Abstract
Autophagy is a highly conserved cellular process by which cytoplasmic components are sequestered in autophagosomes and delivered to lysosomes for degradation. As a major intracellular degradation and recycling pathway, autophagy is crucial for maintaining cellular homeostasis as well as remodeling during normal development, and dysfunctions in autophagy have been associated with a variety of pathologies including cancer, inflammatory bowel disease and neurodegenerative disease. Stem cells are unique in their ability to self-renew and differentiate into various cells in the body, which are important in development, tissue renewal and a range of disease processes. Therefore, it is predicted that autophagy would be crucial for the quality control mechanisms and maintenance of cellular homeostasis in various stem cells given their relatively long life in the organisms. In contrast to the extensive body of knowledge available for somatic cells, the role of autophagy in the maintenance and function of stem cells is only beginning to be revealed as a result of recent studies. Here we provide a comprehensive review of the current understanding of the mechanisms and regulation of autophagy in embryonic stem cells, several tissue stem cells (particularly hematopoietic stem cells), as well as a number of cancer stem cells. We discuss how recent studies of different knockout mice models have defined the roles of various autophagy genes and related pathways in the regulation of the maintenance, expansion and differentiation of various stem cells. We also highlight the many unanswered questions that will help to drive further research at the intersection of autophagy and stem cell biology in the near future.
Introduction
Macroautophagy (herein referred to as autophagy) is an evolutionarily conserved cellular process by which cells sequester a portion of their cytoplasm and organelles into double-membraned vesicles called autophagosomes that subsequently fuse with lysosomes for degradation of the enclosed materials.Citation1 Autophagosome formation is controlled by a series of protein complexes acting sequentially, including the ULK1-ATG13-RB1CC1/FIP200-C12orf44/ATG101 complex for autophagy induction, the class III phosphatidylinositol (PtdIns) 3-kinase complex (including BECN1, ATG14/ATG14L/Barkor, PIK3R4/VPS15, PIK3C3/VPS34 and AMBRA1) for initiation of autophagosomes, and the ATG12–ATG5-ATG16L1 complex and the MAP1LC3A/LC3 (ATG8 homolog)–phosphatidylethanolamine (PE) complex (both ubiquitin-like conjugation systems) for the extension and closure of the autophagosome double membranes (). Besides these core components, a number of other autophagy-related (ATG) genes characterized in yeast, and their mammalian orthologs, are also required for autophagy.Citation2 In addition, recent studies using proteomics approaches identified many other proteins in extensive networks that connect various cellular processes and signaling pathways to the autophagy machinery.Citation3
Figure 1. The process and regulation of autophagosome formation in mammalian cells. Mammalian autophagosome formation is induced by the activation of the ULK1 complex (comprising ULK1, ATG13, RB1CC1/FIP200 and C12orf44/ATG101), which is promoted by suppression of MTOR complex 1 (MTORC1) under starvation conditions or AMPK (both directly as well as by repressing MTORC1) in low-energy states. Once the activated ULK1 complex translocates to part of the endoplasmic reticulum (or possibly other intracellular membranes), it coordinates with the class III phosphatidylinositol-3-phosphate (PtdIns3P) kinase complex (including PIK3C3, BECN1, PIK3R4/p150, AMBRA1, UVRAG, ATG14 and SH3GLB1) for the initiation of autophagosomes upon formation of PtdIns3P and recruitment of double ZFYVE 1. Two ubiquitin-like systems including the ATG12–ATG5-ATG16L1 complex (containing ATG5, ATG7, ATG10, ATG12 and ATG16L1) and the LC3–phosphatidylethanolamine (PE) conjugation system (including ATG3, ATG4, ATG7 and MAP1LC3) will then promote the elongation of phagophore membranes for the formation of autophagosomes.
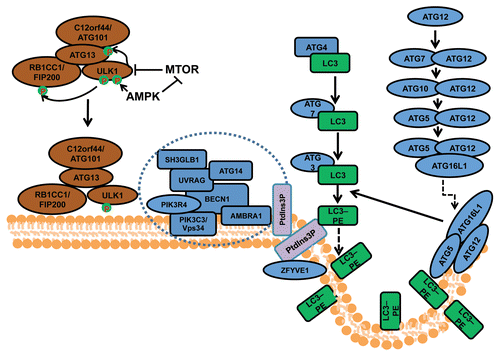
Upon autophagosome maturation and the fusion of its outer membrane with the lysosome membrane, the contents as well as the inner membrane of autophagosomes are degraded to generate amino acids and other cellular building blocks for recycling by the cell. Besides this recycling function in response to nutrient or energy starvation, autophagy is increasingly recognized as a quality control mechanism for both proteins and organelles.Citation2,Citation4,Citation5 Autophagy is induced as a result of various intrinsic and extrinsic cellular stress conditions, such as endoplasmic reticulum stress, reactive oxygen species (ROS) accumulation, hypoxia and bacterial infections. Activation of autophagy serves to clear the damaged protein aggregates, and impaired organelles as well as intracellular pathogens. Therefore, autophagy is crucial for maintaining cellular homeostasis as well as remodeling during normal development, and dysfunctions in autophagy have been associated with a variety of diseases such as cancer, inflammatory bowel diseases and neurodegenerative diseases.Citation4,Citation5 A hallmark of cellular defects in neurodegenerative diseases is the accumulation of ubiquitinated protein aggregates (or inclusion bodies), which is normally cleared by the ubiquitin-proteasome degradation system as well as autophagy for larger aggregates.Citation6-Citation8 The accumulation of large protein aggregates has been linked to increased neuronal apoptosis and axonal degeneration, accounting for the phenotypes in neurodegenerative diseases. In cancer, defective autophagy has been linked to increased DNA damage and gene mutations, leading to increased tumorigenesis,Citation9 as well as to the reduced proliferation of tumor cells during cancer progression and metastasis.Citation10,Citation11
In contrast to the body of data derived from studies of somatic cells and disease models, the role of autophagy in the maintenance and function of stem cells is poorly understood. Both embryonic and various tissue stem cells play essential roles in development, tissue renewal and certain disease processes. Many stem cells are also long lived and persist throughout the adult life of an organism, thus the quality control mechanisms and maintenance of cellular homeostasis would be crucial for the maintenance of these cells. Thus, autophagy is expected to play an important role in the normal function of stem cells and associated diseases. Indeed, consideration of the available data suggests that the unique properties of stem cells (self-renewal, pluripotency, differentiation and quiescence) are dependent on the activation of the autophagic process.Citation12,Citation13 First, the processes of self-renewal and differentiation both require a strict control of cellular remodeling that involves protein turnover and lysosomal degradation of organelles.Citation14 Second, the elimination and turnover of damaged macromolecules and organelles are essential to preserve the pluripotency of long-lived stem cells during their lengthy periods of quiescence.Citation15,Citation16 Third, basal autophagy is a homeostatic process that mediates quality control, clearance of altered and damaged intracellular proteins and organelles, and cellular remodeling through degradation of structural components. Not surprisingly, there is now accumulating evidence for the active role of autophagy in the regulation of embryonic stem cells (ESCs), several tissue stem cells, in particular hematopoietic stem cells (HSCs), as well as a number of cancer stem cells (CSCs). In addition to serving as an important mechanism for stem cell maintenance and self-renewal, autophagy has been implicated in the regulation of differentiation of various stem cells and their progenies, such as clearance of mitochondria and other organelles during erythrocyte maturation reported decades agoCitation17,Citation18 and recently validated by gene knockout studies.Citation19
In this review, we describe a growing body of knowledge encompassing a range of stem cell systems that have significantly advanced our understanding of autophagy in stem cell biology. We discuss how recent studies of multiple knockout mice models have defined the functions of the autophagy pathway and specific autophagy proteins in ESCs, HSCs and other stem cells. In each system, autophagy has been suggested to regulate the maintenance, expansion and differentiation of stem cells, although different cellular mechanisms may be involved. Given the special capacity of stem cells in perpetuating themselves through self-renewal and generating mature cells through differentiation, the recent advances suggest the emerging concepts that autophagy may function to balance the quiescence, self-renewal and differentiation of stem cells in normal physiological and various stress conditions.
Autophagy in Embryonic Stem Cells
ESCs are pluripotent stem cells present in the early embryo with the capacity to undergo long-term renewal and differentiate into the primary germ layers: ectoderm, endoderm and mesoderm. Investigations using this cell type will allow us to acquire an understanding of the role that autophagy plays in early stages of mammalian development. Much of our knowledge concerning the molecular aspects of autophagy is founded on studies in the yeast Saccharomyces cerevisiae that led to the identification of the autophagy-related (ATG) genes (see ref. Citation1 for review). These genes provided the essential tools for investigating the mechanism, regulation and role of autophagy. The prediction that a number of these ATG genes existed as homologs in higher eukaryotes prompted molecular studies in mammalian cells. The first detailed molecular study into autophagy in a mammalian cell setting was performed using mouse embryonic stem cells.Citation20 This study showed that bulk turnover of proteins labeled with [14C] amino acids can be induced by subjecting wild-type mouse ESCs to amino acid starvation. This bulk protein turnover is significantly reduced (> 50%) in mouse ESC (mESC) atg5−/− cells indicating the importance of autophagy in protein turnover. However, the absence of the ATG5 protein in these cells does not appear to affect their growth rate or colony morphology under replete culture conditions. Similarly, mES becn1−/− cells, lacking expression of the homolog of VPS30/ATG6, show no evidence of a growth defect under nonstarvation conditions,Citation21 although in a later study it was reported that these cells are unable to form embryoid bodies (see below).Citation22 Mice lacking expression of other individual autophagy-related genes including Atg7,Citation23 Atg9,Citation24 Atg3,Citation25 Atg16l1,Citation26 Becn1,Citation21 and Ambra1Citation27 have been generated, but investigations of ESCs isolated from these mice have not been reported in any detail.
Autophagy is essential for early but not late differentiation and development of embryos
Development and differentiation processes are often accompanied by considerable remodeling of cells and tissues, and it has been suggested that autophagy plays a key role in this process providing both recycled material for building new structures while removing the old material.Citation28 Since ESCs give rise to each of the three germ layers, it might be reasonable to expect that autophagic activity would be indispensable for normal differentiation and development of the embryo. However, it is not clear from the available evidence that this is the case.
It has been established that autophagy is essential for the very early stages of embryogenesis. Fertilized mouse oocytes lacking ATG5 (by oocyte-specific conditional knockout of the Atg5 gene, thus also removing maternal ATG5 protein) do not proceed beyond the 4- to 8-cell stage if they were fertilized by Atg5-null sperm, and therefore fail to form the blastocysts and the inner cell mass.Citation29 Using GFP-LC3 as a marker, autophagy was found to be significantly upregulated in wild-type fertilized rat oocytes immediately (4 h) after fertilization of the oocyte by the spermatoazoa.Citation29 Autophagic levels were subsequently suppressed between the 1- and 2-cell stage but elevated at the 4-cell stage. The levels of autophagy in cells in the later stages of development including the blastocyst stage containing the inner-cell mass from which the ESCs were derived were not reported.Citation29 It is not clear why the very early stages are dependent on autophagic activity for survival. Presumably autophagy is required for controlling levels of key regulatory protein complexes or perhaps to provide substrates for cellular energy homeostasis prior to pre-implantation, after which cells have access to trans-placental nutrients.
The role of autophagy in ESC function in later stages of embryo development is less clear according to experimental observations of mice lacking expression of a number of different ATGs. atg3−/−,Citation25 atg5−/−,Citation30 atg7−/−,Citation23 atg9−/−24 or atg16l1−/−26 mice give birth to phenotypically normal litters at Mendelian frequencies without any apparent anatomical abnormalities, suggesting that autophagy does not appear to play a pivotal role in the timing and coordination of differentiation in the developing embryo. These autophagy-deficient embryos are able to develop beyond the 8-cell stage (and indeed give rise to birth of live pups) due to the presence of maternally inherited ATG proteins in the oocyte cytoplasm. However, compared with pups of wild-type litters, autophagy-deficient pups have reduced birth weight and typically die within 1–2 d of birth possibly due to suckling defects caused by deficient neurological development.
Although the results of investigations using autophagy-deficient mice suggest that autophagy is dispensable for embryogenesis, it is possible that cellular and tissue defects arising from autophagic deficiency are more subtle, and have yet to be identified in the embryo. For example, atg5−/− mice develop neuronal inclusions and have decreased adipose mass. Autophagy plays an important role in quality control of cell components. It is possible that the lack of autophagy in ESCs contributes to the accumulation of damaged cellular components during embryogenesis, the implications of which are manifested later in life, and in terminally differentiated cells. Tissue-specific knockouts of different ATG genes give rise to a range of phenotypes, many of which relate to the unwanted accumulation of aggregates and damaged organelles such as mitochondria (reviewed in ref. Citation2) .
It is possible that other quality control pathways such as the ubiquitin-proteasome system (UPS) are to some extent able to compensate for the absence of autophagic activity in ESCs. Human ESCs (hESCs) exhibit high proteasome activity that is downregulated upon differentiation, suggesting that high proteasome activity is an intrinsic characteristic of hESC identity.Citation31 Furthermore, hESCs lose their high proteasome activity in a continuous and progressive manner during the differentiation process, and differentiated cells showed increased levels of polyubiquitinated proteins. However, in another study it was reported that proteins damaged by carbonylation or formation of advanced glycation end products accumulate in murine ESCs but are cleared upon differentiation, an event that correlates with increased proteasome activity.Citation32 It is possible that increased autophagic activity observed upon differentiation contributes to the removal of such damaged proteins. Further studies are required to investigate the relationship between the UPS and autophagy in ESCs.
In contrast to other ATG genes, Becn1−/− mice exhibit a profound developmental delay at E6.5 with the amniotic fold failing to developCitation33 and consequently die at an early embryonic stage. Widespread cell death is present in null embryos at E7.5. It has been suggested that BECN1 through its binding partners and control of the PIK3C3 lipid kinase is involved in membrane trafficking events other than autophagy, thereby explaining the more severe phenotype of becn1−/− miceCitation34 compared with other ATG knockout mice. Becn1+/− mice survive, but develop tumors indicating that Becn1 can also function as a haploinsufficient tumor suppressor gene. AMBRA1 is a positive regulator of BECN1-dependent autophagy. However, a functional deficiency of AMBRA1 in mouse embryos does not phenocopy BECN1 deficiency, but rather leads to severe neural tube defects, accumulation of ubiquitinated proteins, unbalanced cell proliferation and excessive apoptotic cell death, suggesting that AMBRA1 may regulate target genes other than Becn1 or that BECN1 may have additional roles at later developmental stages.
Autophagy is required for embryoid body formation
mESCs deficient in ATG5 progress normally through embryonic development. However, there is some evidence from studies using an in vitro model of development that suggests autophagy may be important under particular circumstances. In one study it was reported that when compared with wild-type mESCs, autophagy-deficient mESCs cultured outside of the blastocyst exhibit altered behavior.Citation22 Wild-type mESCs cultured in the absence of feeder cells and leukemia inhibitory factor (LIF) are able to form undifferentiated cell aggregates that develop into simple embryoid bodies (EBs) that contain an outer layer of primitive endoderm cells and an inner solid core of ectodermal cells. Cystic EBs are formed when the inner ectodermal cells undergo programmed cell death. These events mimic cavitation in early embryo development (PCD).
In this model of early embryo development, autophagy in wild-type ESCs is detectable throughout development, but is greatest at times when programmed cell death is maximal during cavitation. Compared with wild-type ESCs, atg5−/− and becn1−/− ES cells cultures in vitro fail to undergo cavitation. The results of further experiments indicated that dying cells do not express the engulfment signals for phagocytosis including phosphatidyl or lysophosphatdylcholine. EBs derived from these atg5−/− and becn1−/− cells have a defect in ATP production which, when reversed using methylpyruvate, restores cavitation and removal of apoptotic corpses. Methylpyruvate is a cell-permeable form of pyruvate that can feed in to the mitochondrial tricarboxylic acid cycle. These results suggest that the cells in EBs rely on autophagy for energy homeostasis presumably through the production of amino acids.
The role of autophagy for energy homeostasis in normal mammalian post-implantation embryo development may be limited, given the reliable supply of trans-placental nutrients. This notion is supported by the results of experiments investigating development in other organisms including insects such as the fruit fly Drosophila melanogaster, which do not have a maternal nutrient supply. In these species autophagy-deficient embryos suffer severe developmental defects.Citation35 Nevertheless, the lowered birth weight of autophagy-deficient mice suggests a disturbance in their metabolism.
The majority of studies investigating autophagy in ESCs have been performed using mice. However, hESC lines that constitutively express GFP-LC3 (HES3-GFP-LC3) have been developed and validated for monitoring autophagic events.Citation36 Since GFP-LC3 expression is maintained after teratoma formation, these cell lines represent a useful tool for investigating autophagy during early human embryogenesis. The results of studies using these cells suggest links between autophagy and differentiation. HES3-GFP-LC3 cells deprived of mouse embryonic fibroblast (MEF)-secreted maintenance factors by culturing in unconditioned medium for 3 d undergo spontaneous differentiation, and exhibit a significant increase in the number of fluorescent puncta compared with cells maintained in conditioned medium.Citation36 Acute induction of differentiation using the TGFβ/TGFβ receptor II inhibitor SB431542 leads to a rapid increase in fluorescent puncta as early as 2 h after addition of the inhibitor. The inhibitor does not induce autophagy in differentiated cell types. Although unclear as to whether autophagy acts upstream or concomitant with differentiation, acute induction by SB431542 suggests a regulatory upstream role for degradation of pluripotency-regulating protein complexes.
The available evidence suggests that MTOR plays an important role in the regulation of pluripotency and self-renewal in hESCs. MTOR is also a central player in the regulation of autophagy. Inhibition of MTOR with rapamycin or depletion of MTOR transcript in hESCs leads to a significant reduction in levels of the pluripotency regulation transcription factors (POU5F1/Oct4 and SOX2), promotion of mesoderm and endoderm activities and decreased proliferation.Citation37 Disrupting the kinase activity of MTOR in mESCs results in reduced cell size and a proliferation arrest.Citation38 The regulation of MTOR appears to differ according to the differentiation pathway. A transcriptome analysis of hESCs differentiating into neural cells indicated that transcripts associated with MTOR are upregulated.Citation39 Although not reported in these studies, autophagic activity would be expected to be significantly upregulated upon MTOR inhibition. It is possible that autophagy plays a role, albeit indispensable, in the loss of pluripotency and self-renewal properties of ESCs. The targets for degradation by autophagy remain to be identified.
Mitochondria and mitophagy in ESCs
Macroautophagy was originally thought to be restricted to the bulk turnover of cellular components. However, it is now apparent that a number of different cellular components can be targeted by the autophagic machinery in a selective manner using different receptor complexes.Citation40 Studies in ESCs on the different selective forms of autophagy are limited to a few reports, and include the elimination of mitochondria (mitophagy)Citation41,Citation42 and midbodies (midbophagy).Citation43 It is likely that other selective forms of autophagy occur in ESCs but have yet to be reported.
In mammalian cells the mitochondrion is responsible for satisfying the majority of ATP requirements. Our knowledge of mitochondria in stem cells is limited.Citation44 Observations of mitochondria in a number of human and mouse ESC lines using electron microscopy show the presence of few mitochondria that also have poorly developed cristae, the site of oxidative phosphorylation, and ATP production. Fluorescence microscopy shows mitochondria to be located in small perinuclear groups. The energy demands of stem cells are thought to be largely met by the action of glycolysis.Citation45 The proliferative capacity of mouse ESCs indeed closely correlates with high activity of different glycolytic enzymes, elevated glycolytic flux and low mitochondrial oxygen consumption. However, Zhou et al. showed that hESCs rather exhibit aerobic glycolysis,Citation46 a phenomenon often seen in highly proliferative cells such as cancer cells and during embryogenesis,Citation47,Citation48 and that the uncoupling of glycolysis and oxidative phosphorylation in hESCs is caused by the expression of UCP2, presumably inhibiting the entry of pyruvate into the TCA cycle via pyruvate dehydrogenase. These data challenge the commonly held belief that mitochondria in hESCs are not metabolically active because they exhibit only few and underdeveloped cristae.Citation49-Citation51
Maintaining integrity, function and regulation of mitochondrial dynamics is important for all cells, and mitophagy contributes to their quality control. Mitophagy is tightly controlled in ESCs. BNIP3L/NIX, a BCL2 family member, is required for the elimination of mitochondria by mitophagy during erythroid maturation. Although mESC bnip3l−/− cells appear normal, mice derived from the cell are anemic.Citation52 The results of a recent study provide evidence that mitophagy can be induced under particular circumstances in mESCs, and may play an important role in quality control in stem cells. In this study it was found that downregulation of the growth factor, augmentation of liver regeneration (GFER) protein, a portion of which is located in the inter-membrane space of the mitochondrion, results in loss of mitochondrial membrane potential, excessive fragmentation and elimination of damaged mitochondria by mitophagy.Citation41,Citation42 These ESCs also have significantly reduced levels of pluripotency marker gene expression. It is suggested that GFER modulates the mitochondrial fission GTPase DNMIL/DRP1, a protein responsible for promoting mitochondrial fragmentation. Depletion of GFER in differentiated cells does not affect mitochondrial functionCitation42 suggesting that the role of GFER is restricted to ESCs. The organization and function of mitochondria in ESCs are distinct from their differentiated counterparts, and maintaining the “status quo” appears to be vital for their survival. Mitophagy could also be induced in ESCs by inhibition of complex I of the respiratory chain using rotenone, and mild inhibition of complex III enhances pluripotency of ESC, suggesting that maintenance of mitochondrial integrity is important in ESCs.Citation53
The trigger for loss of pluripotency markers in cells deficient in GFER is not clear, but it is possibly related to changes in mitochondrial dynamics. Mitochondria in normal ESCs undergo dramatic changes in appearance and function during differentiation, and mitophagy may serve in a surveillance role ensuring alterations in mitochondrial behavior is suppressed in the absence of developmental signals.
It has been suggested that the low numbers of mitochondria and the immature morphology of mitochondria in ESCs may be required to maintain the “stemness” of ESCs.Citation41,Citation42 Mitochondrial mass, mitochondrial DNA and ATP content of cells increase significantly when hESCs undergo differentiation.Citation54 Increased mitochondrial activity is accompanied by increased production of ROS as a by-product of oxidative phosphorylation. Antioxidant enzyme production including mitochondrial and cytosolic dismutases, catalase and peroxiredoxins are also increased in differentiating cells.Citation54 Nevertheless, ROS is elevated suggesting these cells are at risk from damage by ROS. Since autophagy is a mechanism for removing cellular constituents that have sustained damage, it is likely that autophagy would be upregulated in ESCs in response to increased mitochondrial activity and ROS production. It is also possible that increased ROS is required for signaling and control of events during differentiation.Citation55 Accordingly, in addition to a role for energy homeostasis, autophagy (mitophagy) is likely to be responsible for quality control of mitochondria in ESCs. Given the pluripotent nature of ESCs, such quality control would be essential, which needs to be further investigated during ESC renewal and differentiation if these cells are to be used for therapeutics.
Midbophagy
The existence of multiple forms of autophagy (including nonspecific autophagy) implies that each is separately regulated in a temporal and spatial manner. Accordingly, mitophagy is activated to maintain the quality of the mitochondrial population, and other forms of autophagy may be activated under different conditions. The removal of midbodies by midbophagy occurs as ESCs undergo differentiation.Citation43 Midbodies are organelles that form between daughter cells during cytokinesis and are required for their final separation. They selectively accumulate in stem cells and induced pluripotent stem cells (iPSCs) in vitro and in vivo. It is suggested that their accumulation may function to maintain or enhance the pluripotency of stem cells. Correct control of midbophagy would be essential to maintain the pluripotent state of ESCs. It is possible that induction of bulk autophagy by starvation or treatment with rapamycin stimulates differentiation through removal of midbodies.
Role of Autophagy in Hematopoietic Stem Cells
Hematopoietic stem cells are perhaps the best studied stem cells in the body due to relatively easy access to populations in the bone marrow and blood, combined with well-established assays in vitro and vivo. The hematopoietic system develops in a hierarchical fashion; a small number of long-term stem cells that are relatively quiescent divide to become progenitors that proliferate and differentiate into mature blood cell lineages that are produced in large numbers every day (). The lympho-hematopoietic system is composed largely of cells with short life spans and therefore requires continuous replenishment. In contrast, the cellular source of these blood cell lineages, the adult hematopoietic stem cells (HSCs) are long-lived and not depleted during the lifetime of the organism.
Figure 2. Autophagy in stemness of hematopoietic stem cells. During hematopoiesis, differentiated blood cells are generated from hematopoietic stem cells (HSCs). Long-term HSCs develop into short-term HSCs, multi- and then oligopotent progenitors and lineage-restricted progenitors, which give rise to the differentiated blood cells. These include erythrocytes, platelets, granulocytes, macrophages, dendritic cells and the lymphocytes T, B and NK cells. Autophagy is thought to play a role in self-renewal and quiescence in HSCs, the two hallmarks of stemness. Moreover, during differentiation autophagy is hypothesized to help multipotency and remodeling. Last, evidence is increasing that autophagy is required to maintain the healthy differentiated lineages.
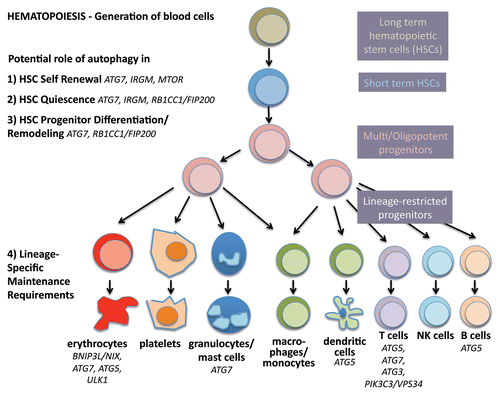
Over the last decade, much progress has been made toward murine and human HSC isolation and identification, based mainly on their characteristic cell surface proteins that are either present (ATXN1/Sca-1 and KIT/c-Kit) or absent (lineage markers such as ITGAM/MAC-1 for mature myeloid cells, CD8 for T cells, PTPRC/B220 for B cells) on immature cells. True long-term murine HSCs are lin-, KIT+, ATXN1+, and further identified by SLAM family markers (CD48- and SLAMF1/CD150+), while lin- CD34+ CD38- enriches them in humans. HSCs reside in a specific hypoxic microenvironment or niche within the bone marrow, the majority in a quiescent state (reviewed in ref. Citation56). In response to intrinsic or extrinsic cues regarding the need for mature blood cells, HSCs can be stimulated to regenerate another HSC (self-renewal) or in asymmetric cell division generate a progenitor cell that can then produce the mature hematopoietic cell lineages (differentiation/multipotency). In summary, and like other stem cells, true HSCs strike a fine balance between quiescence, self-renewal and differentiation. When this balance is not executed properly, the consequences are severe, including either biased differentiation and thereby depletion or overrepresentation of some lineages (e.g., anemia, lymphopenia, myeloproliferation,) and/or hematopoietic malignancies (e.g., leukemias); it is well accepted that transformation events may occur as early as the stem or progenitor cell stage.
There are very few studies that have investigated autophagy in HSCs, but at least one of them has indicated that autophagy is highly active in HSCs in humans.Citation57 Furthermore, murine HSCs, in contrast to their short-lived myeloid progeny, robustly induce autophagy after ex vivo cytokine withdrawal and in vivo calorie restriction, driven by the transcription regulator FOXO3.Citation58 For this review, our prevailing hypothesis is that autophagy is essential to strike the balance between quiescence, self-renewal and differentiation in HSCs.
Autophagy in quiescence of HSCs
The majority of HSCs reside in the G0/G1 phase of the cell cycleCitation59 and are in a state of reversible cell cycle arrest known as quiescence. Quiescent cells are particularly hardy and able to survive long periods of nutrient starvation; entry into quiescence is often associated with metabolic changes (reviewed in ref. Citation60). While proliferating cells devote much of their metabolic capacity to biosynthesis in order to create the material necessary to form a new cell, quiescent cells are relieved of this expensive metabolic requirement. HSCs downregulate protein synthesis and activate pathways that sustain them during periods of nondivision. In addition to cyclin-dependent kinase inhibitors, regulation of quiescence is achieved through the PtdIns3K-MTOR pathway.Citation61-Citation64 Indeed, increased MTOR activity has deleterious effects on HSCs, including excessive HSC proliferation leading to leukemias, followed by stem cell depletion and failure to repopulate the hematopoietic lineages.
The following indirect evidence points toward the idea that autophagy is required for the maintenance of quiescence in HSCs:
First, the slow metabolic turnover of quiescent HSCs means that damaged mitochondria and proteins cannot be easily passed on to daughter cells and thereby diluted, so autophagy may be essential for the required increase in catabolic rate. Interestingly, the UPS, the other major degradation system in the cell, has also been implicated in leukemogenesis. CBL/C-CBL, FBXW7/FBW-7, VHL and MDM2 are molecules from the UPS pathway that are often modified in hematopoietic malignancies. The deletion of genes encoding these protens in mice leads to deleterious HSC phenotypes (reviewed in ref. Citation65).
Second, quiescence is accompanied by changes in metabolism, in which autophagy could play a role due to its role in the clearance of mitochondria (mitophagy). Only a few studies have addressed how HSCs generate ATP.Citation66 However, high levels of autophagy,Citation57,Citation67 low mitochondrial content in HSCsCitation68 and the fact that hypoxia, common in the oxygen-limited niche environment, switches cells to a glycolytic metabolism all point toward the idea that quiescent HSCs primarily use glycolysis rather than oxidative phosphorylation to meet energy requirements.Citation66,Citation69
Third, we know from other cell types that mitophagy controls ROS levels. Increased ROS promote HSCs’ exit out of the quiescent stateCitation69 and mice with loss of autophagy in HSCs develop myeloproliferation,Citation67 a sign of loss of quiescence. Interestingly, ATG7 has recently been shown to bind and modulate TP53/p53, thereby controlling exit out of the cell cycle in response to metabolic stress. Indeed, starved murine fibroblasts lacking ATG7 fail to undergo cell cycle arrest.Citation70
Induction of quiescence and slow cell cycle turnover help ensure maintenance and preservation of long-lived stem cells. This also ensures that HSCs age more slowly, undergoing fewer cycles of replication leading to a minimum of telomere shortening.Citation71 However, cell damage and telomere erosion is not completely prevented in HSCs, despite the slow turnover and the fact that stem cells are one of the few cell types in the body to express telomerase.Citation72 Similar to other cells, entering senescence is important for stem cells to avoid accumulation of damaged proteins, lipids and nucleic acids, and may prevent transformation. Clearly, maintaining cell health and preventing stem cell aging is vital for hematopoietic health, and autophagy’s role in degrading damaged molecules and organelles may be essential to keep cells young. Importantly, autophagy has been inextricably linked to aging: (1) autophagy levels generally decrease with age in multiple cell types including hematopoietic cells,Citation73 (2) loss of function mutations in ATG genes decreases life span in yeast, (3) calorie restriction and rapamycin increases life span in C. elegans and rats, and (4) tissue-specific loss of autophagy genes in mice leads to age-related diseases such as neurodegeneration, diabetes and cancer.Citation5 HSCs from aged mice show a reduced repopulating activity per cell, attenuated self-renewal and homing abilities, myeloid skewing of differentiation, and an increased level of stress-induced apoptosis.Citation74 This phenotype is largely reminiscent of the one found in HSCs from mice deficient for ATG7 or RB1CC1/FIP200.Citation67,Citation75,Citation76 Furthermore, Chen et al. showed that HSCs can be rejuvenated through inhibition of MTOR signaling, restoring self-renewal and hematopoiesis in aged mice and enabling effective vaccination against flu.Citation77
Although it has not been shown directly that autophagy plays a role in HSC senescence, links exist and it is clear that prevention of senescence holds the key to prolonged HSC self-renewal capacity and maintaining hematopoietic homeostasis during the lifetime of an animal or human.
Autophagy in self-renewal of HSCs
Life-long maintenance of HSCs is achieved by ensuring a balance between differentiation and self-renewal. Not much is known about the molecular mechanisms of self-renewal except that control is exerted by the WNT pathway and BMI1, a member of the polycomb protein group. In bmi1−/− mice, HSC numbers are markedly reduced, and stem cell-associated survival gene and proliferation-modulating gene Cdkn2a (encoding P16INK4A and P19ARF in overlapping reading frames) is controlled by BMI1.Citation78 Moreover, recent evidence suggests that accumulation of DNA damage restricts the self-renewal capacity of human HSCsCitation79 with some molecular aspects of the pathway just being elucidated in mice.Citation80 Interestingly, according to this recent study, by committing to differentiate, the lymphoid-biased HSCs forgo self-renewal.Citation80
To date, serial transplantation is the best way to test the ability of HSCs to undergo self-renewal. According to this assay, HSCs from the original donor have self-renewal ability if the recipient of a bone-marrow transplant, Recipient 1 (whose own HSCs were completely ablated before the transplant), serves as a successful donor for subsequent (serial) transplants (Recipient 2, etc.). The number of serial transplants that the original donor's bone marrow can perform successfully is a measure of the self-renewal capacity of the donor’s HSCs.Citation81 To ascertain a role for autophagy in self-renewal, this assay should ideally be performed on autophagy-depleted HSCs. The fact that HSCs without essential autophagy genes fail to survive the first transplantation suggests that self-renewal requires autophagy.Citation67,Citation75 In a surrogate in vitro assay, the serial colony-forming assay, atg7−/− HSCs show a serious loss of colony formation on replating.Citation67 Similarly, human adult HSCs fail to form colonies in colony-forming assays when autophagy is inhibited by 3-methyladenine (3-MA) or ATG5 siRNA.Citation57 The authors of this study conclude diminished self-renewal capacity of HSCs in the absence of autophagy; however, without replating, it is not clear whether colonies formed from self-renewed stem cells, so strictly speaking replating would have been necessary to draw this conclusion.
In summary, as a self-renewing population that may have prolonged periods of quiescence, HSCs must possess robust defense and repair mechanisms in order to avoid damage. By contrast, differentiation of hematopoietic cells leads within days or weeks to cell death (i.e., in RBCs, neutrophils, platelets, and some types of myeloid cells), thereby eliminating long-term damage effects. In long-term self-renewing HSCs, such mechanisms have been shown to include senescence, apoptosis and premature differentiation.Citation69 Increasing evidence suggests that autophagy should be added to this list.
Autophagy in differentiation and multipotency of HSCs
As opposed to the UPS, autophagy can degrade cytoplasm in bulk, including damaged organelles such as mitochondria, ER and ribosomes. One of the best-studied examples for autophagy’s role in differentiation is the clearance of mitochondria in the developing red blood cell (RBC). As RBCs mature they degrade organelles and proteins, leaving only hemoglobin behind, allowing cells to shrink and squeeze through the smallest capillaries. While the nucleus is expelled from differentiating RBCs, clearance of mitochondria is mediated by autophagy. Mitophagy impairment via loss of either the autophagosome-targeting molecule BNIP3L,Citation52 ULK1 (a homolog of yeast Atg1, part of the initial signaling complex for autophagyCitation19) or ATG7, an essential molecule for the elongation of the autophagosome, results in serious RBC developmental and functional defects, including mitochondrial retention.Citation82,Citation83 This is the first convincing example in which autophagy aids execution of cellular differentiation requiring massive remodeling to accommodate specialized cellular functions. Interestingly, GATA1, the master regulator of hematopoiesis and erythropoiesis, was found to control the expression of several autophagy genes (MAP1LC3B and its homologs) as well as genes that control lysosomal biogenesis.Citation84
The bone marrow niche where HSCs reside is usually low in oxygen, and HSCs appear to avoid using oxidative phosphorylation for their energy supply, keeping the niche low in ROS.Citation85 Accordingly, normal HSCs have few mitochondria, and increased mitochondrial biogenesis results in defective HSC maintenance. The transition from quiescence to proliferation/differentiation is accompanied by increased MTOR activity, resulting in increased metabolic rate and ROS.Citation85,Citation86 Antioxidants or rapamycin restore the self-renewal ability of ROShigh HSCs.Citation77,Citation86 Interestingly, HSCs can be identified by their low mitochondrial metabolism using Rhodamine123, a vital dye based on mitochondrial membrane potential.Citation87 The migration of HSCs from the hypoxic niche to a microenvironment that is rich in oxygen and increased ROS is thought to promote their differentiation toward the myeloid lineage.Citation88 Interestingly, increased differentiation toward the myeloid lineage is a hallmark of both the aged and autophagy-deficient hematopoietic system.
Indeed, ROS accumulate in ATG7-deficient hematopoietic stem cells, possibly due to the generation of mitochondrial superoxides produced by spent mitochondria that are not being cleared away. Mice develop myelodysplastic syndrome with accumulation of myeloid cells; this may be a consequence of increased ROS levels biasing differentiation toward the myeloid lineage.Citation67 Similarly, a defect in RB1CC1/FIP200, a component of the ULK1-ATG13-RB1CC1/FIP200-C12orf44/ATG101 complex, leads to increased myeloid lineage cells.Citation75 In both of these models autophagy was deleted in the hematopoietic lineage alone (using the promoter Vav or Tie-2, respectively). Whether osteoclasts or macrophages or the HSC themselves (all present in the bone marrow niche) are responsible for providing a ROS rich environment remains to be shown. It may turn out to be similar, however, to the metabolic interactions between breast tumor cells and stroma demonstrated by Lisanti et al., where levels of autophagy and ROS in the tumor stroma have an impact on tumor progression (reverse Warburg effect).Citation89
Last, many publications point toward the idea that mouse models with modified signaling upstream of the MTOR pathway (constitutively active AKT1 or PTEN deficient) and in theory with decreased autophagy develop myeloid proliferation with increased LY6G+/Gr1+ ITGAM+/MAC-1+ myeloid cells,Citation63,Citation90 similar to the hematopoietic Atg7 knockout model. Conversely, the deletion of the MTORC1 component RAPTOR, therefore theoretically causing an increase in autophagy, results in a decrease of this myeloid population.Citation91 However, it remains to be shown definitively that loss or gain of autophagy contributes to this phenotype, as MTOR inhibition signals for many other important cellular functions such as inhibition of protein translation, mitochondrial biogenesis, cell growth, motility and proliferation.
In summary, there is strong evidence that autophagy plays a role in remodeling during hematopoietic cell differentiation and in the multipotency of HSCs.
Autophagy in Neural Stem Cells
Although it was initially described as a cellular response to starvation conditions in many organisms, autophagy at basal levels (i.e., independent of nutrient stress) has been increasingly recognized as an important mechanism to maintain the cellular homeostasis, particularly in postmitotic cells such as neurons. Indeed, the first observation of the autophagy process, that is, self-eating of the cytoplasm by lysosomes, was made by electron microscopy in neurons almost half a century ago.Citation92,Citation93 Along with ubiquitin-proteasome degradation machinery, autophagy has been proposed to play a protective role against the development of a number of neurodegenerative diseases.Citation6-Citation8 Indeed, neural-specific conditional knockout of essential autophagy genes such as Atg5, Atg7 or Rb1cc1/Fip200 result in abnormal accumulation of ubiquitinated protein aggregates, SQSTM1/p62 and damaged mitochondria, increased apoptosis and neurodegeneration,Citation94-Citation96 providing direct support for a role of basal autophagy in protecting against neurodegenerative diseases.
Despite extensive studies of autophagy in neurodegenerative diseases, surprisingly little is known about the potential role of autophagy in the regulation of neuronal stem cells (NSCs), which, unlike terminally differentiated and quiescent neurons, are capable of expansion through self-renewal and proliferation during neurogenesis as well as differentiation into other neural lineages. Like HSCs, NSCs have been extensively characterized by the use of a combination of markers as well as other approaches such as long-term label retention by the relatively quiescent NSCs residing within the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone of the hippocampal dentate gyrus in adult brain.Citation97,Citation98 Similar to HSCs, niches for NSCs are also hypoxic (e.g., the subgranular zone of the dentate gyrusCitation99) perhaps in order to maintain the relatively slow cycling property of these cells. Therefore, one hint of possible roles of autophagy in NSCs could be through regulation of ROS level and oxidative damage, which might otherwise be elevated in the hypoxic environment. Consistent with such an idea, deletion of the gene encoding the transcription factor PRDM16 results in an increased ROS production and abrogated NSC self-renewal, which can be rescued upon treatment with the antioxidant N-acetyl cysteine.Citation100 Moreover, although in other cells, recent studies suggested that hypoxia inducible factor 1, α subunit (basic helix-loop-helix transcription factor; HIF1A)-dependent expression of BNIP3 promotes mitophagy to control excess ROS production and ROS-induced cell death under conditions of prolonged hypoxia.Citation101,Citation102 In addition, inactivation of transcription factors FOXO1, FOXO3 and FOXO4 (either in combinationCitation103 or FOXO3 onlyCitation104) leads to defective self-renewal and differentiation of NSCs, accompanied with increased ROS, and such phenotypes are rescued by N-acetyl cysteine.Citation103 As recent studies suggested that FOXO1 can control autophagy in cancer cellsCitation105 and that FOXO3 regulates autophagy in muscles,Citation106,Citation107 these findings raised the interesting possibility that possible autophagy defects in these mice might contribute to the ROS elevation and NSC phenotypes caused by FOXO-deficiency.
Although autophagy may regulate NSCs in a similar manner as HSCs by controlling ROS levels, it is not clear whether the elevated ROS upon autophagy inhibition affect self-renewal of NSCs and HSCs through similar mechanisms. Whereas the increased ROS has been shown to deplete HSCs through promoting exit of the quiescent state, a recent study suggested that a high level of ROS is required for self-renewal of NSCs based on the observation of higher endogenous ROS levels in NSCs within the SVZ.Citation108 Nevertheless, there is considerable heterogeneity of the level of ROS in the SVZ, raising the interesting possibility that NSCs with the relatively higher level of ROS are in the proliferative or “activated” state whereas those with lower levels are the quiescent NSCs. It will be important to examine directly the potential role of autophagy in the self-renewal of NSCs by deletion of essential autophagy genes in these cells, and assuming that elevated ROS will be observed as in the case of HSCs, also to resolve the potentially complex regulation of NSCs and their progenies by ROS in both positive and negative manners.
In contrast to the lack of direct studies on self-renewal of NSCs, a recent study showed increased expression of Atg7, Becn1, Map1lc3a and Ambra1, all involved in autophagy, during neurogenesis in mouse olfactory bulbs, implicating a role for autophagy in NSC differentiation.Citation109 Indeed, neuronal differentiation was found to be impaired in Ambra1 knockout mice as shown by decreased expression of several neural markers as well as the previous observation of severe neural tube defects during embryogenesis.Citation27 Consistent with these findings in vivo, increased autophagy was also observed in neuronal differentiation in cultured NSC/progenitors in vitro, which could be markedly inhibited by 3-MA or wortmannin. Moreover, both Ambra1 haploinsufficient and Atg5-null olfactory bulb cells exhibit decreased neuronal differentiation in vitro. Another recent report showed that treatment of chick embryos with 3-MA significantly changes the spatiotemporal expression pattern of several neural markers and decreases the size of the acoustic-vestibular ganglion, suggesting a potential role of autophagy in chicken otic neurogenesis. In both of these studies, restoration of ATP levels by addition of methylpyruvate, a permeable analog for the citric acid cycle, partially rescued the differentiation defects, suggesting a role for autophagy in energy supply to promote neuronal differentiation. It was also reported that induction of autophagy by MTORC1 inhibition with rapamycin potentiates dbcAMP-induced differentiation of NG108–15 neuroblastoma cells, which could be abrogated by inhibition of the autophagy pathway by 3-MA or silencing Becn1, providing further support that autophagy may promote neuronal differentiation.Citation110 Therefore, similar to observations showing altered differentiation in HSCs with loss of RB1CC1/FIP200 or Atg7Citation67,Citation75 or preadipocytes lacking Atg5 or Atg7Citation111,Citation112 as well as reduced terminal differentiation of autophagy-deficient reticulocytes,Citation19,Citation52,Citation113 autophagy plays a critical role in the promotion of neuronal differentiation of NSCs.
Autophagy in Cardiac Stem Cells
A role for autophagy in cardiac stem cell differentiation was initially recognized by Fen Wang and his colleagues.Citation114 Tissue-specific ablation of FGF receptors and FGF receptor substrate 2α (FRS2) in heart progenitor cells leads to cardiac premature differentiation in mice. The differentiation defect is associated with abnormal autophagy activity, indicating that autophagy is required for FGF to regulate cardiac differentiation. Whole embryo and embryoid body culture were performed to investigate the underlying mechanisms. The group discovered that inhibiting autophagy either by small molecules or siRNA suppressed cardiac differentiation of embryoid body and embryo explants. In addition, suppressing autophagy activity blocked the enhancement of differentiation by FGFR inhibitors, while increasing autophagy activity abolished the suppression activity of FGF2 treatment. Thus, it is concluded that FGF signaling inhibits cardiac stem cell differentiation via suppressing autophagy activity. More detailed analysis indicates that FGF signaling suppresses autophagy activity through promoting AKT and MAPK pathways. Further study from whole-heart culture shows autophagy also inhibits cardiomyocyte maturation. These findings raise a number of interesting questions:
First, how does autophagy promote cardiac stem cell differentiation? Autophagy has been demonstrated to mediate the elimination of organelles during cell differentiation, such as erythrocyte and T cell maturation. Whether cardiac stem cells use the same mechanism remains to be answered.
Second, are there any other signal pathways involved in autophagy regulation during cardiac stem cell differentiation? WNT signaling has been proposed to be the upstream regulator of FGF signaling during cardiac development and it also inhibits cardiac differentiation.Citation115,Citation116 GSK3 is a part of the canonical CTNNB1/β-catenin-WNT pathway. One recent study demonstrated that a GSK3-KAT5/TIP60-ULK1 signal directly regulates autophagy.Citation117 Thus, it is possible that autophagy mediates WNT signaling during cardiac development.
This question can be posed in more general terms: What are the mechanisms underlying the transcriptional and epigenetic regulation of autophagy and whether the regulation is organ- or cell-type specific? Indeed, accumulating data implicate an important role of epigenetic factors in regulating autophagy in various pathological conditions.Citation1 Chemical or genetic inhibition of histone deacetylases (HDACs) leads to autophagy induction. HDAC regulates the specification of P19 embryonic carcinoma cells into cardiac lineage.Citation118 In the future, more detailed studies on the regulation of autophagy would be essential to understand the specification and differentiation of cardiac stem cells.
Third, does autophagy play a role in cardiac stem cell proliferation? The process of self-renewal requires the coordination of cell-cycle progression and cell-fate determination. Indeed, increased cardiac differentiation is associated with decreased proliferation.Citation114 It would be of great interest to investigate whether autophagy directly regulates cardiac stem cell proliferation.
It should be noted that the recent work discussed above analyzed the function of autophagy in embryonic cardiac stem cells. Since FGF signaling also regulates the differentiation of adult cardiac stem cells, although in an opposite way,Citation119 it would be interesting to investigate whether autophagy performs the same function in adult cardiac stem cells. Likewise, as cardiac stem cells are also capable of differentiating into smooth muscle cells and endothelial cells besides cardiomyocytes, it would also be interesting to examine whether autophagy is required for the differentiation of cardiac stem cells into the other two cell types.
Autophagy and Mesenchymal Stem Cells
Mesenchymal stem cells (MSCs) are a diverse group of multipotent precursors capable of differentiating to mesenchymal lineages, including adipose, bone, cartilage and muscle.Citation120 Although originally isolated from human bone marrow, MSCs have also been identified in many other adult tissues including muscles, adipose tissues, kidney, pancreas, brain and liver.Citation120 It is important to note that little is known about the MSCs in vivo regarding their identity and function. In addition, MSCs cultured by current methods are a heterogeneous population.Citation121 Therefore, it is disputable whether it is correct to use the term “mesenchymal stem cells.” Instead, MSCs are often called multipotent mesenchymal stromal cells.Citation120,Citation122 Currently, the role of autophagy in MSCs is largely unknown. The very limited knowledge about autophagy and MSCs is entirely derived from a very few studies using bone marrow MSCs. Recently it was shown that primary human bone marrow MSCs exhibit a high level of constitutive autophagy, whereas this basal autophagy decreases after these cells have differentiated into osteoblasts.Citation123 In agreement with this observation, we found that primary mouse bone marrow plastic-adherent stromal cells isolated from GFP-LC3 transgenic miceCitation124 have high levels of GFP-LC3 puncta. However, these GFP-LC3 puncta disappeared after these cells differentiated into osteoblast-like cells (Liu et al., unpublished). To date, the role of constitutive autophagy in bone marrow MSCs is unknown. Another recent report demonstrated that autophagy protects primary rat bone marrow MSCs from apoptosis under hypoxia/serum deprivation.Citation125 It was shown that compared with the hypoxia/serum deprivation control group, 3-MA-treated rat bone marrow MSCs had higher rate of apoptosis.Citation125 In agreement with this, another study showed that autophagy is essential for human bone marrow MSCs (cell line) survival during serum starvation, and these MSCs can release anti-apoptotic or pro-survival factors during serum deprivation to facilitate solid tumor survival and growth.Citation126
Besides this sporadic knowledge in MSCs, there is emerging evidence for the important role of autophagy in MSCs-derived cells including adipocytes, chondrocytes and osteoblasts/osteocytes. It has been shown that adipose-specific deletion of Atg7 leads to decreased adipose mass and enhanced insulin sensitivity, and autophagy is important in normal adipogenesis.Citation82,Citation111 Massive autophagy is activated when wild-type primary MEFs are induced for adipocyte differentiation. Importantly, the autophagy-deficient primary atg5−/− MEFs exhibit dramatically reduced efficiency in adipogenesis.Citation112 On the one hand, silencing of Becn1 results in enhanced chondrocyte death.Citation127 On the other hand, treatment with 3-MA renders the chondrocytes refractory to killing, suggesting that sustained autophagy promotes cell death. Thus, autophagy may play both a cytoprotective and a death-promoting role in chondrocytes.Citation127 Some autophagy proteins including ULK1, BECN1 and MAP1LC3A are constitutively expressed in normal human articular cartilage, whereas the expression is reduced in osteoarthritis chondrocytes and cartilage, suggesting a possible role of autophagy in the development of osteoarthritis.Citation128 It has been shown that osteocytes also utilize autophagy for their homeostasis. Glucocorticoid treatment can induce the development of autophagy, preserving osteocyte viability depending on glucocorticoid dose.Citation129,Citation130 Osteoblasts are the precursor of osteocytes and are responsible for bone formation. As osteoblasts actively produce mineralized matrix during bone formation, it is conceivable that autophagy is activated to meet the high-energy demands of active osteoblasts. Indeed, we can now demonstrate that suppression of autophagy by deleting RB1CC1/FIP200 in osteoblasts leads to severely compromised bone development in mice. In vitro studies show that the consequence of RB1CC1/FIP200 deletion is compromised osteoblast terminal differentiation by limiting osteoblast condensational growth. Furthermore, autophagy inhibitors 3-MA and chloroquine mimic the effects of Rb1cc1/Fip200 deletion on osteoblast condensation and terminal differentiation (Liu et al., unpublished).
Taken together, current data demonstrate the activation of autophagy in cultured bone marrow MSCs. However, the detailed functions of autophagy in MSC biology such as stemness maintenance, self-renewal and differentiation are largely unknown. Recent studies reveal the role of autophagy in the terminal differentiation of MSC-derived cell lineages including osteoblast, osteocyte, chondrocyte and adipocyte. The detailed cellular and molecular mechanisms for the regulation of these cells during differentiation demand future investigation.
Autophagy in Cancer Stem Cells
CSCs (also termed tumor-initiating cells or tumor-propagating cells) are a subpopulation of cancer cells that have some characteristics of stem cells, being capable of self-renewal and differentiation that is responsible for the heterogeneity in bulk tumors.Citation131-Citation134 Recent studies suggest that autophagy also plays a crucial role in the origin, maintenance and systemic distribution of CSCs besides its many functions in normal embryonic and tissue stem cells.
Hypoxia- and starvation-related autophagy: A cytoprotective adaptive mechanism of cancer stem cells against microenvironmental stresses
The physiology of the microenvironment in solid epithelial tumors is characterized by lower oxygen levels (hypoxia), higher lactate levels, extracellular acidosis and depletion of nutrients (e.g., glucose and glutamine) compared with normal tissues.Citation135-Citation137 These changes are usually called tumor microenvironment stresses, and some of these stresses, particularly hypoxia, play critical roles during the evolution of the tumor stromal microenvironment and formation of the putative CSC niches.Citation138,Citation139 The ability of an undifferentiated hypoxic microenvironment to provide essential cellular interactions and environmental signals for the preferential maintenance of CSCs may therefore result in the specific expansion of CSC pools vs. non-CSC tumor cells. Recent studies have explored the hypothesis that hypoxia could induce tumor cell autophagy through hypoxia-inducible factors as a cytoprotective adaptive response. Because hypoxia-mediated autophagy promotes tumor cell survival in response to antiangiogenic therapies, and antiangiogenic agents have been recently shown to increase the population of CSCs via HIF1A,Citation140,Citation141 it might be tempting to suggest that autophagy is instrumental in hypoxia-driven CSC stimulation. Unfortunately, few studies have illustrated how autophagy could causally allow a co-promotion of enhanced tolerance of CSCs to biophysical stressful microenvironmental conditions, maintenance of CSC functionality and the expansion of CSC populations.
Beyond evading biophysical constraints, starving CSCs must use alternative sources of energy via activation of catabolic processes that permit them to recycle intracellular components to maintain metabolic homeostasis, quiescence and cell viability during periods of metabolic stress. Furthermore, CSC functioning and viability depend on their ability to ensure complementary bioenergetic sources at times other than during the development and growth of premalignant, pre-invasive lesions. Migrating CSCs experience hypoxia immediately after extravasation at secondary organs, and metastatic CSCs eventually become oxygen- and nutrient-starved, once the secondary tumors outstrip the blood supply.Citation142-Citation144 These considerations support the notion of protein catabolism functions of autophagy in protecting CSCs under starvation conditions. A causal relationship between activation of autophagy and enhanced cell survival of CSCs was initially suggested by the specific spatio-temporal upregulation pattern observed for BECN1 in certain pre-malignant lesions of the breast. In human comedo-ductal carcinoma in situ (DCIS), BECN1 is notably upregulated at the viable rim of intraductal cells within the hypoxic ductal niche.Citation143,Citation144 The expression levels of the autophagic effector MAP1LC3A are also increased in DCIS lesions vs. normal breast tissues.Citation145 Remarkably, a BECN1-related enhancement of the autophagic flux appears to occur in DCIS tumor-founding progenitor cells because pre-malignant, cytogenetically abnormal DCIS spheroids-forming cells directly isolated from DCIS lesions exhibit an increased expression of autophagy-associated proteins that persists in culture as well as in tumors generated by malignant precursor cells in immunosuppressed mice. Treatment with chloroquine is sufficient to completely suppress the generation of DCIS spheroids/3D structures and ex vivo invasion of autologous breast stroma, induce apoptosis and eliminate cytogenetically abnormal spheroid-forming cells in the organ culture, and abrogate xenograft tumor formation via reduced expression of autophagy-associated proteins,Citation144 confirming that autophagy is necessary for the survival of CSC-like precursor cells pre-existing in pre-malignant lesions.
Starvation-independent activation of autophagy: From adaptive to intrinsic metabolic feature in cancer stem cells
Homeostatic autophagy could operate in a starvation-independent manner to constitute an intrinsic metabolic feature of CSC cellular states. Comparing mammospheres from patients and well-characterized human breast cancer cell lines, autophagic flux is significantly higher in the mammospheres than the adherent cells, both at basal levels and under starvation conditions.Citation146,Citation147 Breast cancer cells bearing CSC-like phenotypes appear to constitutively display higher autophagic fluxes than non-CSC cells because both basal and starvation-induced autophagy are higher in aldehyde dehydrogenase 1 (ALDH1)-positive subpopulations derived from mammospheres than in the bulk tumor cell population.Citation146,Citation147 The specific depletion of BECN1 notably reduces both the size and formation efficiency of mammospheres, thus confirming and expanding the suggestion that the expression status of BECN1 may be pivotal for the tumorigenicity of breast CSCs.Citation146,Citation147 Given that depletion of BECN1 in CSCs also reduces tumor development in nude mice and that decreased survival in autophagy-deficient cells during anoikis does not contribute to any deficiency in mammosphere formation, it might be reasonable to conclude that autophagy is necessarily required for the maintenance and expansion of breast CSC populations.
Autophagy and the glycolytic metabotype in CSCs: Friends or enemies?
An interesting question that remains to be answered relates to the possible active contribution of autophagy to the metabolic shift of cancer cells to enhanced glycolysis (the Warburg effect)Citation148-Citation150 during malignant transformation and acquisition of stemness by CSC-like cell populations. If enhanced glycolysis indeed plays a causal role in the gain of stem-like properties by protecting tumor-initiating cells from the pro-senescent effects of mitochondrial respiration-induced oxidative stress, the ability of autophagy to functionally engage the glycolytic metabotype may generate a cellular state that is metabolically endowed to immortalization.
Friends: Autophagy as a facilitator of the Warburg effect
A Warburg-like glycolytic metabolism, and its facilitation by autophagy may accompany the acquisition of stemness and perhaps, both the occurrence and maintenance of CSC cellular states. Some malignant cells, particularly those driven by the Kras oncogene, appear to depend on elevated levels of autophagy for survival, even in the absence of external stressors.Citation151 Kras [Kras(V12)]-induced malignant transformation is accompanied by upregulation of Atg5 and Atg7, and autophagic vacuole formation;Citation152 pharmacological inhibition of autophagic activity and genetic ablation of Atg5 or Atg7 completely block Kras-induced anchorage-independent cell growth in soft agar and tumor formation in nude mice, partially establishing a causal role of autophagy induction in malignant transformation.Citation152 In autophagy-deficient MEFs expressing oncogenic Kras, there is a decrease in both glucose uptake and in the glycolytic flux, thus suggesting dependency on autophagy for the glycolytic capacity of oncogenically transformed cells.Citation153 Remarkably, it has been recently confirmed that a significant impairment of glucose uptake and intracellular lactate production and decreased sensitivity to reduced glucose availability occur in Rb1cc1/fip200-null primary MEFs transformed by Kras.Citation10 Loss of autophagy, therefore, leads to deficient glycolysis and appears to intrinsically contribute to the decreased proliferation of mammary tumor cells. Further studies are required to clarify whether the inhibition of mammary tumor initiation, progression and metastasis imposed by an increased accumulation of both total and healthy mitochondria mass in autophagy-null tumors can be exclusively explained in terms of the necessary and/or sufficient “autophagy/glycolysis addiction” of CSCs.
Another body of evidence is related to the ability of autophagy to actively refuel the high-consuming, glycolysis-driven, anabolic phenotype. If the Warburg-like glycolytic metabotype can be correctly redefined in terms of the obligatory dependence of cancer cells to rapidly produce other metabolic end products in an unrestricted manner (i.e., independently of the generally starved microenvironmental conditions), the ability of autophagy to generate an internal nutrient pool that is recycled back and reused to synthesize essential cellular components can be expected to pleiotropically synergize with the anabolic side of the Warburg effect. Importantly, glycolysis can produce ATP at a higher rate without the undesirable presence of endogenous ROS and, by promoting flux through the pentose phosphate pathway, glycolysis can also support antioxidant defenses by inducing NADPH production, which is required for generating the key antioxidant enzyme, reduced glutathione.Citation154,Citation155 It should be noted that systemic dissemination of CSCs occurs at earlier stages of tumor development than previously thought, and fewer chromosomal aberrations are found in these disseminated CSCs compared with the matched primary tumors.Citation156 Because failure to maintain autophagy-driven generation of metabolites of low molecular weight for continued use in the energetic and biosynthetic metabolism of the cells associates with increased DNA damage, gene amplification and aneuploidy, the ability of autophagy to alleviate cellular energy and anabolic deficits may significantly limit genomic instability during early tumorigenesis,Citation157,Citation158 thus functioning as a driving force of more stable CSC-like cellular states.
Enemies: Autophagy as an antagonist of the Warburg effect
Phenotypically diverse cancer cells can be efficiently reversed back to a less-tumorigenic iPSC-like stage of common ancestry that appears to be fully capable of originating nontumorigenic ontogenies.Citation159-Citation162 If mitochondrial restructuring and bioenergetics intimately reflect the molecular dynamics fundamental for the resetting and redirection of cell fate (i.e., the glycolytic metabotype supports the anabolic and catabolic requirements of pluripotent cell homeostasis, whereas redirection of pluripotency into defined lineages requires mitochondrial biogenesis and maturation of efficient oxidative energy generation and distribution networks), the generation of terminally differentiated phenotypes following (re)-expression of pluripotency-specific genes might imply that activation of a glycolytic Warburg effect in malignant cells apparently impedes the activation of CSC-like properties, including tumorigenesis. In this alternate scenario, does autophagy promote mitochondria-driven oxidative phosphorylation (OXPHOS)? Autophagy is constitutively activated in most Kras-mutated pancreatic intraepithelial neoplasms and high-grade pancreatic ductal carcinomas, but not in normal pancreatic ductal epithelium or low-grade pancreatic intraepithelial neoplasms.Citation163 Genetic and pharmacological inhibition of autophagy attenuates growth and tumorigenicity of pancreatic cancer cells through a mechanism involving a significant decrease in OXPHOS and tricarboxylic acid intermediates. In agreement with a putative pro-mitochondrial OXPHOS activity of autophagy, oncogenic upregulation of autophagy can support cell survival and transformation primarily through the maintenance of mitochondrial metabolic function and energy levels.Citation164
Regardless of the pro- or anti-glycolytic activity of oncogene-driven autophagy, both the activation of some oncogenes and/or the loss of some tumor suppressor genes might impose a metabolic insult on CSCs that depletes energy sources, thereby making them indirectly dependent on autophagy either to empower the Warburg-like glycolytic metabotype or preserve mitochondrial function and to directly provide catabolically-derived metabolic substrates.
Mitophagy: A direct way to connect autophagy with the metabolic reprogramming of CSCs
The cellular energy deficit triggered by cellular states of glucose deficiency imposed by oncogenic transformation (e.g., Kras) can induce a decline in mitochondrial respiration that is not related to changes in mitochondrial biogenesis; instead, it is inversely associated with the increased formation of acidic vesicles enclosing mitochondria, a process accompanied by the induction of autophagy-related proteins, such as BECN1, ATG5, LC3-II and vacuolar ATPases (i.e., mitophagy).Citation164 Conversely, genetic ablation and pharmacological inhibition of autophagy promote the efficient recovery of respiratory protein expression and respiratory activity. Mitophagy might also have an impact on mitochondrial dynamics to segregate the mitochondria destined for clearance through autophagy, which may result in the loss of mitochondrial function and the accelerated onset of the glycolytic metabolism.Citation165 Our group has explored if somatic cells reprogrammed to iPSCs bio-energetically utilize mitophagy. Interestingly, pharmacological promotion of mitochondrial fusion using mdivi-1 (for mitochondrial division inhibitor) at concentrations that rapidly induce the formation of mitophagy-refractory mitochondrial net-like or collapsed perinuclear mitochondrial structures largely impede somatic cell reprogramming to pluripotency.Citation166 Although these findings strongly suggest that mitophagy is functionally integrated into the transcriptional network that specified the unique pluripotency of stem cells, future studies should elucidate whether the ability to directly shift the oxidative:glycolytic production ratios closer to those of pluripotent cells can molecularly explain the impact of autophagy-regulated mitochondria fusion-fission dynamics on the acquisition and maintenance of CSC cellular states ().
Figure 3. Autophagy/mitophagy and the CSC cellular state: Two models. Mitochondria can be eliminated through nonspecific autophagic processes or selectively removed by mitophagy. The former is generally related to metabolic responses that are imposed by a lack of nutrients. Mitophagy, however, permits a tight adjustment in the number of cellular mitochondria by regulating the selective degradation of damaged, dysfunctional and superfluous mitochondria and adjust to changing physiological demands. Mitophagy, therefore, can play a crucial role in adapting the number and quality of mitochondria to new microenvironmental conditions. (A) Cell-autonomous model. Upregulation of mitophagy leading to significant reductions in both the number and the size of mitochondria (i.e., the “mitochondrial phenotype” that is commonly associated with stem cells) concurrently enhances a bioenergetic shift from somatic oxidative mitochondria toward an alternative glycolytic phenotype (“direct Warburg effect”), an energetic infrastructure associated with the transcriptional networks responsible for stemness and pluripotency. (B) Non-cell-autonomous model (“autophagic tumor stroma model of cancer metabolism”). Cancer cells’ mitochondrial oxidative phosphorylation (OXPHOS) can induce oxidative stress in adjacent fibroblasts (cancer-associated fibroblasts [CAFs] via H2O2 and reactive oxygen species (ROS), resulting in the onset of a myofibroblastic autophagic phenotype in CAFs. The CAFs autophagic phenotype leads to a loss of mitochondria via mitophagy, forcing CAFs to undergo aerobic glycolysis (“reverse Warburg effect”). The products of aerobic glycolysis (such as L-lactate and ketones) are then reused by cancer cells for OXPHOS, resulting in increased mitochondrial mass in cancer cells. The utilization of the high-energy metabolites L-lactate and ketones in cancer cells can promote the transcriptional activation of CSC-like phenotypes.
![Figure 3. Autophagy/mitophagy and the CSC cellular state: Two models. Mitochondria can be eliminated through nonspecific autophagic processes or selectively removed by mitophagy. The former is generally related to metabolic responses that are imposed by a lack of nutrients. Mitophagy, however, permits a tight adjustment in the number of cellular mitochondria by regulating the selective degradation of damaged, dysfunctional and superfluous mitochondria and adjust to changing physiological demands. Mitophagy, therefore, can play a crucial role in adapting the number and quality of mitochondria to new microenvironmental conditions. (A) Cell-autonomous model. Upregulation of mitophagy leading to significant reductions in both the number and the size of mitochondria (i.e., the “mitochondrial phenotype” that is commonly associated with stem cells) concurrently enhances a bioenergetic shift from somatic oxidative mitochondria toward an alternative glycolytic phenotype (“direct Warburg effect”), an energetic infrastructure associated with the transcriptional networks responsible for stemness and pluripotency. (B) Non-cell-autonomous model (“autophagic tumor stroma model of cancer metabolism”). Cancer cells’ mitochondrial oxidative phosphorylation (OXPHOS) can induce oxidative stress in adjacent fibroblasts (cancer-associated fibroblasts [CAFs] via H2O2 and reactive oxygen species (ROS), resulting in the onset of a myofibroblastic autophagic phenotype in CAFs. The CAFs autophagic phenotype leads to a loss of mitochondria via mitophagy, forcing CAFs to undergo aerobic glycolysis (“reverse Warburg effect”). The products of aerobic glycolysis (such as L-lactate and ketones) are then reused by cancer cells for OXPHOS, resulting in increased mitochondrial mass in cancer cells. The utilization of the high-energy metabolites L-lactate and ketones in cancer cells can promote the transcriptional activation of CSC-like phenotypes.](/cms/asset/16f41840-49cf-489b-8c82-33b315c95b9d/kaup_a_10924132_f0003.gif)
Mitophagy and the “reverse Warburg effect”: Connecting autophagic stroma with cancer stem cells
Both autophagy inhibition and promotion appear to similarly impair the occurrence of cancer cells with tumor-initiating capacities. In normal tissues, autophagy-mediated damage mitigation may efficiently suppress tumorigenesis; conversely, macromolecular recycling may support CSC survival by buffering bioenergetic demands under stressful metabolic and microenvironmental conditions.Citation167-Citation169 Therefore, activation of autophagy in normal tissues operates as a bona fide tumor suppressive mechanism, whereas autophagy inhibition may be extremely beneficial for anti-CSC therapy in established tumors. However, both autophagy inhibitors (e.g., chloroquine) and autophagy promoters (e.g., MTOR inhibitors) block tumorigenesis and cancer progression by eliminating CSCs.Citation143,Citation144,Citation170,Citation171 Although the simplest solution to this “autophagy paradox” could be just to claim that different types of tumors undergo different bioenergetic adjustments, the answer may also lie with the interaction between tumor cells and adjacent, autophagic, stromal cells. First, it has been observed that the stromal fibroblasts associated with breast cancer epithelial cells are glycolytic.Citation172,Citation173 The conversion of cancer-associated fibroblasts to glycolytic metabolism is induced by hydrogen peroxide secreted from the adjacent cancer cells, which results in loss of CAV1 from the fibroblasts.Citation174 Second, a loss of stromal CAV1 causes the induction of mitophagy, which actively reduces the number of mitochondria in the stromal fibroblasts, thus reducing mitochondrial function and switching the tumor stroma metabolism to glycolysis. As a consequence, the autophagic/mitophagic tumor stroma generates recycled nutrients that can be used as chemical building blocks by anabolic epithelial cancer cells.Citation175 Indeed, the increased glycolysis due to enhanced mitochondrial turnover generates excessive stromal cell lactate and ketones, which are secreted into the intracellular space.Citation176 The cancer cells then take up the high-energy metabolites (lactate and ketones) and use them to feed cancer cell mitochondrial energy production and generate mitochondrial precursors for the biogenesis of new cancer cells. Lactate and ketones from the autophagic tumor stroma increase the transcriptional expression of gene profiles normally associated with stemness, including genes commonly upregulated in embryonic stem cells, and may therefore promote the dynamic appearance of CSC cellular states, resulting in significant decreases in patient survival.Citation177 This host-parasite autophagic/mitophagic relationship from the tumor stroma to the epithelial cancer cells, which has been designated “the autophagic tumor stromal model of cancer cell metabolism,” “battery-operated tumor growth,” “stromal-epithelial metabolic coupling” and the “reverse Warburg effect”Citation177-Citation181 (), functionally resolves the “autophagy paradox” because the systemic induction of autophagy prevents epithelial cancer cells from using recycled nutrients, whereas the systemic inhibition of autophagy prevents stromal cells from producing recycled nutrients, both effectively “starving” cancer cells.
The autophagy-regulated migratory/invasive phenotype in cancer stem cells
CSCs can be generated by the microenvironment through induction of CSC molecular features in more differentiated tumor cells. In addition to a role in CSC maintenance, the microenvironment is hypothesized to be involved in metastasis by the induction of the epithelial-to-mesenchymal transition (EMT) phenomenon, leading to the dissemination and invasion of tumor cells. Not surprisingly, EMT and CSC-like features have been linked to one another.Citation182-Citation186 In breast cancer, the undifferentiated CD44+CD24-/low antigenic state commonly attributed to breast cancer subpopulations with CSC properties is highly enriched with EMT transcriptional factors and mesenchymal markers.Citation187,Citation188 Our group recently explored whether autophagy might be important not only for survival but also for the maintenance of a migratory, invasive, treatment-resistant CD44+CD24-/low mesenchymal cellular state.Citation189,Citation190 Impairment of the autophagic flux drastically decreases the number of breast cancer cells bearing CD44+CD24-/low cell surface antigens and appears to impede the ontogeny of generating the invasive CD44+CD24-/low mesenchymal phenotype by preventing the full acquisition of a post-EMT status. The essential autophagy gene Atg12 might be pivotal for the autophagy-regulated migratory phenotype because its genetic ablation activates the transcription of the epithelial marker CD24 while concomitantly repressing the gene coding for the mesenchymal filament vimentin.Citation189 The deficiency of ATG12 in glioma cancer cells, while failing to significantly have an impact on many tumor cell functions in 2D cultures (e.g., proliferation or cell viability), notably reduces their capacity to invade an organotypic matrix consisting of human fibroblasts embedded in polymerized collagen.Citation191,Citation192 Further supporting a previously unrecognized role of autophagy in the metastatic potential of migratory CSCs, the autophagy-associated genes DRAM1 and SQSTM1 regulate the motility and invasion of glioblastoma stem cells.Citation193 The fact that a number of autophagy regulators are highly expressed in tumor cells carrying a mesenchymal signature, which intrinsically defines aggressiveness and metastatic invasion, might suggest that autophagy positively acts in concert with EMT-related processes to reduce proliferation in favor of enhanced cell motility, thereby providing a selective advantage during early stages of cancer metastasis, while supporting the survival of disseminated cells with CSC-like properties.
Targeting autophagy in cancer stem cells: A therapeutic corollary
Much work remains before developing an approach for pharmacologically altering CSC-related autophagy in a clinical setting.Citation194-Citation196 First, our current ability to accurately measure basal autophagic flux or determine whether autophagy is activated or inhibited in patient samples is very limited and of unproven utility in human patients. Similarly, we are awaiting more tests to identify and isolate CSCs in epithelial tumors; circulating CSCs directly obtained from the portal vein and peripheral blood of cancer patients could represent a powerful biomarker for patient monitoring during autophagy-based, CSC-targeted therapies. Second, all human clinical trials currently exploring autophagy inhibition as a therapeutic strategy use chloroquine or the derivative hydroxychloroquine due to its long track record of safety in human patients.Citation197 However, whether chloroquine and its derivatives represent the most efficacious drugs for inhibiting autophagy is debatable. Hydroxychloroquine when combined with autophagy inducers, such as chemotherapy (i.e., temozolamide), proteasome inhibitors (i.e., bortezomib), MTOR inhibitors (i.e., temsirolimus) and/or radiation, has low response rates in preliminary clinical trials, indicating that hydroxychloroquine is not a potent autophagy inhibitor at clinically tolerable doses. The fact that an encouraging achievement of stable disease in 76% of advanced melanoma patients has been observed when combining hydroxychloroquine with temsirolimus clearly motivates the development of new, more potent, autophagy inhibitors (e.g., Lys05, Spautin-1).Citation195 Ultimately, we may be able to develop very effective anticancer treatment modalities by combining a certain class of autophagy inhibitors with molecular-targeted drugs directed against specific cellular targets in CSCs. In this scenario, autophagy-based, “customized” combinatorial approaches will likely provide new powerful methods to better manage CSC-driven cancer origin and evolution in the near future.
Concluding Remarks
The past decade has witnessed significant growth in interest regarding stem cells and autophagy, but research investigating the intersection of these two exciting areas is very much in its infancy. Thus, we can expect tremendous growth in our understanding of the role of autophagy in various stem cells and more importantly of the mechanisms by which the autophagy pathway and different autophagy proteins control and balance the quiescence, self-renewal and differentiation of stem cells. Given the nature of these stem cell-specific characteristics, much of the future research will necessarily be performed using in vivo model systems. So far, conditional knockout of specific autophagy genes in mouse models have provided the strongest direct evidence for autophagy in different stem cells.Citation67,Citation75 It is anticipated that studies using other genetic models such as the fly and worm will also likely be fruitful based on the power of genetics to look for extensive gene interactions as well as the availability of well-defined stem cells in these systems. Likewise, future investigations into autophagy in stem cell functions will be facilitated by development of assays in cultured cells (especially for human stem cells) that allow monitoring of selective autophagy targets such as the mitochondrion in live cells.
Besides increasing our understanding of basic stem cell biology, studies of autophagy in stem cells also have significant therapeutic implications. Embryonic stem cells hold promise as therapeutic tools. However, if the potential is to be realized it is important that ESCs are maintained in a viable, pluripotent state and free of genetic damage. Although the lack of autophagy does not appear to affect the ability of ES cells to form full-term embryos, the lack of autophagic activity appears to have an effect on cells cultured in vitro under particular circumstances and stresses (e.g., inability to cavitate). It is not clear what the individual stressors are under these circumstances, but they may relate to the absence of particular growth factors and/or correct growth conditions. Autophagy may play an important role in protecting ESCs against unwanted outcomes either during renewal or differentiation in vitro either through the provision of an energy source or the elimination of cellular damage including DNA and defective mitochondria. It remains to be determined as to whether interference with autophagy during the early stages of ES cell expansion or differentiation has long-term consequences for the mature organism. If the hypothesis is correct that mitophagy plays a role in eliminating substandard mitochondria from ES and early progenitor cells, one would predict that tissues originating from the autophagy-deficient ES cells would have increased loads of mtDNA mutations and/or display premature aging phenotypes. With the advent of iPSC technology in which pluripotent stem cells destined for regenerative medicine applications are generated from individuals of advanced age (autophagic/mitophagic) quality control processes would need to be carefully examined and controlled to ensure long-term safety of any cellular grafts. Similarly, further studies of autophagy in tissue stem cells such as HSCs may shed light on the pathogenesis of hematopoietic disorders, serve as a basis for the development of new therapeutic strategies for such disorders, prove useful for the expansion of HSCs in vitro as a possible replacement for blood transfusion, and provide vital insight into general stem cell biology.
Recent studies suggested radiation therapy-induced autophagy contributes to the radio-resistance of glioma stem cells.Citation198 Interestingly, the autophagy inhibitor bafilomycin A1 and silencing of Atg5 and Becn1 sensitizes the CD133+ glioma stem cells to γ-radiation and significantly decreases the viability of the irradiated cells and their ability to form neurospheres. Therefore, modulation of autophagy could be used in future therapies for cancer. However, we should acknowledge that a close affinity of normal tissue stem cells and CSCs in terms of autophagic fluxes might underlie a cautionary note for off-target effects of autophagy-based anti-CSC therapies. Discerning the specific autophagy requirements for normal tissue stem cells and CSCs may be an essential requisite when assessing autophagy targets for discriminatory targeting of CSCs with minimal sequelae. Only through a rigorous scrutiny of the nuances of the autophagy machinery in normal tissue stem cells and CSCs can we clarify the pathogenic mechanisms of autophagy and reveal the autophagic processes exploited by cancer cells to co-opt stem cell traits. Other interesting questions for future studies on the role of autophagy in CSCs include: Do CSCs necessarily require the activation of the autophagy machinery to ensure survival and/or functioning? Does autophagy endow tumor cells with self-renewal capabilities? Does autophagy constitute a shared metabolic feature of the stemness signatures among all of the intra-individual and inter-individual heterogeneity of CSCs cellular states? It is time for the autophagy communityCitation199 to make the responses as unambiguous as possible.
| Abbreviations: | ||
| 3-MA | = | 3-methyladenine |
| ATG | = | autophagy-related |
| CSCs | = | cancer stem cells |
| DCIS | = | ductal carcinoma in situ |
| EBs | = | embryoid bodies |
| EMT | = | epithelial-to-mesenchymal transition |
| ESCs | = | embryonic stem cells |
| hESCs | = | human ESCs |
| HDACs | = | histone deacetylases |
| HSCs | = | hematopoietic stem cells |
| iPSC | = | induced pluripotent stem cell |
| LIF | = | leukemia inhibitory factor |
| MEFs | = | mouse embryonic fibroblasts |
| mESC | = | mouse ESC |
| MSCs | = | mesenchymal stem cells |
| NSCs | = | neuronal stem cells |
| ROS | = | reactive oxygen species |
| SVZ | = | subventricular zone |
| ULK | = | Unc51-like kinase |
| UPS | = | ubiquitin-proteasome system |
Acknowledgments
J.L.G. is supported by NIH grants (primarily GM052890 for work related to this review). A.K.S. is funded by the Oxford Biomedical Research Center, NIHR. Work at Dr. Menendez’s Laboratory is financially supported through funding from the Instituto de Salud Carlos III [Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria (FIS), Spain, Grants CP05-00090 and PI06-0778 and RD06-0020-0028], the Fundación Científica de la Asociación Española Contra el Cáncer (AECC, Spain) and the Ministerio de Ciencia e Innovación (SAF2009-11579, Plan Nacional de I+D+ I, MICINN, Spain). Alejandro Vazquez-Martin received the Sara Borrell post-doctoral contract [CD08/00283, Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria (FIS), Spain]. F.L is supported by NIAMS of the NIH under Award Number R01AR062030.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823 - 30; http://dx.doi.org/10.1038/ncb0910-823; PMID: 20811354
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68 - 76; http://dx.doi.org/10.1038/nature09204; PMID: 20562859
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728 - 41; http://dx.doi.org/10.1016/j.cell.2011.10.026; PMID: 22078875
- Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell 2011; 146:682 - 95; http://dx.doi.org/10.1016/j.cell.2011.07.030; PMID: 21884931
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443:780 - 6; http://dx.doi.org/10.1038/nature05291; PMID: 17051204
- Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Hematology Am Soc Hematol Educ Program 2006; 2006:1 - 12, 505-6; http://dx.doi.org/10.1182/asheducation-2006.1.1; PMID: 17124032
- Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 2007; 27:19 - 40; http://dx.doi.org/10.1146/annurev.nutr.27.061406.093749; PMID: 17311494
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961 - 7; http://dx.doi.org/10.1038/nrc2254; PMID: 17972889
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011; 25:1510 - 27; http://dx.doi.org/10.1101/gad.2051011; PMID: 21764854
- Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev 2011; 25:1999 - 2010; http://dx.doi.org/10.1101/gad.17558811; PMID: 21979913
- Phadwal K, Watson AS, Simon AK. Tightrope act: autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell Mol Life Sci 2013; 70:89 - 103; PMID: 22669258
- Vessoni AT, Muotri AR, Okamoto OK. Autophagy in stem cell maintenance and differentiation. Stem Cells Dev 2012; 21:513 - 20; http://dx.doi.org/10.1089/scd.2011.0526; PMID: 22066548
- Zeng M, Zhou JN. Roles of autophagy and mTOR signaling in neuronal differentiation of mouse neuroblastoma cells. Cell Signal 2008; 20:659 - 65; http://dx.doi.org/10.1016/j.cellsig.2007.11.015; PMID: 18207367
- Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol 2006; 4:e83; http://dx.doi.org/10.1371/journal.pbio.0040083; PMID: 16509772
- Frankel LB, Wen J, Lees M, Høyer-Hansen M, Farkas T, Krogh A, et al. microRNA-101 is a potent inhibitor of autophagy. EMBO J 2011; 30:4628 - 41; http://dx.doi.org/10.1038/emboj.2011.331; PMID: 21915098
- Tooze J, Davies HG. Cytolysomes in amphibian erythrocytes. J Cell Biol 1965; 24:146 - 50; http://dx.doi.org/10.1083/jcb.24.1.146; PMID: 14286289
- Kent G, Minick OT, Volini FI, Orfei E. Autophagic vacuoles in human red cells. Am J Pathol 1966; 48:831 - 57; PMID: 5937781
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008; 112:1493 - 502; http://dx.doi.org/10.1182/blood-2008-02-137398; PMID: 18539900
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657 - 68; http://dx.doi.org/10.1083/jcb.152.4.657; PMID: 11266458
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809 - 20; PMID: 14638851
- Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007; 128:931 - 46; http://dx.doi.org/10.1016/j.cell.2006.12.044; PMID: 17350577
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425 - 34; http://dx.doi.org/10.1083/jcb.200412022; PMID: 15866887
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A 2009; 106:20842 - 6; http://dx.doi.org/10.1073/pnas.0911267106; PMID: 19926846
- Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell 2008; 19:4762 - 75; http://dx.doi.org/10.1091/mbc.E08-03-0309; PMID: 18768753
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008; 456:264 - 8; http://dx.doi.org/10.1038/nature07383; PMID: 18849965
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447:1121 - 5; PMID: 17589504
- Mizushima NA. A(β) generation in autophagic vacuoles. J Cell Biol 2005; 171:15 - 7; http://dx.doi.org/10.1083/jcb.200508097; PMID: 16216920
- Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science 2008; 321:117 - 20; http://dx.doi.org/10.1126/science.1154822; PMID: 18599786
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032 - 6; http://dx.doi.org/10.1038/nature03029; PMID: 15525940
- Vilchez D, Boyer L, Morantte I, Lutz M, Merkwirth C, Joyce D, et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 2012; 489:304 - 8; http://dx.doi.org/10.1038/nature11468; PMID: 22972301
- Hernebring M, Brolén G, Aguilaniu H, Semb H, Nyström T. Elimination of damaged proteins during differentiation of embryonic stem cells. Proc Natl Acad Sci U S A 2006; 103:7700 - 5; http://dx.doi.org/10.1073/pnas.0510944103; PMID: 16672370
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003; 100:15077 - 82; http://dx.doi.org/10.1073/pnas.2436255100; PMID: 14657337
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol 2010; 20:355 - 62; http://dx.doi.org/10.1016/j.tcb.2010.03.002; PMID: 20356743
- Denton D, Chang TK, Nicolson S, Shravage B, Simin R, Baehrecke EH, et al. Relationship between growth arrest and autophagy in midgut programmed cell death in Drosophila. Cell Death Differ 2012; 19:1299 - 307; http://dx.doi.org/10.1038/cdd.2012.43; PMID: 22555456
- Tra T, Gong L, Kao LP, Li XL, Grandela C, Devenish RJ, et al. Autophagy in human embryonic stem cells. PLoS One 2011; 6:e27485; http://dx.doi.org/10.1371/journal.pone.0027485; PMID: 22110659
- Zhou J, Su P, Wang L, Chen J, Zimmermann M, Genbacev O, et al. mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc Natl Acad Sci U S A 2009; 106:7840 - 5; http://dx.doi.org/10.1073/pnas.0901854106; PMID: 19416884
- Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, et al. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol 2004; 24:6710 - 8; http://dx.doi.org/10.1128/MCB.24.15.6710-6718.2004; PMID: 15254238
- Fathi A, Hatami M, Hajihosseini V, Fattahi F, Kiani S, Baharvand H, et al. Comprehensive gene expression analysis of human embryonic stem cells during differentiation into neural cells. PLoS One 2011; 6:e22856; http://dx.doi.org/10.1371/journal.pone.0022856; PMID: 21829537
- Mijaljica D, Nazarko TY, Brumell JH, Huang WP, Komatsu M, Prescott M, et al. Receptor protein complexes are in control of autophagy. Autophagy 2012; 8:1701 - 5; http://dx.doi.org/10.4161/auto.21332; PMID: 22874568
- Todd LR, Damin MN, Gomathinayagam R, Horn SR, Means AR, Sankar U. Growth factor erv1-like modulates Drp1 to preserve mitochondrial dynamics and function in mouse embryonic stem cells. Mol Biol Cell 2010; 21:1225 - 36; http://dx.doi.org/10.1091/mbc.E09-11-0937; PMID: 20147447
- Todd LR, Gomathinayagam R, Sankar U. A novel Gfer-Drp1 link in preserving mitochondrial dynamics and function in pluripotent stem cells. Autophagy 2010; 6:821 - 2; http://dx.doi.org/10.4161/auto.6.6.12625; PMID: 20581476
- Kuo TC, Chen CT, Baron D, Onder TT, Loewer S, Almeida S, et al. Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat Cell Biol 2011; 13:1214 - 23; http://dx.doi.org/10.1038/ncb2332; PMID: 21909099
- Lonergan T, Bavister B, Brenner C. Mitochondria in stem cells. Mitochondrion 2007; 7:289 - 96; http://dx.doi.org/10.1016/j.mito.2007.05.002; PMID: 17588828
- Kondoh H, Lleonart ME, Nakashima Y, Yokode M, Tanaka M, Bernard D, et al. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid Redox Signal 2007; 9:293 - 9; http://dx.doi.org/10.1089/ars.2006.1467; PMID: 17184172
- Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J 2012; 31:2103 - 16; http://dx.doi.org/10.1038/emboj.2012.71; PMID: 22446391
- Fritz V, Fajas L. Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene 2010; 29:4369 - 77; http://dx.doi.org/10.1038/onc.2010.182; PMID: 20514019
- Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010; 329:1492 - 9; http://dx.doi.org/10.1126/science.1188015; PMID: 20847263
- St John JC, Ramalho-Santos J, Gray HL, Petrosko P, Rawe VY, Navara CS, et al. The expression of mitochondrial DNA transcription factors during early cardiomyocyte in vitro differentiation from human embryonic stem cells. Cloning Stem Cells 2005; 7:141 - 53; http://dx.doi.org/10.1089/clo.2005.7.141; PMID: 16176124
- Chung S, Arrell DK, Faustino RS, Terzic A, Dzeja PP. Glycolytic network restructuring integral to the energetics of embryonic stem cell cardiac differentiation. J Mol Cell Cardiol 2010; 48:725 - 34; http://dx.doi.org/10.1016/j.yjmcc.2009.12.014; PMID: 20045004
- Prigione A, Adjaye J. Modulation of mitochondrial biogenesis and bioenergetic metabolism upon in vitro and in vivo differentiation of human ES and iPS cells. Int J Dev Biol 2010; 54:1729 - 41; http://dx.doi.org/10.1387/ijdb.103198ap; PMID: 21305470
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454:232 - 5; http://dx.doi.org/10.1038/nature07006; PMID: 18454133
- Varum S, Momcilović O, Castro C, Ben-Yehudah A, Ramalho-Santos J, Navara CS. Enhancement of human embryonic stem cell pluripotency through inhibition of the mitochondrial respiratory chain. Stem Cell Res 2009; 3:142 - 56; http://dx.doi.org/10.1016/j.scr.2009.07.002; PMID: 19716358
- Cho YM, Kwon S, Pak YK, Seol HW, Choi YM, Park J, et al. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem Biophys Res Commun 2006; 348:1472 - 8; http://dx.doi.org/10.1016/j.bbrc.2006.08.020; PMID: 16920071
- Sauer H, Wartenberg M. Reactive oxygen species as signaling molecules in cardiovascular differentiation of embryonic stem cells and tumor-induced angiogenesis. Antioxid Redox Signal 2005; 7:1423 - 34; http://dx.doi.org/10.1089/ars.2005.7.1423; PMID: 16356105
- Eliasson P, Jönsson JI. The hematopoietic stem cell niche: low in oxygen but a nice place to be. J Cell Physiol 2010; 222:17 - 22; http://dx.doi.org/10.1002/jcp.21908; PMID: 19725055
- Salemi S, Yousefi S, Constantinescu MA, Fey MF, Simon HU. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res 2012; 22:432 - 5; http://dx.doi.org/10.1038/cr.2011.200; PMID: 22184008
- Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013; 494:323 - 7; http://dx.doi.org/10.1038/nature11895; PMID: 23389440
- Lemischka IR, Raulet DH, Mulligan RC. Developmental potential and dynamic behavior of hematopoietic stem cells. Cell 1986; 45:917 - 27; http://dx.doi.org/10.1016/0092-8674(86)90566-0; PMID: 2871944
- Valcourt JR, Lemons JM, Haley EM, Kojima M, Demuren OO, Coller HA. Staying alive: metabolic adaptations to quiescence. Cell Cycle 2012; 11:1680 - 96; http://dx.doi.org/10.4161/cc.19879; PMID: 22510571
- Haneline LS, White H, Yang FC, Chen S, Orschell C, Kapur R, et al. Genetic reduction of class IA PI-3 kinase activity alters fetal hematopoiesis and competitive repopulating ability of hematopoietic stem cells in vivo. Blood 2006; 107:1375 - 82; http://dx.doi.org/10.1182/blood-2005-05-1985; PMID: 16239435
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006; 441:475 - 82; http://dx.doi.org/10.1038/nature04703; PMID: 16598206
- Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006; 441:518 - 22; http://dx.doi.org/10.1038/nature04747; PMID: 16633340
- Gan B, Sahin E, Jiang S, Sanchez-Aguilera A, Scott KL, Chin L, et al. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci U S A 2008; 105:19384 - 9; http://dx.doi.org/10.1073/pnas.0810584105; PMID: 19052232
- Moran-Crusio K, Reavie LB, Aifantis I. Regulation of hematopoietic stem cell fate by the ubiquitin proteasome system. Trends Immunol 2012; 33:357 - 63; http://dx.doi.org/10.1016/j.it.2012.01.009; PMID: 22349458
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010; 7:380 - 90; http://dx.doi.org/10.1016/j.stem.2010.07.011; PMID: 20804973
- Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med 2011; 208:455 - 67; http://dx.doi.org/10.1084/jem.20101145; PMID: 21339326
- Robinson LA, Fleming WH, Galbraith TA. Intrapleural doxycycline control of malignant pleural effusions. Ann Thorac Surg 1993; 55:1115 - 21, discussion 1121-2; http://dx.doi.org/10.1016/0003-4975(93)90017-C; PMID: 8494419
- Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011; 9:298 - 310; http://dx.doi.org/10.1016/j.stem.2011.09.010; PMID: 21982230
- Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012; 336:225 - 8; http://dx.doi.org/10.1126/science.1218395; PMID: 22499945
- Liu L, Rando TA. Manifestations and mechanisms of stem cell aging. J Cell Biol 2011; 193:257 - 66; http://dx.doi.org/10.1083/jcb.201010131; PMID: 21502357
- Allsopp RC, Morin GB, DePinho R, Harley CB, Weissman IL. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood 2003; 102:517 - 20; http://dx.doi.org/10.1182/blood-2002-07-2334; PMID: 12663456
- Phadwal K, Alegre-Abarrategui J, Watson AS, Pike L, Anbalagan S, Hammond EM, et al. A novel method for autophagy detection in primary cells: impaired levels of macroautophagy in immunosenescent T cells. Autophagy 2012; 8:677 - 89; http://dx.doi.org/10.4161/auto.18935; PMID: 22302009
- Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med 2011; 208:2691 - 703; http://dx.doi.org/10.1084/jem.20111490; PMID: 22110168
- Liu F, Lee JY, Wei H, Tanabe O, Engel JD, Morrison SJ, et al. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010; 116:4806 - 14; http://dx.doi.org/10.1182/blood-2010-06-288589; PMID: 20716775
- Liu F, Guan JL. FIP200, an essential component of mammalian autophagy is indispensible for fetal hematopoiesis. Autophagy 2011; 7:229 - 30; http://dx.doi.org/10.4161/auto.7.2.14125; PMID: 21088496
- Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal 2009; 2:ra75; http://dx.doi.org/10.1126/scisignal.2000559; PMID: 19934433
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003; 423:302 - 5; http://dx.doi.org/10.1038/nature01587; PMID: 12714971
- Yahata T, Takanashi T, Muguruma Y, Ibrahim AA, Matsuzawa H, Uno T, et al. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 2011; 118:2941 - 50; http://dx.doi.org/10.1182/blood-2011-01-330050; PMID: 21734240
- Wang J, Sun Q, Morita Y, Jiang H, Gross A, Lechel A, et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell 2012; 148:1001 - 14; http://dx.doi.org/10.1016/j.cell.2012.01.040; PMID: 22385964
- Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet 2008; 9:115 - 28; http://dx.doi.org/10.1038/nrg2269; PMID: 18202695
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A 2009; 106:19860 - 5; PMID: 19910529
- Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A 2010; 107:832 - 7; http://dx.doi.org/10.1073/pnas.0913170107; PMID: 20080761
- Kang YA, Sanalkumar R, O’Geen H, Linnemann AK, Chang CJ, Bouhassira EE, et al. Autophagy driven by a master regulator of hematopoiesis. Mol Cell Biol 2012; 32:226 - 39; http://dx.doi.org/10.1128/MCB.06166-11; PMID: 22025678
- Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007; 110:3056 - 63; http://dx.doi.org/10.1182/blood-2007-05-087759; PMID: 17595331
- Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med 2008; 205:2397 - 408; http://dx.doi.org/10.1084/jem.20081297; PMID: 18809716
- Kim M, Cooper DD, Hayes SF, Spangrude GJ. Rhodamine-123 staining in hematopoietic stem cells of young mice indicates mitochondrial activation rather than dye efflux. Blood 1998; 91:4106 - 17; PMID: 9596656
- Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 2009; 461:537 - 41; http://dx.doi.org/10.1038/nature08313; PMID: 19727075
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984 - 4001; http://dx.doi.org/10.4161/cc.8.23.10238; PMID: 19923890
- Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, Paktinat M, et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010; 115:1406 - 15; http://dx.doi.org/10.1182/blood-2009-06-229443; PMID: 20008787
- Hoshii T, Tadokoro Y, Naka K, Ooshio T, Muraguchi T, Sugiyama N, et al. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J Clin Invest 2012; 122:2114 - 29; http://dx.doi.org/10.1172/JCI62279; PMID: 22622041
- De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol 1966; 28:435 - 92; http://dx.doi.org/10.1146/annurev.ph.28.030166.002251; PMID: 5322983
- Dixon JS. “Phagocytic” lysosomes in chromatolytic neurones. Nature 1967; 215:657 - 8; http://dx.doi.org/10.1038/215657a0; PMID: 6050233
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/10.1038/nature04723; PMID: 16625205
- Liang CC, Wang C, Peng X, Gan B, Guan J-L. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem 2010; 285:3499 - 509; http://dx.doi.org/10.1074/jbc.M109.072389; PMID: 19940130
- Doetsch F, Caillé I, Lim DA, García-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999; 97:703 - 16; http://dx.doi.org/10.1016/S0092-8674(00)80783-7; PMID: 10380923
- Palmer TD, Takahashi J, Gage FH. The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci 1997; 8:389 - 404; http://dx.doi.org/10.1006/mcne.1996.0595; PMID: 9143557
- Mazumdar J, O’Brien WT, Johnson RS, LaManna JC, Chavez JC, Klein PS, et al. O2 regulates stem cells through Wnt/β-catenin signalling. Nat Cell Biol 2010; 12:1007 - 13; http://dx.doi.org/10.1038/ncb2102; PMID: 20852629
- Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol 2010; 12:999 - 1006; http://dx.doi.org/10.1038/ncb2101; PMID: 20835244
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006; 3:177 - 85; http://dx.doi.org/10.1016/j.cmet.2006.02.002; PMID: 16517405
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283:10892 - 903; http://dx.doi.org/10.1074/jbc.M800102200; PMID: 18281291
- Paik JH, Ding Z, Narurkar R, Ramkissoon S, Muller F, Kamoun WS, et al. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 2009; 5:540 - 53; http://dx.doi.org/10.1016/j.stem.2009.09.013; PMID: 19896444
- Renault VM, Rafalski VA, Morgan AA, Salih DA, Brett JO, Webb AE, et al. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 2009; 5:527 - 39; http://dx.doi.org/10.1016/j.stem.2009.09.014; PMID: 19896443
- Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol 2010; 12:665 - 75; http://dx.doi.org/10.1038/ncb2069; PMID: 20543840
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6:458 - 71; http://dx.doi.org/10.1016/j.cmet.2007.11.001; PMID: 18054315
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472 - 83; http://dx.doi.org/10.1016/j.cmet.2007.11.004; PMID: 18054316
- Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2011; 8:59 - 71; http://dx.doi.org/10.1016/j.stem.2010.11.028; PMID: 21211782
- Vázquez P, Arroba AI, Cecconi F, de la Rosa EJ, Boya P, de Pablo F. Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 2012; 8:187 - 99; http://dx.doi.org/10.4161/auto.8.2.18535; PMID: 22240590
- Chin TY, Kao CH, Wang HY, Huang WP, Ma KH, Chueh SH. Inhibition of the mammalian target of rapamycin promotes cyclic AMP-induced differentiation of NG108-15 cells. Autophagy 2010; 6:1139 - 56; http://dx.doi.org/10.4161/auto.6.8.13564; PMID: 20935515
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, et al. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 2009; 119:3329 - 39; PMID: 19855132
- Baerga R, Zhang Y, Chen PH, Goldman S, Jin S. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 2009; 5:1118 - 30; http://dx.doi.org/10.4161/auto.5.8.9991; PMID: 19844159
- Takano-Ohmuro H, Mukaida M, Kominami E, Morioka K. Autophagy in embryonic erythroid cells: its role in maturation. Eur J Cell Biol 2000; 79:759 - 64; http://dx.doi.org/10.1078/0171-9335-00096; PMID: 11089924
- Zhang J, Liu J, Huang Y, Chang JY, Liu L, McKeehan WL, et al. FRS2α-mediated FGF signals suppress premature differentiation of cardiac stem cells through regulating autophagy activity. Circ Res 2012; 110:e29 - 39; http://dx.doi.org/10.1161/CIRCRESAHA.111.255950; PMID: 22207710
- Qyang Y, Martin-Puig S, Chiravuri M, Chen S, Xu H, Bu L, et al. The renewal and differentiation of Isl1+ cardiovascular progenitors are controlled by a Wnt/beta-catenin pathway. Cell Stem Cell 2007; 1:165 - 79; http://dx.doi.org/10.1016/j.stem.2007.05.018; PMID: 18371348
- Kwon C, Qian L, Cheng P, Nigam V, Arnold J, Srivastava D. A regulatory pathway involving Notch1/beta-catenin/Isl1 determines cardiac progenitor cell fate. Nat Cell Biol 2009; 11:951 - 7; http://dx.doi.org/10.1038/ncb1906; PMID: 19620969
- Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 2012; 336:477 - 81; http://dx.doi.org/10.1126/science.1217032; PMID: 22539723
- Karamboulas C, Swedani A, Ward C, Al-Madhoun AS, Wilton S, Boisvenue S, et al. HDAC activity regulates entry of mesoderm cells into the cardiac muscle lineage. J Cell Sci 2006; 119:4305 - 14; http://dx.doi.org/10.1242/jcs.03185; PMID: 17038545
- Rosenblatt-Velin N, Lepore MG, Cartoni C, Beermann F, Pedrazzini T. FGF-2 controls the differentiation of resident cardiac precursors into functional cardiomyocytes. J Clin Invest 2005; 115:1724 - 33; http://dx.doi.org/10.1172/JCI23418; PMID: 15951838
- Nombela-Arrieta C, Ritz J, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol 2011; 12:126 - 31; http://dx.doi.org/10.1038/nrm3049; PMID: 21253000
- Pevsner-Fischer M, Levin S, Zipori D. The origins of mesenchymal stromal cell heterogeneity. Stem Cell Rev 2011; 7:560 - 8; http://dx.doi.org/10.1007/s12015-011-9229-7; PMID: 21437576
- Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell 2012; 10:709 - 16; http://dx.doi.org/10.1016/j.stem.2012.05.015; PMID: 22704511
- Oliver L, Hue E, Priault M, Vallette FM. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev 2012; 21:2779 - 88; http://dx.doi.org/10.1089/scd.2012.0124; PMID: 22519885
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101 - 11; http://dx.doi.org/10.1091/mbc.E03-09-0704; PMID: 14699058
- Zhang Q, Yang YJ, Wang H, Dong QT, Wang TJ, Qian HY, et al. Autophagy activation: a novel mechanism of atorvastatin to protect mesenchymal stem cells from hypoxia and serum deprivation via AMP-activated protein kinase/mammalian target of rapamycin pathway. Stem Cells Dev 2012; 21:1321 - 32; http://dx.doi.org/10.1089/scd.2011.0684; PMID: 22356678
- Sanchez CG, Penfornis P, Oskowitz AZ, Boonjindasup AG, Cai DZ, Dhule SS, et al. Activation of autophagy in mesenchymal stem cells provides tumor stromal support. Carcinogenesis 2011; 32:964 - 72; http://dx.doi.org/10.1093/carcin/bgr029; PMID: 21317300
- Bohensky J, Shapiro IM, Leshinsky S, Terkhorn SP, Adams CS, Srinivas V. HIF-1 regulation of chondrocyte apoptosis: induction of the autophagic pathway. Autophagy 2007; 3:207 - 14; PMID: 17224629
- Caramés B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum 2010; 62:791 - 801; http://dx.doi.org/10.1002/art.27305; PMID: 20187128
- Xia X, Kar R, Gluhak-Heinrich J, Yao W, Lane NE, Bonewald LF, et al. Glucocorticoid-induced autophagy in osteocytes. J Bone Miner Res 2010; 25:2479 - 88; http://dx.doi.org/10.1002/jbmr.160; PMID: 20564240
- Jia J, Yao W, Guan M, Dai W, Shahnazari M, Kar R, et al. Glucocorticoid dose determines osteocyte cell fate. FASEB J 2011; 25:3366 - 76; http://dx.doi.org/10.1096/fj.11-182519; PMID: 21705669
- Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med 2011; 17:313 - 9; http://dx.doi.org/10.1038/nm.2304; PMID: 21386835
- Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 2012; 21:283 - 96; http://dx.doi.org/10.1016/j.ccr.2012.03.003; PMID: 22439924
- Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell 2012; 10:717 - 28; http://dx.doi.org/10.1016/j.stem.2012.05.007; PMID: 22704512
- Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat 2011; 126:355 - 64; http://dx.doi.org/10.1007/s10549-010-0924-x; PMID: 20458531
- Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer 2008; 8:56 - 61; http://dx.doi.org/10.1038/nrc2255; PMID: 18059462
- Gillies RJ, Gatenby RA. Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis?. J Bioenerg Biomembr 2007; 39:251 - 7; http://dx.doi.org/10.1007/s10863-007-9085-y; PMID: 17624581
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis?. Nat Rev Cancer 2004; 4:891 - 9; http://dx.doi.org/10.1038/nrc1478; PMID: 15516961
- Lin Q, Yun Z. Impact of the hypoxic tumor microenvironment on the regulation of cancer stem cell characteristics. Cancer Biol Ther 2010; 9:949 - 56; http://dx.doi.org/10.4161/cbt.9.12.12347; PMID: 20581454
- Quail DF, Taylor MJ, Postovit LM. Microenvironmental regulation of cancer stem cell phenotypes. Curr Stem Cell Res Ther 2012; 7:197 - 216; http://dx.doi.org/10.2174/157488812799859838; PMID: 22329582
- Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A 2012; 109:2784 - 9; http://dx.doi.org/10.1073/pnas.1018866109; PMID: 22308314
- Piao Y, Liang J, Holmes L, Zurita AJ, Henry V, Heymach JV, et al. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol 2012; 14:1379 - 92; http://dx.doi.org/10.1093/neuonc/nos158; PMID: 22965162
- Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007; 129:465 - 72; http://dx.doi.org/10.1016/j.cell.2007.04.019; PMID: 17482542
- Espina V, Mariani BD, Gallagher RI, Tran K, Banks S, Wiedemann J, et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PLoS One 2010; 5:e10240; http://dx.doi.org/10.1371/journal.pone.0010240; PMID: 20421921
- Espina V, Liotta LA. What is the malignant nature of human ductal carcinoma in situ?. Nat Rev Cancer 2011; 11:68 - 75; http://dx.doi.org/10.1038/nrc2950; PMID: 21150936
- Mitchener JS, Shelburne JD, Bradford WD, Hawkins HK. Cellular autophagocytosis induced by deprivation of serum and amino acids in HeLa cells. Am J Pathol 1976; 83:485 - 92; PMID: 937509
- Gong C, Song E, Codogno P, Mehrpour M. The roles of BECN1 and autophagy in cancer are context dependent. Autophagy 2012; 8:1853 - 5; http://dx.doi.org/10.4161/auto.21996; PMID: 22960473
- Warburg O. On the origin of cancer cells. Science 1956; 123:309 - 14; http://dx.doi.org/10.1126/science.123.3191.309; PMID: 13298683
- Warburg O. On respiratory impairment in cancer cells. Science 1956; 124:269 - 70; PMID: 13351639
- Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol 1927; 8:519 - 30; http://dx.doi.org/10.1085/jgp.8.6.519; PMID: 19872213
- Bayley JP, Devilee P. The Warburg effect in 2012. Curr Opin Oncol 2012; 24:62 - 7; http://dx.doi.org/10.1097/CCO.0b013e32834deb9e; PMID: 22123234
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 2011; 22:165 - 78; http://dx.doi.org/10.1091/mbc.E10-06-0500; PMID: 21119005
- Pani G, Galeotti T, Chiarugi P. Metastasis: cancer cell’s escape from oxidative stress. Cancer Metastasis Rev 2010; 29:351 - 78; http://dx.doi.org/10.1007/s10555-010-9225-4; PMID: 20386957
- Pani G, Giannoni E, Galeotti T, Chiarugi P. Redox-based escape mechanism from death: the cancer lesson. Antioxid Redox Signal 2009; 11:2791 - 806; http://dx.doi.org/10.1089/ars.2009.2739; PMID: 19686053
- Hüsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008; 13:58 - 68; http://dx.doi.org/10.1016/j.ccr.2007.12.003; PMID: 18167340
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51 - 64; http://dx.doi.org/10.1016/j.ccr.2006.06.001; PMID: 16843265
- Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy 2007; 3:28 - 31; PMID: 16969128
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21:1621 - 35; http://dx.doi.org/10.1101/gad.1565707; PMID: 17606641
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21:1367 - 81; http://dx.doi.org/10.1101/gad.1545107; PMID: 17510285
- Mahalingam D, Kong CM, Lai J, Tay LL, Yang H, Wang X. Reversal of aberrant cancer methylome and transcriptome upon direct reprogramming of lung cancer cells. Sci Rep 2012; 2:592; http://dx.doi.org/10.1038/srep00592; PMID: 22912920
- Ramos-Mejia V, Fraga MF, Menendez P. iPSCs from cancer cells: challenges and opportunities. Trends Mol Med 2012; 18:245 - 7; http://dx.doi.org/10.1016/j.molmed.2012.04.001; PMID: 22521522
- Zhang X, Cruz FD, Terry M, Remotti F, Matushansky I. Terminal differentiation and loss of tumorigenicity of human cancers via pluripotency-based reprogramming. Oncogene 2012; In press http://dx.doi.org/10.1038/onc.2012.237; PMID: 22777357
- Folmes CD, Nelson TJ, Dzeja PP, Terzic A. Energy metabolism plasticity enables stemness programs. Ann N Y Acad Sci 2012; 1254:82 - 9; http://dx.doi.org/10.1111/j.1749-6632.2012.06487.x; PMID: 22548573
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25:717 - 29; http://dx.doi.org/10.1101/gad.2016111; PMID: 21406549
- Dang CV. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res 2010; 70:859 - 62; http://dx.doi.org/10.1158/0008-5472.CAN-09-3556; PMID: 20086171
- Chen T, Shen L, Yu J, Wan H, Guo A, Chen J, et al. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell 2011; 10:908 - 11; http://dx.doi.org/10.1111/j.1474-9726.2011.00722.x; PMID: 21615676
- Abollo-Jiménez F, Jiménez R, Cobaleda C. Physiological cellular reprogramming and cancer. Semin Cancer Biol 2010; 20:98 - 106; http://dx.doi.org/10.1016/j.semcancer.2010.02.002; PMID: 20188173
- Chen HY, White E. Role of autophagy in cancer prevention. Cancer Prev Res (Phila) 2011; 4:973 - 83; http://dx.doi.org/10.1158/1940-6207.CAPR-10-0387; PMID: 21733821
- Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest 2009; 119:1109 - 23; http://dx.doi.org/10.1172/JCI35660; PMID: 19363292
- Calabretta B, Salomoni P. Inhibition of autophagy: a new strategy to enhance sensitivity of chronic myeloid leukemia stem cells to tyrosine kinase inhibitors. Leuk Lymphoma 2011; 52:Suppl 1 54 - 9; http://dx.doi.org/10.3109/10428194.2010.546913; PMID: 21250825
- Martelli AM, Evangelisti C, Follo MY, Ramazzotti G, Fini M, Giardino R, et al. Targeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in cancer stem cells. Curr Med Chem 2011; 18:2715 - 26; http://dx.doi.org/10.2174/092986711796011201; PMID: 21649579
- Wallace DC. Mitochondria and cancer. Nat Rev Cancer 2012; 12:685 - 98; http://dx.doi.org/10.1038/nrc3365; PMID: 23001348
- Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle 2010; 9:4297 - 306; http://dx.doi.org/10.4161/cc.9.21.13817; PMID: 21051947
- Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010; 9:3485 - 505; http://dx.doi.org/10.4161/cc.9.17.12721; PMID: 20861672
- Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011; 10:1271 - 86; http://dx.doi.org/10.4161/cc.10.8.15330; PMID: 21512313
- Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle 2012; 11:2285 - 302; http://dx.doi.org/10.4161/cc.20718; PMID: 22684298
- Castello-Cros R, Bonuccelli G, Molchansky A, Capozza F, Witkiewicz AK, Birbe RC, et al. Matrix remodeling stimulates stromal autophagy, “fueling” cancer cell mitochondrial metabolism and metastasis. Cell Cycle 2011; 10:2021 - 34; http://dx.doi.org/10.4161/cc.10.12.16002; PMID: 21646868
- Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging (Albany NY) 2010; 2:185 - 99; PMID: 20442453
- Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal 2012; 16:1264 - 84; http://dx.doi.org/10.1089/ars.2011.4243; PMID: 21883043
- Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer 2005; 5:744 - 9; http://dx.doi.org/10.1038/nrc1694; PMID: 16148886
- Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality?. Nat Med 2009; 15:1010 - 2; http://dx.doi.org/10.1038/nm0909-1010; PMID: 19734877
- Hollier BG, Evans K, Mani SA. The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J Mammary Gland Biol Neoplasia 2009; 14:29 - 43; http://dx.doi.org/10.1007/s10911-009-9110-3; PMID: 19242781
- Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 2008; 3:e2888; http://dx.doi.org/10.1371/journal.pone.0002888; PMID: 18682804
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003; 100:3983 - 8; http://dx.doi.org/10.1073/pnas.0530291100; PMID: 12629218
- Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007; 11:259 - 73; http://dx.doi.org/10.1016/j.ccr.2007.01.013; PMID: 17349583
- Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle 2011; 10:3871 - 85; http://dx.doi.org/10.4161/cc.10.22.17976; PMID: 22127234
- Amaravadi RK. Autophagy and tumor cell invasion. Cell Cycle 2012; 11:3718 - 9; http://dx.doi.org/10.4161/cc.22147; PMID: 22982997
- Macintosh RL, Timpson P, Thorburn J, Anderson KI, Thorburn A, Ryan KM. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle 2012; 11:2022 - 9; http://dx.doi.org/10.4161/cc.20424; PMID: 22580450
- Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013; 32:699 - 712; PMID: 22525272
- Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, et al. Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res 2009; 69:8844 - 52; http://dx.doi.org/10.1158/0008-5472.CAN-08-4401; PMID: 19903843
- Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008; 100:672 - 9; http://dx.doi.org/10.1093/jnci/djn123; PMID: 18445819
- Suzuki HI, Kiyono K, Miyazono K. Regulation of autophagy by transforming growth factor-β (TGF-β) signaling. Autophagy 2010; 6:645 - 7; http://dx.doi.org/10.4161/auto.6.5.12046; PMID: 20458184
- Tu YF, Kaipparettu BA, Ma Y, Wong LJ. Mitochondria of highly metastatic breast cancer cell line MDA-MB-231 exhibits increased autophagic properties. Biochim Biophys Acta 2011; 1807:1125 - 32; http://dx.doi.org/10.1016/j.bbabio.2011.04.015; PMID: 21570379
- Caja L, Bertran E, Campbell J, Fausto N, Fabregat I. The transforming growth factor-beta (TGF-β) mediates acquisition of a mesenchymal stem cell-like phenotype in human liver cells. J Cell Physiol 2011; 226:1214 - 23; http://dx.doi.org/10.1002/jcp.22439; PMID: 20945437
- Mancias JD, Kimmelman AC. Targeting autophagy addiction in cancer. Oncotarget 2011; 2:1302 - 6; PMID: 22185891
- McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A 2012; 109:8253 - 8; http://dx.doi.org/10.1073/pnas.1118193109; PMID: 22566612
- Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011; 147:223 - 34; http://dx.doi.org/10.1016/j.cell.2011.08.037; PMID: 21962518
- Amaravadi R. Autophagy can contribute to cell death when combining targeted therapy. Cancer Biol Ther 2009; 8:130 - 3; PMID: 19901522
- Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer 2009; 125:717 - 22; http://dx.doi.org/10.1002/ijc.24402; PMID: 19431142
- Klionsky DJ. The autophagy community. Autophagy 2012; 8:1003; http://dx.doi.org/10.4161/auto.20666; PMID: 22647354