Abstract
The NLR (nucleotide-binding domain leucine-rich repeat containing) proteins serve as regulators of inflammatory signaling pathways. NLRX1, a mitochondria-localized NLR protein, has been previously shown to negatively regulate inflammatory cytokine production activated via the MAVS-DDX58 (RIG-I) pathway. The literature also indicates that DDX58 has a negative impact upon autophagy. Consistent with the inhibitory role of NLRX1 on DDX58, our recent study indicates a role of NLRX1 in augmenting virus-induced autophagy. This effect is through its interaction with another mitochondrial protein TUFM (Tu translation elongation factor, mitochondrial, also known as EF-TuMT, COXPD4, and P43). TUFM also reduces DDX58-activated cytokines but augments autophagy. Additionally it interacts with ATG12–ATG5-ATG16L1 to form a molecular complex that modulates autophagy. The work shows that both NLRX1 and TUFM work in concert to reduce cytokine response and augment autophagy.
The mitochondrion has recently surfaced as an important organelle where host antiviral functions take place. Our study reveals that a previously identified mitochondrial modulator of interferon (IFN) production, NLRX1, modulates autophagy in addition to mitigating the interaction between the RNA helicase, DDX58 [DEAD (Asp-Glu-Ala-Asp) box polypeptide 58] and the type I IFN-activating mitochondrial antiviral signaling protein (MAVS, also known as IPS-1, VISA and CARDIFF). Its mode of function is opposite of that previously found for DDX58, which antagonizes the function of the autophagy-related proteins ATG12–ATG5. However, a direct interaction between NLRX1 and ATG12–ATG5 was not identified. A quantitative high throughput proteomic approach uncovered another intrinsic mitochondrial protein, TUFM. The study uncovered a function of TUFM that is also similar to NLRX1 in inhibiting type I IFN and promoting virus-mediated autophagy. In addition, TUFM interacts with ATG12–ATG5 and ATG16L1, thus serving as a tether between NLRX1 and the autophagic machinery in response to viral infection.
The mitochondrial adaptor MAVS effectively transduces the sensing of intracellular microbial RNA to nuclear translocation of NFKB and IRF3, which initiates the transcription of type I IFN. Besides its potent effect in limiting viral replication and spreading, type I IFN also drives the expression of ISGs (interferon-stimulated genes). Given the potency of this machinery, its activation is kept in-check by a group of proteins, including intrinsic mitochondrial proteins or proteins temporarily recruited to mitochondria. A key interaction that leads to MAVS activation and IFN production is the interaction of MAVS with the RNA helicase/receptors, DDX58 or IFIH1/MDA-5. However, MAVS and DDX58 also serve functions that lie outside of IFN induction. The former has been shown in several studies to mediate virally-induced apoptosis, whereas the latter has been found to reduce autophagy.
Recent studies have shown that autophagy is important in antiviral innate immunity. Autophagy could either directly have an impact upon the life cycle of intracellular viruses or modulate antiviral type I IFN signaling. Unlike type I IFN production, which normally contains a viral infection, the function of autophagy could be either antiviral or proviral depending on the viruses and host cell types. As part of the intracellular membrane systems, the exchange of materials from the autophagosomes with other subcellular compartments is crucial in delivering viral PAMPs (pathogen associated molecular patterns) to the endosome of plasmacytoid dendritic cells to activate endosomal TLRs (Toll-like Receptors). Additionally, autophagy can also compartmentalize and degrade viral products to limit their replication. In contrast, autophagy is used by a number of viruses for their proper replication. Encephalomyocarditis virus, chikungunya virus, dengue virus, poliovirus, coxsackievirus B3, and hepatitis C virus have all been shown to rely on autophagic function for their replication. It is noteworthy that autophagy was shown to quench type I IFN generation in MEFs (mouse embryonic fibroblasts). ATG12–ATG5 inhibits type I IFN by directly associating with MAVS; and the absence of ATG5 leads to compromised autophagy and subsequent accumulation of mitochondrial MAVS and reactive oxygen species, which partially results in elevated type I IFN generation. In addition to MEFs, autophagy defects in human hepatocytes also result in enhanced type I IFN signaling in response to hepatitis C virus. Hence the RIG-I-like receptor family (RLR) type I IFN pathway is intertwined with autophagy to modulate host innate antiviral responses.
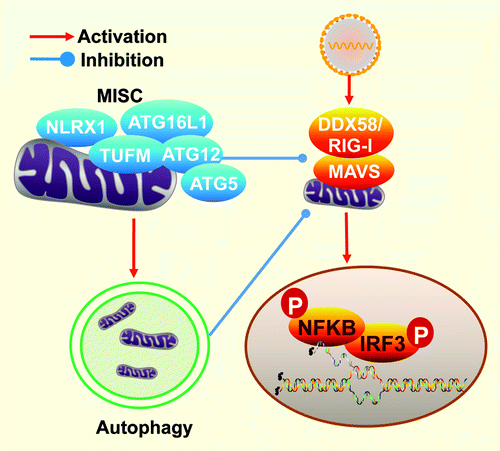
NLR proteins exhibit a wide spectrum of functions including inflammasome-based IL1B and IL18 processing, modulation of NFKB and MAPK signaling, major histocompatibility complex transcription and the modulation of the RLR pathway. In our study, we show that NLRX1 possesses opposing roles in the regulation of type I IFN and autophagy. While the protein has been previously shown to negatively impact DDX58-induced type I IFN response, this work shows that it is also required for virally-induced autophagy. Recent evidence suggests that other NLR members, including NLRC4 (also known as IPAF), NOD2 and NLRP4, also intersect with the autophagic machinery. Given that NLRX1 resides in a large molecular mass, a quantitative high-throughput mass spectrometric approach was employed to identify NLRX1-interacting partners that might explain the mechanism by which NLRX1 augments autophagy. Among the interactome, we identified TUFM as a mitochondrial protein that potently associates with NLRX1, and ATG12–ATG5 and ATG16L1. TUFM is known to be important in mitochondrial protein elongation, but its bacterial counterpart serves as a PAMP that can activate the plant immune response, albeit the eukaryotic form no longer has the immune-activating domain. Similar to NLRX1, overexpression and RNA interference targeting TUFM showed that the protein substantially inhibits RLR activation/type I IFN production, but augments autophagy. Reduced type I IFN and enhanced autophagy result in increased viral titer. These findings collectively revealed a mitochondrial complex comprised of NLRX1 and TUFM which have the dual functions of reducing type I IFN and enhancing autophagy responses to viral infections (). It is likely that both of these functions are important for maintaining a homeostatic state, but are hijacked by viruses to reduce host antiviral responses.
Figure 1. This illustration shows a mitochondrial immune signaling complex (MISC) that centers on NLRX1 and TUFM with the latter recruiting ATG12–ATG5 and ATG16L1. It shows the independent dual functions in directly inhibiting DDX58 and promoting autophagy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.