Abstract
BNIP3 (BCL2/adenovirus E1B 19 kDa interacting protein 3) is an atypical BH3-only protein that is induced by hypoxia-inducible factor 1 (HIF1) under hypoxia. BNIP3 is primarily regulated at the transcriptional level. However, little is known about the underlying mechanism of BNIP3 degradation. In this study, we found that BNIP3 was downregulated when hypoxia was accompanied by amino acid starvation. The BNIP3 downregulation did not occur at the transcription level and was independent of HIF1A. BNIP3 was primarily degraded by the proteasome, but BNIP3 was subjected to both proteasomal and autophagic degradation in response to starvation. The autophagic degradation of BNIP3 was dependent on ATG7 and MAP1LC3. We determined that autophagic degradation of BNIP3 was specifically regulated by ULK1 via the MTOR-AMPK pathway. Moreover, we confirmed that BNIP3 could play a protective role in tumor cells under hypoxia, and the treatment with Torin1, an MTOR inhibitor, decreased the BNIP3 level and enhanced the death of hypoxic tumor cells.
Introduction
In physiological conditions, the tumor microenvironment is characterized by reduced oxygen and nutrient levels.Citation1 Significant regions of a tumor are located far from blood vessels, resulting in a gradient of O2 and nutrients.Citation2 The low levels of oxygen and nutrients in the tumor microenvironment enhance autophagy signaling in the tumor tissue.Citation1 In the same tumor, hypoxia-induced autophagy may act like a double-edged sword, with cell death and survival occurring side by side.Citation3
Autophagy is a highly conserved process in which proteins, organelles and pathogens are sequestered in a double-membrane vesicle and delivered to the lysosome for degradation.Citation4 A series of protein complexes composed of autophagy-related gene (ATG) products is required for the formation of autophagosomes. The ULK1/2 complex (UNC-51-like kinases 1 and 2, the mammalian orthologs of yeast Atg1) is an essential positive regulator of autophagy initiation.Citation5 When nutrients are abundant, the mechanistic target of rapamycin (MTOR) complex 1 (MTORC1) binds to and phosphorylates both ULK1/2 and another complex member, ATG13. These phosphorylation events inhibit autophagy initiation. During starvation, MTORC1 dissociates from the ULK1/2 complex, freeing ULK1/2 to trigger autophagosome nucleation and elongation.Citation6,Citation7 It was originally assumed that AMP-activated protein kinase (AMPK) indirectly activates the ULK1/2 complex by inhibiting MTORC1. However, recent studies suggest a new mechanism in which AMPK directly phosphorylates and activates ULK1/2, which subsequently induces autophagy.Citation8 Following ULK1/2 complex activation, BECN1 and the phosphatidylinositol 3-kinase, catalytic subunit type 3 (PIK3C3) mediate autophagosome nucleation, and then autophagosomes mature through two ubiquitin-like conjugation systems.Citation9 The autophagosomes subsequently fuse with lysosomes, which decompose intracellular cargo with degradative enzymes. The degraded products are released into cytosol, where they are available to participate in synthetic and metabolic pathways.
BNIP3 (BCL2 and adenovirus E1B 19-kDa-interacting protein 3) is a protein with homology to BCL2 in its BH3 domain.Citation10 BNIP3 contains several defined domains such as the PEST domain (amino acids 54 to 81), a BH3 domain (amino acids 104 to 119) and a transmembrane domain (TM, amino acids 164 to 184), MAP1LC3-interacting region (LIR, amino acids 18 to 21).Citation11,Citation12 Because the promoter of BNIP3 contains a hypoxia response element (HRE) and appears to be a direct target of transcriptional activation by HIF1, BNIP3 expression is strongly activated by hypoxia.Citation13,Citation14 BNIP3 is a potent inducer of autophagy in many different cell types.Citation15-Citation18 However, the functional role of BNIP3-mediated autophagy is unclear. BNIP3 induces cell death in malignant glioma cells and cardiac myocytes,Citation17,Citation19 but BNIP3-induced autophagy during hypoxia can be a survival mechanism in cancer cells.Citation15,Citation18 Although the functional role of BNIP3-mediated autophagy appears to differ according to the cell type and the context, BNIP3 clearly plays a key role in the pathogenesis of many diseases, such as heart failure and cancer.Citation20
We investigated the degradation mechanism of BNIP3 during hypoxia followed by starvation and found an alternative mechanism of BNIP3 degradation. Our results suggest that BNIP3 could be a preferable substrate for autophagy as well as the ubiquitin-proteasome system (UPS), and that BNIP3 degradation by autophagy was dependent on ULK1 via the AMPK-MTORC1 pathway. Our study demonstrates that BNIP3 might play a protective role in SK-Hep-1 and HeLa cells under hypoxia and that the autophagic removal of BNIP3 by the treatment of Torin1 could restrict the survival of cancer cells.
Results
BNIP3 is downregulated in response to amino acid starvation
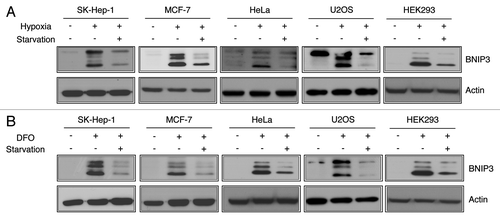
The physiological hypoxia condition is defined by the lack of two factors, oxygen and nutrients.Citation3 In addition, oxygen deficiency precedes nutrient deprivation in physiological circumstances.Citation21 To investigate how BNIP3 is regulated under hypoxic-starved conditions, we exposed SK-Hep-1 cells to normoxia (21% O2) or hypoxia (1% O2) for 24 h and then subjected the cells to amino acid deprivation with Earle’s Balanced Salt Solution (EBSS) for an additional 4 h. The western blot analysis clearly showed that the protein level of BNIP3 increased in response to hypoxia (). However, the BNIP3 level was markedly reduced by starvation following hypoxia. To examine whether these responses were specific to SK-Hep-1 cells, we investigated BNIP3 expression in several other human cell lines, such as MCF-7, HeLa, U2OS and HEK293 cells under the same conditions. We found that the level of BNIP3 was dramatically decreased by starvation in all these cell lines. In order to exclude the effect of reoxygenation during the media change for starvation, we exposed cells to normal oxygen conditions after hypoxia. We found that the BNIP3 protein level was retained until 2 h after reoxygenation indicating that the short-term exposure to oxygen during the media change could not affect the BNIP3 expression (Fig. S1). We further confirmed the decrease in BNIP3 under hypoxia-mimicking conditions using deferoxamine (DFO). The five tested cell lines were treated with 100 μM DFO for 24 h followed by starvation. A western blot analysis showed that starvation under hypoxia-mimicking conditions also markedly decreased BNIP3 levels (). These data demonstrate that BNIP3 was downregulated in response to starvation regardless of O2 concentration.
Figure 1. BNIP3 is induced by hypoxia and downregulated by starvation. (A) Human hepatocellular carcinoma cells (SK-Hep-1), breast cancer cells (MCF-7), cervical cancer cells (HeLa), osteoblast cells (U2OS) and human embryonic kidney cells (HEK293) were respectively incubated under normoxia (21% O2), hypoxia (1% O2) for 24 h or hypoxia followed by amino acid starvation for an additional 4 h. Changes in BNIP3 levels were analyzed by western blotting. (B) Those five different human cell lines were treated with 100 μM deferoxamine (DFO) for 24 h in the normoxic condition followed by amino acid starvation for 4 h.
BNIP3 downregulation is due to lysosomal degradation
Because amino acid starvation decreased the protein level of BNIP3, we investigated the mRNA level of BNIP3 following starvation by qPCR analysis. We found that the BNIP3 mRNA level increased by 2.7-fold under hypoxia compared with normoxia, whereas it was not decreased by starvation in hypoxic conditions (). To further clarify whether the decrease of BNIP3 level was independent of the HIF1A stability and activity, we analyzed the protein levels of HIF1A and lactate dehydrogenase A (LDHA), which could be induced by HIF1A.Citation22 Although the expression levels of HIF1A, LDHA and BNIP3 were provoked by hypoxia, the protein levels of HIF1A and LDHA were not affected by starvation, which was in contrast to the BNIP3 protein level (). To further confirm whether this differential effect was dependent on HIF1A activation, we performed an overexpression experiment. Flag-tagged BNIP3 was overexpressed under normal oxygen conditions, which was followed by challenge with amino acid starvation. Consistent with earlier results, BNIP3 was downregulated in response to starvation independent of hypoxia ().
Figure 2. Amino acid deprivation does not alter the mRNA level of BNIP3 but significantly decreases its protein level through lysosomal degradation. (A) SK-Hep-1 cells were exposed to normoxia (21% O2), hypoxia (1% O2) or hypoxia followed by starvation for 4 h, respectively. After starvation, total RNA was isolated and analyzed to quantify the BNIP3 mRNA levels by real-time quantitative PCR. (B) The protein levels of HIF1A, LDHA and BNIP3 were analyzed by western-blot analysis. (C) Flag-tagged BNIP3 was transiently expressed in SK-Hep-1 cells, which were subjected to starvation for 4 h. BNIP3 was detected by western blot, using an anti-Flag antibody. (D) SK-Hep-1 cells were incubated for 24 h under normoxic or hypoxic conditions with or without additional starvation in the presence or absence of 100 μM cycloheximide (CHX) and subjected to western blot analysis. CHX-induced translational inhibition was confirmed by monitoring the level of TP53. The protein level of BNIP3 was analyzed by densitometry using the ImageJ software program. (E) SK-Hep-1 cells were incubated for 24 h under normoxic or hypoxic conditions. The cells were then treated with 20 μM MG132, 10 mM 3MA or 10 nM BafA1 in the presence or absence of amino acids for an additional 4 h. (F) SK-Hep-1 cells were exposed to hypoxia for 24 h and treated with 20 μM MG132 and/or 10 nM BafA1 under starved conditions for additional 4 h of hypoxia. Inhibition of autophagy was confirmed by western blot analysis of SQSTM1 and MAP1LC3B. Inhibition of proteasome was assessed by TP53 expression level. The protein level of BNIP3 was analyzed by densitometry using the ImageJ software program. The data are shown as means ± SD for three independent experiments. *p < 0.005, t test; n.s., not significant.
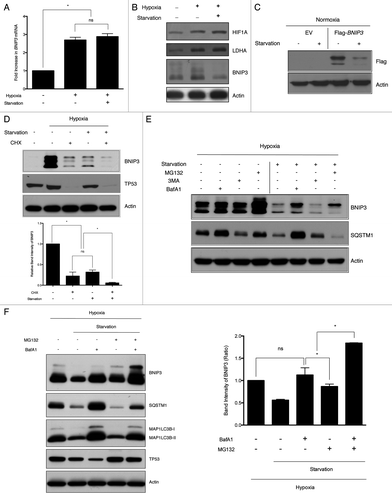
When we treated SK-Hep-1 cells with 100 μM cycloheximide (CHX) and starved the cells, BNIP3 levels reduced to 22% and 32% of that induced by hypoxia, respectively. Noticeably, starvation together with the CHX treatment further decreased BNIP3 to 6% of the level induced by hypoxia, demonstrating that the starvation-induced downregulation of BNIP3 was due to its rapid degradation (). To further investigate BNIP3 degradation, we treated cells with MG132, 3-methyladenine (3MA) or bafilomycin A1 (BafA1) under hypoxia in either fed or starved condition. Under hypoxic conditions, the inhibition of the proteasome stabilized BNIP3. BNIP3 expression appeared to be marginally increased by treatment with either 3MA or BafA1, indicating that BNIP3 was preferentially removed by proteasomes under hypoxic conditions (). However, when hypoxia was accompanied by starvation, the MG132 treatment only partially attenuated BNIP3 degradation. In contrast, the BafA1 treatment also blocked the BNIP3 degradation. When both MG132 and BafA1 were included simultaneously, the BNIP3 protein level was increased by 80% compared with the individual treatment indicating the additive effect of MG132 and BafA1 (). These data suggest that both lysosomal and proteasomal degradation were involved in the decrease of the BNIP3 level under the hypoxia plus starvation condition.
Lysosomal degradation of BNIP3 is mediated by macroautophagy
During starvation, lysosomal degradation primarily progresses through autophagy.Citation23 At least three distinct autophagic pathways exist: macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy.Citation24 Therefore, to investigate which type of autophagy is involved in BNIP3 degradation, we depleted LAMP2 using siRNA to block CMA. Under hypoxia followed by starvation, BNIP3 degradation was not blocked by the knockdown of LAMP2, indicating that CMA was not involved in BNIP3 degradation (). Because autophagosome mediation is the main discrepancy between macroautophagy and CMA or microautophagy, we used a microtubule disruptor, vinblastine, to inhibit macroautophagy flux by blocking the movement of autophagosomes to lysosomes.Citation25 When we disrupted microtubules with 50 μM vinblastine during BNIP3 degradation induced by starvation, the accumulation of MAP1LC3B-II in the presence of vinblastine confirmed that vinblastine successfully inhibited macroautophagy (). Moreover, BNIP3 accumulated and its degradation was dramatically decreased under starvation, suggesting that BNIP3 degradation should be mediated by macroautophagy (hereafter referred to as autophagy).
Figure 3. BNIP3 degradation is mediated by macroautophagy, which is dependent on the ATG7-MAP1LC3 conjugation system. (A) SK-Hep-1 cells were treated with nontargeting (negative control, NC) or LAMP2 siRNAs for 48 h, and subjected to hypoxia for 24 h, followed by amino acid starvation for 4 h. BNIP3 protein levels were then analyzed by western blotting. (B) SK-Hep-1 cells were exposed to hypoxia for 24 h, followed by starvation for 4 h in the presence or absence of 50 μM vinblastine. BNIP3 expression and autophagy inhibition were analyzed by western blot. SK-Hep-1 cells were treated with nontargeting (NC), BECN1 (C) and MAP1LC3B (D) siRNAs for 48 h, followed by hypoxic exposure for 24 h, and then by amino acid starvation for 4 h. Autophagy induction and BNIP3 degradation were analyzed by western blotting. (E) Scrambled shRNA (NC) and ATG7 shRNA (shATG7) were transfected into SK-Hep-1 cells respectively. Knockdown of ATG7 was confirmed by western blot analysis. Two different shRNAs, shATG7(1) and shATG7(2), were used and a clone with more effective knockdown was selected. Stable cell lines were incubated under hypoxia (1% O2) for 24 h with or without additional starvation for 4 h. The expression level of BNIP3 was analyzed by western blotting.
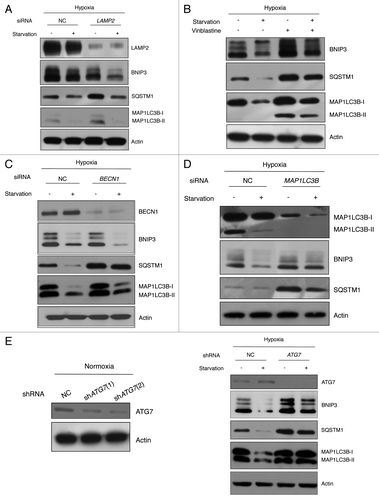
To determine whether autophagy was involved in BNIP3 degradation, autophagy was assessed under conditions that caused BNIP3 degradation. As expected, hypoxia induced autophagy. Moreover, autophagy induction was more distinct in starvation, regardless of the oxygen conditions (Fig. S2). To confirm that the autophagy induction was directly related to BNIP3 degradation, we used siRNAs to knockdown BECN1 and MAP1LC3B, which play essential roles in autophagosome maturation.Citation25 We found that the knockdown of BECN1 or MAP1LC3B effectively blocked autophagy as judged by the expression level of SQSTM1 (). Unexpectedly, BECN1 knockdown did not attenuate BNIP3 degradation (; Fig. S3A), while MAP1LC3B knockdown blocked BNIP3 degradation (). In addition, knockdown of PIK3C3, one of the components in the BECN1 complex, could not block the BNIP3 degradation in response to starvation or Torin1 treatment (Fig. S3B). These results exhibit that MAP1LC3B is required for the BNIP3 degradation. There are three isoforms of MAP1LC3, which are MAP1LC3A, MAP1LC3B and MAP1LC3C.Citation26 We found that the respective knockdown of MAP1LC3A and MAP1LC3B attenuated the BNIP3 degradation. In contrast, MAP1LC3C knockdown did not seem to affect the BNIP3 degradation (Fig. S4). To further confirm the requirement of MAP1LC3 for BNIP3 degradation, we depleted ATG7, an upstream regulator of MAP1LC3.Citation4 SK-Hep-1 cells were transfected with lentiviral shRNAs targeting ATG7. When selected stable cell lines were exposed to hypoxic-starved conditions, ATG7 knockdown blocked BNIP3 degradation, demonstrating that the autophagic degradation of BNIP3 should require ATG7 and MAP1LC3 ().
Autophagic degradation of BNIP3 is not dependent on its association with mitochondria
BNIP3 contains a mitochondrial-targeting TM domain that can associate with the mitochondrial membrane.Citation27,Citation28 To investigate whether membrane-associated BNIP3 could be sequestered to autophagosomes along with mitochondria during autophagy, we constructed a BNIP3 expression vector lacking the TM domain. We overexpressed full-length BNIP3 or TM-deleted BNIP3 (BNIP3∆TM) in SK-Hep-1 cells and then observed the localization of BNIP3 by fluorescence microscopy. Full-length BNIP3 was colocalized with mitochondria, but BNIP3∆TM showed a diffuse localization pattern ().Citation29 To test if BNIP3∆TM can be degraded by starvation-induced autophagy, we transiently transfected cells with an expression vector containing full-length BNIP3 or BNIP3∆TM. After 24 h transfection, the cells were exposed to hypoxia for 24 h, which was followed by starvation for an additional 4 h. Full-length BNIP3 was degraded in response to starvation. Surprisingly, BNIP3∆TM was also degraded by starvation, showing that BNIP3 degradation did not occur simply as a byproduct of mitochondrial autophagy (). Consequently, we investigated whether the BNIP3 degradation was due to autophagy and/or UPS. We found that the degradation of both full-length BNIP3 and BNIP3∆TM were blocked by BafA1 as well as MG132 (Fig. S5). Moreover, full-length BNIP3 and BNIP3∆TM were more accumulated in response to the treatment of both BafA1 and MG132, suggesting that BNIP3 could be degraded by autophagy independent of its localization to mitochondria.
Figure 4. BNIP3 degradation is not dependent on BNIP3 translocation to mitochondria. (A) Full-length BNIP3 or BNIP3 lacking TM domain (BNIP3∆TM) was expressed in SK-Hep-1 cells, and its localization was visualized by immunofluorescence microscopy. Nuclei were stained with Hoechst 33342, and mitochondria were detected with Mito-tracker Red. Full-length BNIP3 and BNIP3∆TM were detected with an anti-Flag antibody and visualized with FITC-conjugated anti-mouse IgG. (B) Flag-tagged BNIP3 and BNIP3∆TM were transiently expressed in SK-Hep-1 cells. At 24 h after transfection, the cells were incubated with or without starvation and subjected to western-blot analysis. (C) Various mitochondrion-associated proteins were analyzed by western blotting following hypoxia or hypoxia plus starvation. Mitochondrial autophagy was assessed according to the expression level of mitochondrial membrane proteins, COX4I1 and TOMM20 (left panel). The data are shown as means ± SD for three independent experiments. *p < 0.0005, t test.
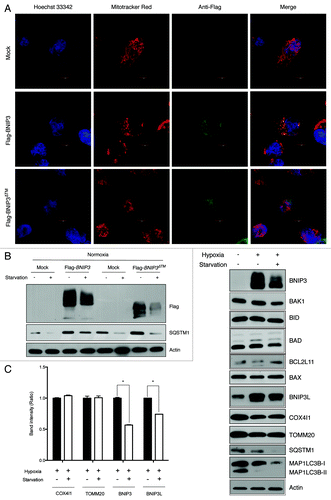
To further confirm that BNIP3 degradation was independent of mitochondrial autophagy, we analyzed several mitochondria-associated proteins and mitochondrial membrane proteins under starvation. In experiments involving the hypoxia-plus starvation conditions, we examined the expression levels of a series of BH3-only proteins like BAK1, BID, BAD, BCL2L11 and BAX proteins that interact with the mitochondrial membrane.Citation30-Citation33 Although we found that BNIP3 was reduced by 43% and BNIP3L by 26% under hypoxia plus starvation, no degradation of other mitochondrial associated proteins was observed, suggesting that starvation-induced BNIP3 degradation should not be related to its association with mitochondria (). Moreover, the protein levels of COX4I1 and TOMM20 in mitochondrial membrane were not changed in response to starvation suggesting that mitophagy might be negligible in our experimental conditions.
Autophagic degradation of BNIP3 is not mediated by interaction with MAP1LC3B or SQSTM1
We investigated whether a specific mediator is involved in the BNIP3 degradation. Even though both BNIP3 and BNIP3L can interact with MAP1LC3B via LIR, recent papers reveal that BNIP3∆TM could not interact with MAP1LC3B.Citation20,Citation34,Citation35 Therefore, we excluded MAP1LC3B as a direct mediator for degradation of BNIP3. Instead of MAP1LC3B, it is plausible that other mediators like SQSTM1 and NBR1 could be involved because SQSTM1 and NBR1 are documented as adaptor proteins for ubiquitinated proteins.Citation36 Therefore, we examined whether BNIP3 could interact with SQSTM1. We found that both BNIP3 and BNIP3∆TM could be ubiquitinated and bound to SQSTM1 (). However, the ubiquitination and subsequent binding to SQSTM1 were markedly reduced under starvation conditions, suggesting that interaction with SQSTM1 might not be related to the BNIP3 degradation. In addition, the knockdown of either SQSTM1 or NBR1 could not block the BNIP3 degradation implying that SQSTM1 or NBR1 might not be required for the BNIP3 degradation ( and ).
Figure 5. BNIP3 degradation is independent of SQSTM1 or NBR1. (A) SK-Hep-1 cells were transfected with empty vector (EV), Flag-BNIP3 or Flag-BNIP3∆TM under normoxia for 24 h and challenged with amino acid starvation in the presence or absence of BafA1 and/or MG132. BNIP3 protein was immunoprecipitated with anti-Flag antibody. Ubiquitination and SQSTM1 interaction were assessed by western-blot analysis. (B) SK-Hep-1 cells were transfected with SQSTM1 for 48 h followed by the overexpression of Flag-BNIP3 or Flag-BNIP3∆TM under normoxia for 24 h and challenged with amino acid starvation or Torin1 treatment. BNIP3 expression was analyzed by western blot using anti-Flag antibody. Autophagy was assessed by the MAP1LC3B expression level. (C) SK-Hep-1 cells were transfected with SQSTM1 or NBR1 siRNA for 48 h and exposed to hypoxia for 24 h. Amino acid starvation were challenged for an additional 4 h, followed by western blot analysis.
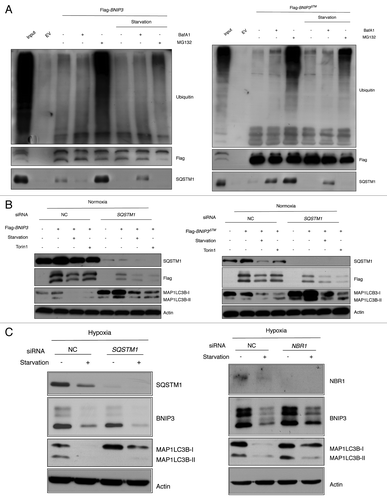
MTORC1 is an upstream regulator of autophagic BNIP3 degradation
Because MTORC1 is the crucial regulator of autophagy in response to amino acid starvation, we investigated whether BNIP3 degradation was mediated by MTORC1 inhibition using Torin1, a specific MTOR inhibitor.Citation37 SK-Hep-1 cells were cultured under hypoxic conditions for 24 h and treated with 50 nM or 250 nM Torin1 for 4 h. As expected, levels of p-RPS6 and p-EIF4EBP1 decreased, indicating that Torin1 effectively inhibited MTORC1 (). The observed degradation of SQSTM1 confirmed that Torin1 induced autophagy at a concentration as low as 50 nM. Noticeably, BNIP3 expression was dramatically downregulated by the Torin1 treatment during hypoxia. This result was confirmed under hypoxia-mimicking conditions by the DFO treatment (Fig. S6A). Treatment with siRNAs targeting MTOR and RPTOR, which are primary components of MTORC1, further confirmed that MTORC1 inhibition led to the autophagic degradation of BNIP3 (). When we measured the autophagic flux, MAP1LC3B-II was highly accumulated in the presence of Torin1 and BafA1 under hypoxia or hypoxia-mimicking conditions, implying that autophagy was effectively induced in response to the Torin1 treatment (; Fig. S6B). In contrast with the Torin1 treatment, the rapamycin treatment had mild effect on the BNIP3 degradation (Fig. S7), although rapamycin inhibits MTORC1 and induces autophagy.Citation37 This discrepancy could be explained by the notion that rapamycin may be a weak inducer of autophagy in mammalian cells.Citation38 To exclude the possibility that the decrease in the BNIP3 level was the result of a translational inhibition by MTORC1 inactivation, we transfected cells with siRNA targeting RPS6KB1 and exposed them to hypoxia for 24 h. Noticeably, the BNIP3 expression level was not decreased after the depletion of RPS6KB1, confirming that BNIP3 was degraded by autophagy that was induced upon the MTORC1 inhibition ().
Figure 6. BNIP3 degradation is mediated by MTORC1 inhibition. (A) SK-Hep-1 cells were cultured under hypoxia (1% O2) for 24 h. Torin1 was added for additional 4 h at the indicated dose, which was analyzed by western blot. The inhibition of MTORC1, BNIP3 degradation and autophagy induction were confirmed by western blot analysis. (B) SK-Hep-1 cells were transfected with MTOR or RPTOR siRNA and then exposed to hypoxia for 24 h followed by western-blot analysis. (C) SK-Hep-1 cells were exposed to hypoxia for 24 h, after which the cells were treated with 250 nM Torin1 in the presence or absence of 10 nM BafA1. The ratio of MAP1LC3B-I to MAP1LC3B-II was assessed by densitometry. (D) Negative control and RPS6KB1 siRNAs were transfected into SK-Hep-1 cells for 48 h. The transfected cells were incubated under normoxia or hypoxia for 24 h and were subjected to western-blot analysis.
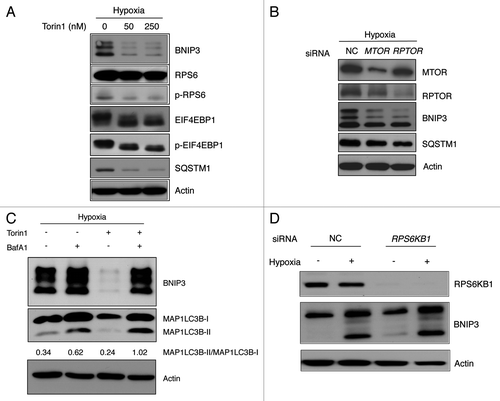
BNIP3 degradation is regulated by ULK1 via MTORC1 and AMPK
MTORC1-regulated autophagy was known to be directly related to ULK1.Citation5 To investigate whether the autophagic degradation of BNIP3 could be regulated by ULK1 through the MTORC1 pathway, we depleted ULK1 using siRNA and exposed SK-Hep-1 and HeLa cells to hypoxia for 24 h. Hypoxia was followed by amino acid starvation or Torin1 treatment for 4 h. A western blot analysis confirmed that BNIP3 degradation decreased with ULK1 depletion, showing that ULK1 could regulate the BNIP3 degradation in response to starvation or MTORC1 inhibition (; Fig. S8A).
Figure 7. BNIP3 degradation is regulated by ULK1 via MTORC1 and AMPK. (A) SK-Hep-1 cells were transfected with ULK1 siRNA for 48 h and exposed to hypoxia for 24 h followed by amino acid starvation or Torin1 treatment for additional 4 h and cells subjected to western-blot analysis. (B) SK-Hep-1 cells were incubated under hypoxic conditions for 24 h and then treated with 250 nM Torin1, 5 mM metformin or 20 μM Compound C for 4 h. MTORC1 inhibition and AMPK activation or inhibition were confirmed by western blotting. ULK1 inhibition was assessed by the level of p-ULK1 (S757), and ULK1 activation by that of p-ULK1 (S555).
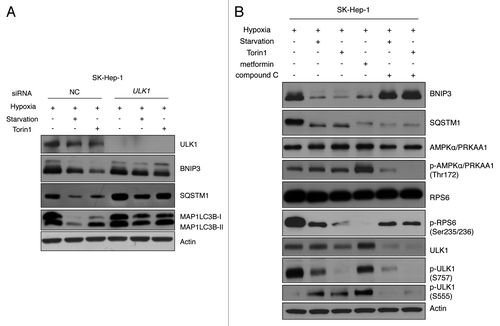
AMPK can indirectly activate ULK1 through MTORC1 inhibition.Citation39 However, recent findings demonstrated that AMPK directly phosphorylates ULK1 at Ser 555, which subsequently activates ULK1 and leads to autophagy activation.Citation6,Citation40 To investigate whether BNIP3 degradation is dependent on ULK1 activation through AMPK, SK-Hep-1 or HeLa cells were incubated under hypoxic conditions; this treatment was followed by starvation and treatment with Torin1 (MTOR inhibitor), metformin (AMPK activator) or compound C (AMPK inhibitor). As expected, MTORC1 inhibition resulted in the decrease of p-ULK1 (S757), indicating the derepression of ULK1,Citation6 which led to BNIP3 degradation (; Fig. S8B). The metformin treatment increased the level of phosphorylated AMPK, indicating that AMPK was activated in SK-Hep-1 cells. AMPK activation also increased the level of p-ULK1 (S555) and subsequently degraded BNIP3 through autophagy (). Unexpectedly, AMPK was not activated by metformin in HeLa cells, in contrast to what was observed in SK-Hep-1 cells; consequently, BNIP3 degradation was not observed (Fig. S8B). This discrepancy can be explained by the lack of STK11 in HeLa cells because metformin activates AMPK through its upstream kinase STK11.Citation41 However, AMPK inhibition by compound C completely reduced the level of p-ULK1 (S555), resulting in the accumulation of BNIP3 in both SK-Hep-1 and HeLa cells. Interestingly, despite MTORC1 inhibition, compound C could attenuate the BNIP3 degradation. Although the metformin-induced reduction of p-ULK1 (S757) was not significant, BNIP3 was still degraded. These results imply that direct ULK1 activation by AMPK may be a prerequisite for BNIP3 degradation.
Loss of BNIP3 reduces cell viability under hypoxic conditions
In order to investigate whether BNIP3 is a pro-death or prosurvival factor under our experimental conditions, SK-Hep-1 and HeLa cells were infected with lentiviral shRNAs for BNIP3. Stable cell lines selected using puromycin were exposed to hypoxia and then subjected to western blot analysis. BNIP3 was found to be efficiently downregulated by shBNIP3 (; Fig. S9A). To assess the effect of BNIP3 knockdown on cell viability, we cultured negative control cells and shBNIP3-treated cells under hypoxic conditions for up to 72 h. The viable cells were counted using the trypan blue assay at specific time points. Surprisingly, the shBNIP3-treated cells proliferated much less than the negative control cells (; Fig. S9B). When we performed the colony formation assay under hypoxic conditions for 7 d, the loss of BNIP3 reduced the colony-forming ability of the SK-Hep-1 cells (). These results indicate that BNIP3 was able to protect cells, at least SK-Hep-1 and HeLa cells, under conditions of hypoxic stress.
Figure 8. The viability of tumor cells is reduced in response to the loss of BNIP3 or Torin1 treatment. (A) Scrambled shRNA and BNIP3 shRNA were transfected into SK-Hep-1 cells. Knockdown of BNIP3 was confirmed by western-blot analysis. (B) Selected stable cell lines were incubated under hypoxic conditions for up to 72 h. The proliferation rate was assessed by counting viable cells at indicated time points. (C) For measurements of colony-forming ability, stable cell lines were plated at a low density (1000 cells/well) in 6-well plates and then incubated under hypoxic conditions for 7 d. Colonies were fixed and stained with crystal violet, and colony numbers were then counted using ImageJ software program (n = 3). (D) SK-Hep-1 cells were cultured under normoxia or hypoxia for 24 h, which was followed by the Torin1 treatment for additional 24 h. Cell death was assessed by the trypan blue assay. (E) BNIP3 degradation, MTORC1 inhibition and autophagy induction were confirmed by western-blot analysis. (F) SK-Hep-1 cells were cultured under normoxic or hypoxic conditions for 24 h and then treated with Torin1 for 24 h. After treatment, the cells were harvested and seeded in soft agar followed by incubation under hypoxia. Two weeks after the incubation, the cells were stained with crystal violet, and the colonies were counted by the ImageJ software program. (G) Schematic diagram of BNIP3 regulation by ULK1-dependent autophagy via MTORC1 and AMPK. The data are shown as means ± SD of three independent experiments performed in triplicate. t test; *p < 0.05, **p < 0.005, ***p < 0.0005, n.s., not significant.
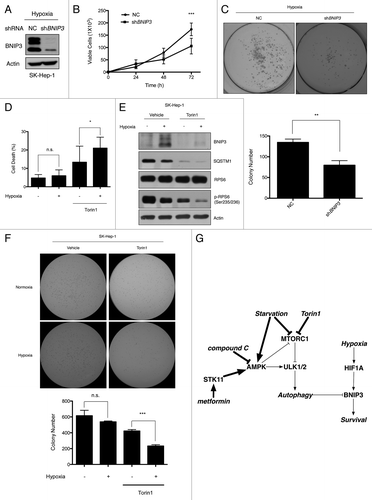
When we exposed SK-Hep-1 and HeLa cells to hypoxia for 24 h and treated them with Torin1 for an additional 24 h, hypoxia did not cause severe cell death, as determined by the trypan blue assay (; Fig. S9C). In contrast, Torin1 induced cell death even under normoxia. Moreover, the cytotoxicity of Torin1 was significantly increased in hypoxic conditions. The results of the western blot analysis showed that Torin1 significantly induces autophagy and completely removes BNIP3 (; Fig. S9D). To further confirm that Torin1 treatment inhibits the long-term survival of tumor cells during hypoxia, we performed a soft agar assay. As shown in and Figure S9E, the number of colonies dramatically decreased in the presence of Torin1 under hypoxia, confirming that the MTORC1 inhibition by Torin1 treatment limited the survival of hypoxic tumor cells.
Discussion
In this study, we found that BNIP3 was induced by hypoxia and degraded by autophagy in various types of human cells, with accompanying amino acid starvation. BNIP3 degradation was specifically triggered by ULK1-mediated autophagy, which could be regulated by MTORC1 and AMPK. Our results also suggest that BNIP3 could protect tumor cells from hypoxic stress, and that MTORC1 inhibition might limit the survival of hypoxic tumor cells.
Although both oxygen and nutrients are deficient under physiological hypoxia,Citation21 most studies on hypoxia have been conducted with the condition that O2 is the only limiting factor.Citation3 Due to a pivotal role in diseases related to hypoxia, there have been many studies on how BNIP3 is regulated.Citation14 However, most reports were about transcriptional regulation of BNIP3. Noticeably, we found that BNIP3 was significantly induced by hypoxia but was downregulated when hypoxia was accompanied by starvation. We further confirmed that the downregulation of BNIP3 did not occur at the transcriptional level, and the reduction of BNIP3 was independent of the HIF1A activity. BNIP3 expression level is reduced by serum deprivation when BNIP3 was overexpressed,Citation42 even though the mechanism underlying the downregulation of BNIP3 remained unclear. We demonstrated that BNIP3 downregulation occurred in its degradation step. It is well known that BNIP3 contains a PEST sequence that targets it for proteasomal degradation, and is rapidly degraded by UPS.Citation43 In this study, we demonstrated that autophagy also took part in the BNIP3 degradation when starvation was accompanied by hypoxia.
BNIP3 remains loosely associated with mitochondrial membrane via its TM domain.Citation28 Therefore, during autophagy, it is possible that membrane-associated BNIP3 can be sequestered to autophagosomes with mitochondria. However, we found that BNIP3∆TM was also degraded by autophagy, implying that BNIP3 degradation occurred regardless of the association with mitochondria. Additionally, several mitochondria-associated proteins and mitochondrial membrane proteins were not significantly reduced under starvation suggesting that mitochondrial autophagy might be negligible and not be related to the BNIP3 degradation in our experimental conditions. Noticeably, BNIP3L, a homologous protein of BNIP3, was also degraded by starvation, suggesting that there might be a specific mediator for degradation of BNIP3 family. Even though we showed that SQSTM1 could bind to both BNIP3 and BNIP3∆TM respectively, knockdown of SQSTM1 or NBR1 was not able to block the BNIP3 degradation. The specific mediator of BNIP3 degradation, if any, remains to be identified.
BECN1 is a well-known regulator of autophagy. However, all pathways of autophagy are not regulated by BECN1.Citation44 In line with this report, we found that BNIP3 degradation could also be mediated by ULK1 in addition to BECN1. Although the knockdown of ATG7 or ULK1 reversed the starvation-induced BNIP3 degradation even in the condition that BECN1 or PIK3C3 was depleted, BECN1 knockdown only exhibited partial effect on both BNIP3 degradation and the inhibition of SQSTM1 degradation (Fig. S10). Therefore, we cannot exclude the possibility that BNIP3 is preferentially degraded by classical autophagy.
ULK1-dependent autophagy is regulated by MTORC1 and AMPK.Citation39 In addition, hypoxia-induced autophagy associated with nutritional stress is suggested to be regulated by the AMPK-MTOR pathway.Citation3 In agreement with these reports, MTORC1 inhibition or AMPK activation resulted in dramatic degradation of BNIP3 through the ULK1 activation under hypoxic conditions. Noticeably, SQSTM1 degradation was not attenuated by the compound C treatment, suggesting that AMPK-mediated degradation of BNIP3 could be a specific process, rather than the result of global autophagy. Under hypoxic conditions, MTORC1 is inactivated while AMPK could be activated.Citation45,Citation46 Therefore, BNIP3 could be degraded even under hypoxia-only conditions, regardless of nutrient availability. However, at least under our experimental conditions, the observed MTORC1 inhibition or AMPK activation was not severe under hypoxia-only conditions (data not shown). Consistently, hypoxia-induced autophagy was not as severe as that caused by starvation or hypoxia followed by starvation (Fig. S2). Therefore, hypoxia-induced autophagy may be well controlled and remain under a certain threshold. Following additional stimuli, such as starvation or the Torin1 treatment, autophagy might exceed the threshold, and BNIP3 could be degraded by autophagy. Indeed, hypoxia is less effective to inhibit MTORC1 in tumor cells than in normal cells,Citation45 and constitutive activation of MTORC1 could prevent hypoxia from inhibiting MTORC1.Citation47 In addition, hypoxia by itself is not enough for AMPK activity to overcome the threshold to turn on autophagy.Citation48 Collectively, our results suggest that BNIP3 degradation should specifically be mediated by ULK1-dependent autophagy under the coordinated regulation of AMPK and MTORC1 as depicted in .
BNIP3 has essentially been known as a pro-death protein.Citation19,Citation43,Citation49 However, recent studies have shown that BNIP3 can protect cells from hypoxic stress. BNIP3 acts as a survival factor in colon and prostate cancer cells by inducing autophagy under hypoxia.Citation15 In mouse embryonic fibroblast, BNIP3 mediates the mitochondrial autophagy to remove reactive oxygen species, and subsequently prevents cell death.Citation18 Recently, Cosse et al. have reported that BNIP3 protects HepG2 cells against etoposide-induced cell death under hypoxia.Citation50 Moreover, it is suggested that hypoxia associated with nutrient limitation could induce cell death, whereas well-controlled hypoxia with no nutrient limitation might be a prosurvival factor.Citation3 In line with these reports, we found that the loss of BNIP3 prevented the proliferation of SK-Hep-1 and HeLa cells under hypoxic conditions, supporting the protective role of BNIP3. We also demonstrated that inhibiting MTORC1 by Torin1 accelerated cell death under hypoxia and limited the long-term survival of tumor cells. Because BNIP3 degradation was evident in response to Torin1 treatment, it is plausible that the detrimental effect of Torin1 might be partially related to BNIP3 degradation. Since Torin1 could exert numerous effects on the cell,Citation37 we cannot rule out the possibility that Torin1-mediated cytotoxicity was not due to the loss of BNIP3. It is also possible that Torin1 could induce excessive autophagy, which might subsequently exceed the threshold leading to cell death. Since autophagy induced by the rapamycin treatment protects cardiac myocytes from BNIP3-mediated cell death,Citation51,Citation52 it remains to be investigated further, to what extent the inhibition of MTORC1 can affect the degradation of BNIP3 by ULK1-dependent autophagy.
In conclusion, we demonstrated that BNIP3 was degraded by starvation-induced autophagy and that the autophagy was dependent on ULK1, which could be tightly regulated by MTORC1 and AMPK. Our results also suggested that BNIP3 could protect tumor cells from hypoxic stress and that MTORC1 inhibition might limit the survival of hypoxic tumor cells. Because BNIP3 can play key roles in the pathogenesis of many diseases, MTORC1- and AMPK-mediated BNIP3 regulation may be a therapeutic option to investigate for the treatment of cancer and heart disease.
Materials and Methods
Additional methods are presented in the Supplementary Information.
Cell culture and reagents
SK-Hep-1, HeLa, MCF-7, U2OS and HEK293 cells were purchased from ATCC. Dulbecco’s modified Eagle's medium (DMEM, 11965-092) and McCoy’s 5A medium (16600-082) were purchased from Invitrogen. SK-Hep-1, HeLa, MCF-7 and HEK293 cells were cultured in DMEM, and U2OS cells were cultured in McCoy’s 5A. In all cases, the culture medium was supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin. The cells were maintained in a humidified atmosphere with 5% CO2 at 37°C. bafilomycin A1 (B1793), 3-methyladenine (M9281), MG132 (C2211), cycloheximide (C7698), rapamycin (R0395), deferoxamine mesylate (D9533) and EBSS (E3024) were obtained from Sigma and LE agarose from Seakem (50004). Torin1 was generously provided by Drs. D. Sabatini (Whitehead Institute) and N. Gray (Harvard Medical School).
Hypoxia and amino acid starvation
Hypoxic culture conditions (1% O2) were achieved by culturing cells in a multi-gas incubator (Sanyo, MCO-5M). For amino acid starvation, cells were washed twice with EBSS followed by media change with EBSS. Consequently, cells were re-exposed to hypoxia for an additional 4 h.
Western blot analysis
Cells were harvested and lysed in the RIPA buffer (Cell Signaling Technology, 9806) containing 20 mM TRIS-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, a phosphatase inhibitor cocktail tablet (04 906 837 001) and a complete protease inhibitor cocktail tablet (04 693 124 001) from Roche. After the lysates were sonicated, the protein concentration was determined by the Bradford assay. Proteins were separated by SDS–PAGE and transferred electrophoretically onto a PVDF membrane (Millipore, IPVH00010). Each antibody incubation was performed overnight at 4°C followed by the secondary antibody treatment for 1 h at room temperature. Information regarding the primary antibody titers, manufacturers and catalog numbers is presented in Table S1. Immunoreactive proteins were visualized using the ECL system (Amersham, RPN3004) or the SuperSignal WestPico chemiluminescent substrate (Pierce, 34080) and then exposed to X-ray film. The ImageJ software program from the NIH was used for the densitometry analysis.
siRNA
All siRNAs including nontargeting siRNA were obtained from Dharmacon. For the siRNA transfection, SK-Hep-1 cells at a concentration of 5 × 104 cells/dish were incubated for 24 h in 6 cm dishes. The cells were then transfected with 100 nM siRNA in Opti-MEM medium (Invitrogen, 11058-021) without FBS using Lipofectamine 2000 (Invitrogen, 11668027) according to the manufacturer’s protocol. The siRNA sequences and catalog numbers are described in Table S2.
shRNA
Lentiviral particles expressing short hairpin RNAs (Mission shRNA) against ATG7 and BNIP3 and nontargeting shRNAs were purchased from Sigma. Lentiviral particles were transfected into SK-Hep-1 and HeLa cells in the presence of 8 μg/ml Polybrene (Sigma, H9268). Twenty-four hours after the transfection, the cells were selected with 2 ug/ml of puromycin for one month. The target sequences and catalog numbers of the hairpin RNAs against ATG7 and BNIP3 and the nontargeting shRNAs are described in Table S3.
Immunofluorescence microscopy
SK-Hep-1 cells were untransfected or transiently transfected with Flag-BNIP3 or BNIP3△TM expression vector as described. After 24 h of transfection, the cells were incubated with DMEM or EBSS for 4 h, and then incubated with MitoTracker Red (Invitrogen, M7512) for 45 min at 37°C. After PBS washing, cells were fixed with 3.7% paraformaldehyde for 30 min at RT and permeabilized with 0.2% Triton X-100 in PBS (PBST). After rinsing, cell were blocked with PBST containing 1% bovine serum albumin (PBST-B) and then incubated with the monoclonal anti-mouse Flag antibody (1:100) in PBST-B overnight at 4°C. After being washed, cells were treated with anti-mouse secondary antibody conjugated to FITC (Sigma, F0257, 1:400) for 1 h at RT. After PBS washing, cells were incubated with Hoest 33342 for nucleus staining. Coverslips were mounted on glass slides and fluorescence images were obtained using Fluoview FV 1000 fluorescence microscope.
Trypan blue assay
The culture medium was removed and replaced with 0.4% trypan blue (Sigma, T8154) in PBS for 5 min at room temperature. The percentage of dead cells was calculated by counting the number of blue cells in a population of clear cells. At least 200 cells were counted for each assay.
Soft agar growth transformation assay
Anchorage-independent growth was assessed by colony formation on soft agar. Briefly, equal volumes of agar (1%, DNA grade) and 2× DMEM (with 20% FBS) were mixed at 40°C to produce a 0.5% base agar in six-well tissue culture plates. Cells (0.1 ml at a concentration of 2 × 105 cells/ml) were suspended in the solution containing 3 ml of 2× DMEM (with 20% FBS) and 3 ml of 0.7% agar. An equal volume (1.5 ml) of this cell suspension was then added to each well (as 0.35% top agar), with a final concentration of 5,000 cells per well. The top agar was then covered with culture medium. The plates were incubated at 37°C under 5% CO2 in a humidified incubator for 3 to 4 weeks, and the medium was changed every 3 to 4 d. Colony formation was observed using light phase-contrast microscopy, and photographs were obtained after staining with 0.5 ml of 0.005% crystal violet in PBS for 45 min at room temperature. The experiments were repeated three times to ensure the reproducibility of our results.
Statistics
Statistical analysis was performed using GraphPad Prism version 5.0 for Mac (GraphPad). All values are expressed as the means ± standard deviation (S.D.). Student’s t test was used to evaluate the significance between two experimental conditions, and p < 0.05 was considered to be statistically significant.
| Abbreviations: | ||
| AMPK | = | AMP-activated protein kinase |
| ATG7 | = | autophagy-related 7 |
| BAD | = | BCL2-associated agonist of cell death |
| BafA1 | = | bafilomycin A1 |
| BAK1 | = | BCL2-antagonist/killer 1 |
| BAX | = | BCL2-associated X protein |
| BCL2L11 | = | BCL2-like 11 (apoptosis facilitator) |
| BID | = | BH3 interacting domain death agonist |
| BECN1 | = | Beclin 1, autophagy-related |
| BNIP3 | = | BCL2/adenovirus E1B 19kDa interacting protein 3 |
| BNIP3L | = | BCL2/adenovirus E1B 19kDa interacting protein 3-like |
| CHX | = | cycloheximide |
| CMA | = | chaperone-mediated autophagy |
| COX4I1 | = | cytochrome c oxidase subunit IV isoform 1 |
| DFO | = | deferoxamine |
| EBSS | = | Earle’s Balanced Salt Solution |
| EIF4EBP1 | = | eukaryotic translation initiation factor 4E binding protein 1 |
| HIF1A | = | hypoxia inducible factor 1, alpha subunit (basic helix-loop-helix transcription factor) |
| I/R | = | ischemia-reperfusion |
| LAMP2 | = | lysosomal-associated membrane protein 2 |
| LDHA | = | lactate dehydrogenase A |
| LIR | = | MAP1LC3-interacting region |
| MAP1LC3 | = | microtubule-associated protein 1 light chain 3 |
| MTOR | = | mechanistic target of rapamycin |
| MTORC1 | = | MTOR complex 1 |
| NBR1 | = | neighbor of BRCA1 gene 1 |
| PIK3C3 | = | phosphatidylinositol 3-kinase, catalytic subunit type 3 |
| RPS6 | = | ribosomal protein S6 |
| RPTOR | = | regulatory associated protein of MTOR, complex 1 |
| RPS6KB1 | = | ribosomal protein S6 kinase, 70kDa, polypeptide 1 |
| SQSTM1 | = | sequestosome 1 |
| STK11/LKB1 | = | serine/threonine kinase 11 |
| TM | = | transmembrane domain |
| TOMM20 | = | translocase of outer mitochondrial membrane 20 homolog (yeast) |
| ULK1 | = | UNC-51-like kinase 1 |
| UPS | = | ubiquitin-proteasome system |
| 3MA | = | 3-methyladenine |
Additional material
Download Zip (29.6 MB)Acknowledgments
We sincerely thank Dr. D. Sabatini (Massachusetts Institute of Technology, MA USA) and Dr. N. Gray (Harvard Medical School, MA USA) for generously providing Torin1 and Dr. J. Jung (University of Southern California, CA USA) for providing the pEGFP-MAP1LC3B vector. We also thank Mr. Jae Hyun Park (IPS-TECH) for technical support on the hypoxia experiment. This work was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korea government (MEST): the National Honor Scientist Support Program (No. 20100020417) and by the WCU program, NRF, MEST, Korea (R31-10105).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/23072
References
- Høyer-Hansen M, Jäättelä M. Autophagy: an emerging target for cancer therapy. Autophagy 2008; 4:574 - 80; PMID: 18362515
- Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 2008; 8:705 - 13; http://dx.doi.org/10.1038/nrc2468; PMID: 19143055
- Mazure NM, Pouysségur J. Hypoxia-induced autophagy: cell death or cell survival?. Curr Opin Cell Biol 2010; 22:177 - 80; http://dx.doi.org/10.1016/j.ceb.2009.11.015; PMID: 20022734
- Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ 2005; 12:Suppl 2 1542 - 52; http://dx.doi.org/10.1038/sj.cdd.4401765; PMID: 16247502
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22:132 - 9; http://dx.doi.org/10.1016/j.ceb.2009.12.004; PMID: 20056399
- Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132 - 41; http://dx.doi.org/10.1038/ncb2152; PMID: 21258367
- Kundu M. ULK1, mammalian target of rapamycin, and mitochondria: linking nutrient availability and autophagy. Antioxid Redox Signal 2011; 14:1953 - 8; http://dx.doi.org/10.1089/ars.2010.3809; PMID: 21235397
- Roach PJ. AMPK -> ULK1 -> autophagy. Mol Cell Biol 2011; 31:3082 - 4; http://dx.doi.org/10.1128/MCB.05565-11; PMID: 21628530
- Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344 - 8; http://dx.doi.org/10.1126/science.1193497; PMID: 21127245
- Boyd JM, Malstrom S, Subramanian T, Venkatesh LK, Schaeper U, Elangovan B, et al. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 1994; 79:341 - 51; http://dx.doi.org/10.1016/0092-8674(94)90202-X; PMID: 7954800
- Ray R, Chen G, Vande Velde C, Cizeau J, Park JH, Reed JC, et al. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem 2000; 275:1439 - 48; http://dx.doi.org/10.1074/jbc.275.2.1439; PMID: 10625696
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010; 11:45 - 51; http://dx.doi.org/10.1038/embor.2009.256; PMID: 20010802
- Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA 2000; 97:9082 - 7; http://dx.doi.org/10.1073/pnas.97.16.9082
- Chinnadurai G, Vijayalingam S, Gibson SB. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene 2008; 27:Suppl 1 S114 - 27; http://dx.doi.org/10.1038/onc.2009.49; PMID: 19641497
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 2009; 29:2570 - 81; http://dx.doi.org/10.1128/MCB.00166-09; PMID: 19273585
- Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ 2007; 14:146 - 57; http://dx.doi.org/10.1038/sj.cdd.4401936; PMID: 16645637
- Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 2005; 24:980 - 91; http://dx.doi.org/10.1038/sj.onc.1208095; PMID: 15592527
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283:10892 - 903; http://dx.doi.org/10.1074/jbc.M800102200; PMID: 18281291
- Gustafsson ÅB. Bnip3 as a dual regulator of mitochondrial turnover and cell death in the myocardium. Pediatr Cardiol 2011; 32:267 - 74; http://dx.doi.org/10.1007/s00246-010-9876-5; PMID: 21210091
- Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, et al. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ 2011; 18:721 - 31; http://dx.doi.org/10.1038/cdd.2010.146; PMID: 21278801
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis?. Nat Rev Cancer 2004; 4:891 - 9; http://dx.doi.org/10.1038/nrc1478; PMID: 15516961
- Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 1996; 271:32529 - 37; http://dx.doi.org/10.1074/jbc.271.51.32529; PMID: 8955077
- Cuervo AM. Cell biology. Autophagy’s top chef. Science 2011; 332:1392 - 3; http://dx.doi.org/10.1126/science.1208607; PMID: 21680833
- Kundu M, Thompson CB. Macroautophagy versus mitochondrial autophagy: a question of fate?. Cell Death Differ 2005; 12:Suppl 2 1484 - 9; http://dx.doi.org/10.1038/sj.cdd.4401780; PMID: 16247496
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- He H, Dang Y, Dai F, Guo Z, Wu J, She X, et al. Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. J Biol Chem 2003; 278:29278 - 87; http://dx.doi.org/10.1074/jbc.M303800200; PMID: 12740394
- Yasuda M, Theodorakis P, Subramanian T, Chinnadurai G. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J Biol Chem 1998; 273:12415 - 21; http://dx.doi.org/10.1074/jbc.273.20.12415; PMID: 9575197
- Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci U S A 2002; 99:12825 - 30; http://dx.doi.org/10.1073/pnas.202474099; PMID: 12226479
- Gang H, Hai Y, Dhingra R, Gordon JW, Yurkova N, Aviv Y, et al. A novel hypoxia-inducible spliced variant of mitochondrial death gene Bnip3 promotes survival of ventricular myocytes. Circ Res 2011; 108:1084 - 92; http://dx.doi.org/10.1161/CIRCRESAHA.110.238709; PMID: 21415393
- Billen LP, Shamas-Din A, Andrews DW. Bid: a Bax-like BH3 protein. Oncogene 2008; 27:Suppl 1 S93 - 104; http://dx.doi.org/10.1038/onc.2009.47; PMID: 19641510
- Danial NN. BAD: undertaker by night, candyman by day. Oncogene 2008; 27:Suppl 1 S53 - 70; http://dx.doi.org/10.1038/onc.2009.44; PMID: 19641507
- Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene 2008; 27:Suppl 1 S2 - 19; http://dx.doi.org/10.1038/onc.2009.39; PMID: 19641503
- Sakamaki J, Daitoku H, Ueno K, Hagiwara A, Yamagata K, Fukamizu A. Arginine methylation of BCL-2 antagonist of cell death (BAD) counteracts its phosphorylation and inactivation by Akt. Proc Natl Acad Sci U S A 2011; 108:6085 - 90; http://dx.doi.org/10.1073/pnas.1015328108; PMID: 21444773
- Ma X, Godar RJ, Liu H, Diwan A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 2012; 8:297 - 309; http://dx.doi.org/10.4161/auto.18658; PMID: 22302006
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 2012; 287:19094 - 104; http://dx.doi.org/10.1074/jbc.M111.322933; PMID: 22505714
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279 - 96; http://dx.doi.org/10.4161/auto.7.3.14487; PMID: 21189453
- Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal 2009; 2:pe24; http://dx.doi.org/10.1126/scisignal.267pe24; PMID: 19383975
- Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy 2009; 5:725 - 6; http://dx.doi.org/10.4161/auto.5.5.8504; PMID: 19395872
- Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2 - 11; http://dx.doi.org/10.1128/MCB.06159-11; PMID: 22025673
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456 - 61; http://dx.doi.org/10.1126/science.1196371; PMID: 21205641
- Rothbart SB, Racanelli AC, Moran RG. Pemetrexed indirectly activates the metabolic kinase AMPK in human carcinomas. Cancer Res 2010; 70:10299 - 309; http://dx.doi.org/10.1158/0008-5472.CAN-10-1873; PMID: 21159649
- Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol 2007; 27:6229 - 42; http://dx.doi.org/10.1128/MCB.02246-06; PMID: 17576813
- Kothari S, Cizeau J, McMillan-Ward E, Israels SJ, Bailes M, Ens K, et al. BNIP3 plays a role in hypoxic cell death in human epithelial cells that is inhibited by growth factors EGF and IGF. Oncogene 2003; 22:4734 - 44; http://dx.doi.org/10.1038/sj.onc.1206666; PMID: 12879018
- Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating?. Nat Rev Mol Cell Biol 2012; 13:7 - 12; http://dx.doi.org/10.1038/nrm3249; PMID: 22166994
- Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008; 8:851 - 64; http://dx.doi.org/10.1038/nrc2501; PMID: 18846101
- Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ 2008; 15:1572 - 81; http://dx.doi.org/10.1038/cdd.2008.84; PMID: 18551130
- Kaper F, Dornhoefer N, Giaccia AJ. Mutations in the PI3K/PTEN/TSC2 pathway contribute to mammalian target of rapamycin activity and increased translation under hypoxic conditions. Cancer Res 2006; 66:1561 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-05-3375; PMID: 16452213
- Chhipa RR, Wu Y, Ip C. AMPK-mediated autophagy is a survival mechanism in androgen-dependent prostate cancer cells subjected to androgen deprivation and hypoxia. Cell Signal 2011; 23:1466 - 72; http://dx.doi.org/10.1016/j.cellsig.2011.04.008; PMID: 21554950
- Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol 2005; 17:617 - 25; http://dx.doi.org/10.1016/j.ceb.2005.10.001; PMID: 16243507
- Cosse J-P, Rommelaere G, Ninane N, Arnould T, Michiels C. BNIP3 protects HepG2 cells against etoposide-induced cell death under hypoxia by an autophagy-independent pathway. Biochem Pharmacol 2010; 80:1160 - 9; http://dx.doi.org/10.1016/j.bcp.2010.07.009; PMID: 20637734
- Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol 2006; 41:256 - 64; http://dx.doi.org/10.1016/j.yjmcc.2006.04.014; PMID: 16769083
- Ding Y, Sun X, Redfield M, Kushwaha S, Xu X. Target of rapamcyin (TOR)-based therapeutics for cardiomyopathy: insights from zebrafish genetics. Cell Cycle 2012; 11:428 - 9; http://dx.doi.org/10.4161/cc.11.3.19164; PMID: 22262179