h in the presence or absence of 75 nM BafA1, added during the final 4 h of incubation. For subsequent experiments involving rapamycin-mediated induction of autophagy in HL-1 cells, a concentration of 1 μM was used. For AC16 cells, a concentration of 1 μM rapamycin was used for all experiments.
Assessment of autophagy by epifluorescence imaging
For assessment of autophagic activity using epifluorescence imaging, cells plated on coverslips in 24-well plates were transfected with GFP-LC3 plasmid (kind gift of Dr. T. Yoshimori, National Institute of Genetics)Citation75 using Lipofectamine 2000 (Invitrogen, 11668-027) following manufacturer’s instructions. To prevent the potential problem of GFP-LC3 aggregate formation,Citation76 transfection was optimized with a low concentration of GFP-LC3 plasmid (0.5 μg for each well of a 24-well plate). Twenty-four hours after transfection, cells were incubated with 1 μM rapamycin for 16 h, in the absence or presence of 75 nM BafA1 added during the final 4 h of incubation. Cells were subsequently fixed in 4% paraformaldehyde and mounted on slides with DAPI (Vector Laboratories, H-1200). Epifluorescence images were taken using Leica DM IRBE epifluorescence microscope (Leica Microsystems) and images were processed using ImageJ software (NIH). Conversion of diffuse cytosolic GFP-LC3-I to punctate autophagic membrane-associated GFP-LC3-II was monitored and considered indicative of autophagic activity in cells. Specifically, the number of puncta in each cell was counted and cells with 30 or more puncta were considered to have induced autophagy.
Western blotting
Cells were lysed using RIPA buffer (Sigma, R0278) and protein concentration determined using Dc assay (Biorad, 500-0113).Citation77 Equal amounts of proteins were then loaded in pre-cast Tris-HCl polyacrylamide gels (Biorad). After electrophoretic separation, proteins were transferred to polyvinylidene difluoride (PVDF) membrane (Biorad, 162-0177) and subsequently blocked for 1h in Starting Block (Thermo Scientific, 37543), followed by overnight incubation with primary antibodies. Membranes were subsequently washed with Tris buffered saline with 0.1% Tween 20 (TBST) and incubated with the appropriate secondary antibodies for 1 h. Membranes were washed again with TBST and chemiluminescent signals developed using ECL Plus reagent (Amersham Biosciences). The signals were captured using ChemiDoc XRS System (Biorad) and digital images analyzed using ImageLab software (Biorad). The primary antibodies used are as follows: rabbit anti-LC3B (Cell Signaling, 2775), rabbit anti-BECN1 (Cell Signaling, 3738), rabbit anti-PARP1 (Cell Signaling, 9542), rabbit anti-CASP3 (Cell Signaling, 9662), rabbit anti-phospho-RPS6 (Cell Signaling, 2211), mouse anti-RPS6 (Cell Signaling, 2317), rabbit anti-ubiquitin (Cell Signaling, 3936), rabbit anti-SQSTM1 (Cell Signaling, 5114), rabbit anti-cyt c (Cell Signaling, 4272), mouse anti-ATG5 (Sigma, A2859), mouse anti-VDAC1 (Mitoscences, MSA03), rabbit anti-TUBB/β-tubulin (Sigma, T2200) and GAPDH-HRP (Sigma, G9295). HRP conjugated secondary antibodies were anti-rabbit (GE-Amersham, NA934V) or anti-mouse (GE-Amersham, NA931V).
For detection of ubiquitinated proteins, cells were lysed and detergent soluble and insoluble fractions separated by centrifugation at 8000 g. The resulting pellets (insoluble fraction) were resuspended in 2× SDS buffer and heated at 100°C for 10 min. Equal volumes of sample lysates were then loaded in each lane. The supernatant (soluble fraction) was processed for immunoblotting as mentioned above.
Assessment of cellular respiration
HL-1 cells were pre-treated with 1 µM rapamycin for 16 h, followed by incubation with 50 µM AMA or vehicle control for an additional 24 h, in the presence of rapamycin. They were subsequently trypsinized and resuspended in complete Claycomb media. The O2 flux, as a measure of routine respiration was subsequently measured in unpermeabilized cells using OROBOROS Oxygraph-2K (Oroboros Instruments GmbH) at 37°C in a constantly stirred chamber. The rates of consumption are expressed as pmol/sec/106 cells.
shRNA knockdown
Stable knockdown of BECN1 protein in AC16 cells were performed using lentivirus-mediated delivery of short hairpin RNA (shRNA). Lentiviral constructs containing BECN1 shRNA clones were obtained from Open Biosystems (Accession number: RHS4533-NM-003766, clone IDs: TRCN0000033549, TRCN0000033550, TRCN0000033551, TRCN0000033552, TRCN0000033553). Scrambled RNA-coding lentiviral vector was obtained from Addgene (Plasmid ID 1864, shRNA coding DNA sequence: CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG). Scrambled or BECN1 shRNA-encoding lentiviral vectors were transfected into 293T cells together with packaging plasmids psPAX2 and pMD2.G, using Lipofectamine 2000 (Invitrogen, 11668-027), following the manufacturer’s instructions. Culture supernatant containing pseudovirus was collected and mixed with polybrene, filtered through 0.45 μm filter and used to transduce AC16 cells. Twenty-four hours after transduction, media was removed and the cells were cultured in the presence of 2 μg/μl puromycin to select lentivirus-transduced cells.
Statistics
All statistical analyses were performed using GraphPad Prism 4 (GraphPad Software, Inc.). For statistical analysis, Student’s t-test and one-way ANOVA were performed, wherever applicable. Statistical significance was set to p < 0.05 and the data are represented as mean ± SE.
| Abbreviations: | ||
| 3-MA | = | 3-methyladenine |
| 8-oxo-dGuo | = | 8-oxo-7,8-dihydro-2′-deoxyguanosine |
| 8-oxo-Guo | = | 8-oxo-7,8-dihydroguanosine |
| Δψm | = | mitochondrial membrane potential |
| AMA | = | antimycin A |
| ETC | = | electron transport chain |
| BafA1 | = | bafilomycin A1 |
| cyt c | = | cytochrome c |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| MTOR | = | mechanistic target of rapamycin |
| MTT | = | 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PARP1 | = | poly (ADP-ribose) polymerase-1 |
| ROS | = | reactive oxygen species |
| TMRM | = | tetramethyl rhodamine methyl ester, TUBB, β-tubulin |
| VDAC 1 | = | voltage-dependent anion channel |
| mCAT | = | catalase targeted to the mitochondria |
Additional material
Download Zip (1.7 MB)Acknowledgments
This work was supported by NIH grants DK090115 (J.-S.K., C.L.), DK079879 (J.-S.K.) and American Heart Greater Southeast Affiliate Fellowship (10PRE4310091) to D.D. We are grateful to Debra Akin (University of Florida, USA) for helping us with autophagy assessment using GFP-LC3 plasmid and to Gauthami Balagopal for her help with general labwork. We thank the Cell and Tissue Analysis Core at the University of Florida for the confocal imaging analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/22971
References
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 2005; 120:483 - 95; http://dx.doi.org/10.1016/j.cell.2005.02.001; PMID: 15734681
- Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J 2005; 19:419 - 21; PMID: 15642720
- Grivennikova VG, Kareyeva AV, Vinogradov AD. What are the sources of hydrogen peroxide production by heart mitochondria?. Biochim Biophys Acta 2010; 1797:939 - 44; http://dx.doi.org/10.1016/j.bbabio.2010.02.013; PMID: 20170624
- Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci 2008; 1147:37 - 52; http://dx.doi.org/10.1196/annals.1427.015; PMID: 19076429
- Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol 2010; 45:466 - 72; http://dx.doi.org/10.1016/j.exger.2010.01.003; PMID: 20064600
- Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem 2012; 287:4434 - 40; http://dx.doi.org/10.1074/jbc.R111.271999; PMID: 21832045
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006; 443:787 - 95; http://dx.doi.org/10.1038/nature05292; PMID: 17051205
- Judge S, Leeuwenburgh C. Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am J Physiol Cell Physiol 2007; 292:C1983 - 92; http://dx.doi.org/10.1152/ajpcell.00285.2006; PMID: 17344313
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A 1997; 94:514 - 9; http://dx.doi.org/10.1073/pnas.94.2.514; PMID: 9012815
- Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H. Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech Ageing Dev 1994; 74:121 - 33; http://dx.doi.org/10.1016/0047-6374(94)90104-X; PMID: 7934203
- Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J 2000; 14:312 - 8; PMID: 10657987
- Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res 2011; 90:234 - 42; http://dx.doi.org/10.1093/cvr/cvr015; PMID: 21257612
- Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia--reperfusion, aging, and heart failure. J Mol Cell Cardiol 2001; 33:1065 - 89; http://dx.doi.org/10.1006/jmcc.2001.1378; PMID: 11444914
- Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009; 119:2789 - 97; http://dx.doi.org/10.1161/CIRCULATIONAHA.108.822403; PMID: 19451351
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823 - 30; http://dx.doi.org/10.1038/ncb0910-823; PMID: 20811354
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11:621 - 32; http://dx.doi.org/10.1038/nrm2952; PMID: 20683470
- Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 2005; 1:131 - 40; http://dx.doi.org/10.4161/auto.1.3.2017; PMID: 16874025
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749 - 60; http://dx.doi.org/10.1038/sj.emboj.7601623; PMID: 17347651
- Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 2010; 6:838 - 54; http://dx.doi.org/10.4161/auto.6.7.12113; PMID: 20505317
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36:585 - 95; http://dx.doi.org/10.1038/ng1362; PMID: 15146184
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992 - 2003; http://dx.doi.org/10.1091/mbc.E08-12-1249; PMID: 19225151
- Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol 2006; 41:256 - 64; http://dx.doi.org/10.1016/j.yjmcc.2006.04.014; PMID: 16769083
- McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 2004; 109:3050 - 5; http://dx.doi.org/10.1161/01.CIR.0000130641.08705.45; PMID: 15184287
- Harada M, Hanada S, Toivola DM, Ghori N, Omary MB. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology 2008; 47:2026 - 35; http://dx.doi.org/10.1002/hep.22294; PMID: 18454506
- Nakayama K, Okamoto F, Harada Y. Antimycin A: isolation from a new Streptomyces and activity against rice plant blast fungi. J Antibiot (Tokyo) 1956; 9:63 - 6; PMID: 13345726
- Potter VR, Reif AE. Inhibition of an electron transport component by antimycin A. J Biol Chem 1952; 194:287 - 97; PMID: 14927618
- Pham NA, Robinson BH, Hedley DW. Simultaneous detection of mitochondrial respiratory chain activity and reactive oxygen in digitonin-permeabilized cells using flow cytometry. Cytometry 2000; 41:245 - 51; http://dx.doi.org/10.1002/1097-0320(20001201)41:4<245::AID-CYTO2>3.0.CO;2-#; PMID: 11084609
- Park WH, Han YW, Kim SH, Kim SZ. An ROS generator, antimycin A, inhibits the growth of HeLa cells via apoptosis. J Cell Biochem 2007; 102:98 - 109; http://dx.doi.org/10.1002/jcb.21280; PMID: 17372917
- Park WH, Han YW, Kim SW, Kim SH, Cho KW, Kim SZ. Antimycin A induces apoptosis in As4.1 juxtaglomerular cells. Cancer Lett 2007; 251:68 - 77; http://dx.doi.org/10.1016/j.canlet.2006.11.002; PMID: 17189668
- Mukhopadhyay P, Rajesh M, Haskó G, Hawkins BJ, Madesh M, Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc 2007; 2:2295 - 301; http://dx.doi.org/10.1038/nprot.2007.327; PMID: 17853886
- Scaduto RC Jr., Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 1999; 76:469 - 77; http://dx.doi.org/10.1016/S0006-3495(99)77214-0; PMID: 9876159
- Hofer T, Seo AY, Prudencio M, Leeuwenburgh C. A method to determine RNA and DNA oxidation simultaneously by HPLC-ECD: greater RNA than DNA oxidation in rat liver after doxorubicin administration. Biol Chem 2006; 387:103 - 11; http://dx.doi.org/10.1515/BC.2006.014; PMID: 16497170
- Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 2009; 16:1040 - 52; http://dx.doi.org/10.1038/cdd.2009.49; PMID: 19407826
- Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem 2006; 281:29776 - 87; http://dx.doi.org/10.1074/jbc.M603783200; PMID: 16882669
- Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC, et al. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol 2009; 296:H470 - 9; http://dx.doi.org/10.1152/ajpheart.01051.2008; PMID: 19098111
- Su H, Wang X. Autophagy and p62 in cardiac protein quality control. Autophagy 2011; 7:1382 - 3; http://dx.doi.org/10.4161/auto.7.11.17339; PMID: 21997373
- Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181 - 97; http://dx.doi.org/10.1016/S0076-6879(08)03612-4; PMID: 19200883
- Larsen KB, Lamark T, Øvervatn A, Harneshaug I, Johansen T, Bjørkøy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy 2010; 6:784 - 93; http://dx.doi.org/10.4161/auto.6.6.12510; PMID: 20574168
- Henics T, Wheatley DN. Cytoplasmic vacuolation, adaptation and cell death: a view on new perspectives and features. Biol Cell 1999; 91:485 - 98; http://dx.doi.org/10.1016/S0248-4900(00)88205-2; PMID: 10572624
- Shang F, Taylor A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic Biol Med 2011; 51:5 - 16; http://dx.doi.org/10.1016/j.freeradbiomed.2011.03.031; PMID: 21530648
- Dudek EJ, Shang F, Valverde P, Liu Q, Hobbs M, Taylor A. Selectivity of the ubiquitin pathway for oxidatively modified proteins: relevance to protein precipitation diseases. FASEB J 2005; 19:1707 - 9; PMID: 16099947
- Gnaiger E. Polarographic oxygen sensors, the oxygraph, and high-resolution respirometry to assess mitochondrial function. In: Dykens; JA, Will; Y e, eds. Drug-Induced Mitochondrial Dysfunction. Hoboken, NJ, USA: John Wiley & Sons, Inc, 2008.
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A 1982; 79:1889 - 92; http://dx.doi.org/10.1073/pnas.79.6.1889; PMID: 6952238
- Stroikin Y, Dalen H, Lööf S, Terman A. Inhibition of autophagy with 3-methyladenine results in impaired turnover of lysosomes and accumulation of lipofuscin-like material. Eur J Cell Biol 2004; 83:583 - 90; http://dx.doi.org/10.1078/0171-9335-00433; PMID: 15679103
- Sohal RS, Sohal BH. Hydrogen peroxide release by mitochondria increases during aging. Mech Ageing Dev 1991; 57:187 - 202; http://dx.doi.org/10.1016/0047-6374(91)90034-W; PMID: 1904965
- Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, et al. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res 1999; 85:357 - 63; http://dx.doi.org/10.1161/01.RES.85.4.357; PMID: 10455064
- Hüttemann M, Lee I, Pecinova A, Pecina P, Przyklenk K, Doan JW. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J Bioenerg Biomembr 2008; 40:445 - 56; http://dx.doi.org/10.1007/s10863-008-9169-3; PMID: 18843528
- Vayssier-Taussat M, Kreps SE, Adrie C, Dall’Ava J, Christiani D, Polla BS. Mitochondrial membrane potential: a novel biomarker of oxidative environmental stress. Environ Health Perspect 2002; 110:301 - 5; http://dx.doi.org/10.1289/ehp.02110301; PMID: 11882482
- Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005; 70:200 - 14; http://dx.doi.org/10.1007/s10541-005-0102-7; PMID: 15807660
- Kuchino Y, Mori F, Kasai H, Inoue H, Iwai S, Miura K, et al. Misreading of DNA templates containing 8-hydroxydeoxyguanosine at the modified base and at adjacent residues. Nature 1987; 327:77 - 9; http://dx.doi.org/10.1038/327077a0; PMID: 3574469
- Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions. J Biol Chem 1992; 267:166 - 72; PMID: 1730583
- Lin HS, Jenner AM, Ong CN, Huang SH, Whiteman M, Halliwell B. A high-throughput and sensitive methodology for the quantification of urinary 8-hydroxy-2′-deoxyguanosine: measurement with gas chromatography-mass spectrometry after single solid-phase extraction. Biochem J 2004; 380:541 - 8; http://dx.doi.org/10.1042/BJ20040004; PMID: 14992687
- Gedik CM, Collins A, ESCODD (European Standards Committee on Oxidative DNA Damage). Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. FASEB J 2005; 19:82 - 4; PMID: 15533950
- Sachs HG, Colgan JA, Lazarus ML. Ultrastructure of the aging myocardium: a morphometric approach. Am J Anat 1977; 150:63 - 71; http://dx.doi.org/10.1002/aja.1001500105; PMID: 930852
- Bova MP, Tam D, McMahon G, Mattson MN. Troglitazone induces a rapid drop of mitochondrial membrane potential in liver HepG2 cells. Toxicol Lett 2005; 155:41 - 50; http://dx.doi.org/10.1016/j.toxlet.2004.08.009; PMID: 15585358
- Masubuchi Y, Kano S, Horie T. Mitochondrial permeability transition as a potential determinant of hepatotoxicity of antidiabetic thiazolidinediones. Toxicology 2006; 222:233 - 9; http://dx.doi.org/10.1016/j.tox.2006.02.017; PMID: 16621215
- Kaufmann P, Török M, Zahno A, Waldhauser KM, Brecht K, Krähenbühl S. Toxicity of statins on rat skeletal muscle mitochondria. Cell Mol Life Sci 2006; 63:2415 - 25; http://dx.doi.org/10.1007/s00018-006-6235-z; PMID: 17013560
- Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA 2007; 297:842 - 57; http://dx.doi.org/10.1001/jama.297.8.842; PMID: 17327526
- Levonen AL, Vähäkangas E, Koponen JK, Ylä-Herttuala S. Antioxidant gene therapy for cardiovascular disease: current status and future perspectives. Circulation 2008; 117:2142 - 50; http://dx.doi.org/10.1161/CIRCULATIONAHA.107.718585; PMID: 18427144
- Tinkel J, Hassanain H, Khouri SJ. Cardiovascular antioxidant therapy: a review of supplements, pharmacotherapies, and mechanisms. Cardiol Rev 2012; 20:77 - 83; PMID: 22293859
- Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal 2006; 8:152 - 62; http://dx.doi.org/10.1089/ars.2006.8.152; PMID: 16487049
- Mendl N, Occhipinti A, Müller M, Wild P, Dikic I, Reichert AS. Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2. J Cell Sci 2011; 124:1339 - 50; http://dx.doi.org/10.1242/jcs.076406; PMID: 21429936
- Ma X, Jin M, Cai Y, Xia H, Long K, Liu J, et al. Mitochondrial electron transport chain complex III is required for antimycin A to inhibit autophagy. Chem Biol 2011; 18:1474 - 81; http://dx.doi.org/10.1016/j.chembiol.2011.08.009; PMID: 22118681
- Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009; 164:541 - 51; http://dx.doi.org/10.1016/j.neuroscience.2009.08.014; PMID: 19682553
- Ravikumar B, Berger Z, Vacher C, O’Kane CJ, Rubinsztein DC. Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet 2006; 15:1209 - 16; http://dx.doi.org/10.1093/hmg/ddl036; PMID: 16497721
- Gershon H, Gershon D. Detection of inactive enzyme molecules in ageing organisms. Nature 1970; 227:1214 - 7; http://dx.doi.org/10.1038/2271214a0; PMID: 5212575
- Miquel J, Tappel AL, Dillard CJ, Herman MM, Bensch KG. Fluorescent products and lysosomal components in aging Drosophila melanogaster. J Gerontol 1974; 29:622 - 37; http://dx.doi.org/10.1093/geronj/29.6.622; PMID: 4214300
- Iwai K, Drake SK, Wehr NB, Weissman AM, LaVaute T, Minato N, et al. Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: implications for degradation of oxidized proteins. Proc Natl Acad Sci U S A 1998; 95:4924 - 8; http://dx.doi.org/10.1073/pnas.95.9.4924; PMID: 9560204
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003; 278:25009 - 13; http://dx.doi.org/10.1074/jbc.M300227200; PMID: 12719433
- Dosenko VE, Nagibin VS, Tumanovska LV, Moibenko AA. Protective effect of autophagy in anoxia-reoxygenation of isolated cardiomyocyte?. Autophagy 2006; 2:305 - 6; PMID: 16874046
- Cohen MV, Downey JM. Adenosine: trigger and mediator of cardioprotection. Basic Res Cardiol 2008; 103:203 - 15; http://dx.doi.org/10.1007/s00395-007-0687-7; PMID: 17999026
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007; 13:619 - 24; http://dx.doi.org/10.1038/nm1574; PMID: 17450150
- Kitsis RN, Peng CF, Cuervo AM. Eat your heart out. Nat Med 2007; 13:539 - 41; http://dx.doi.org/10.1038/nm0507-539; PMID: 17479097
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 2007; 3:323 - 8; PMID: 17387262
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72:248 - 54; http://dx.doi.org/10.1016/0003-2697(76)90527-3; PMID: 942051