Abstract
Mitochondria are highly dynamic organelles of crucial importance to the proper functioning of neuronal, cardiac and other cell types dependent upon aerobic efficiency. Mitochondrial dysfunction has been implicated in numerous human conditions, to include cancer, metabolic diseases, neurodegeneration, diabetes, and aging. In recent years, mitochondrial turnover by macroautophagy (mitophagy) has captured the limelight, due in part to discoveries that genes linked to Parkinson disease regulate this quality control process. A rapidly growing literature is clarifying effector mechanisms that underlie the process of mitophagy; however, factors that regulate positive or negative cellular outcomes have been less studied. Here, we review the literature on two major pathways that together may determine cellular adaptation vs. cell death in response to mitochondrial dysfunction. Mitochondrial biogenesis and mitophagy represent two opposing, but coordinated processes that determine mitochondrial content, structure, and function. Recent data indicate that the capacity to undergo mitochondrial biogenesis, which is dysregulated in disease states, may play a key role in determining cell survival following mitophagy-inducing injuries. The current literature on major pathways that regulate mitophagy and mitochondrial biogenesis is summarized, and mechanisms by which the interplay of these two processes may determine cell fate are discussed. We conclude that in primary neurons and other mitochondrially dependent cells, disruptions in any phase of the mitochondrial recycling process can contribute to cellular dysfunction and disease. Given the emerging importance of crosstalk among regulators of mitochondrial function, autophagy, and biogenesis, signaling pathways that coordinate these processes may contribute to therapeutic strategies that target or regulate mitochondrial turnover and regeneration.
Introduction
In addition to ATP production, mitochondria regulate calcium homeostasis, heme and steroid biosynthesis, and cell survival/death decisions.Citation1,Citation2 For most differentiated tissues, the proper control of mitochondrial content, distribution and activity is key to maintaining normal cellular functions, while mitochondrial metabolism is often dispensable in rapidly growing neoplastic or stem cells.Citation3 The mitochondrial proteome is also strikingly different between tissue types.Citation4,Citation5 For these reasons, there are likely to be significant cell-type specific differences, as well as common pathways, underlying the regulation of mitochondrial structure, function, and content, as well as the fate of cells exhibiting increased mitochondrial autophagy. These factors add complexity to the study of mitophagy regulation, particularly in disease states.
The mitochondrial “lifecycle” begins with the coordinated synthesis of nuclear DNA (nDNA) and mitochondrial DNA (mtDNA)-encoded proteins, together with membrane biosynthesis and the proper targeting and folding of respiratory chain subunits. These processes are grouped under the term “mitochondrial biogenesis,” although the dynamics of the system preclude facile identification of newly made mitochondria. In contrast, the clearance of intact mitochondrial segments from the cytoplasm is readily visualized through the process of macroautophagy. Coordination between these two processes must be required for the proper regulation of mitochondrial quality and quantity, although mechanisms that underlie such coordination remain relatively understudied. The importance of cross-regulation is further highlighted by recent studies indicating that disruption of the anabolic-catabolic balance between mitochondrial biogenesis and mitophagy could delay post-injury recovery and contribute to cell death.Citation6-Citation9 Thus, additional studies aimed at this interface may prove fruitful for our understanding of both healthy and disease states.
In the following sections, we review the major regulatory pathways that have been defined for mitophagy and mitochondrial biogenesis, and discuss how these processes may interact to regulate cell fate decisions. Autophagic stress elicited from an imbalance between autophagy induction and the successful degradation and reutilization of degraded components can convert a fundamentally adaptive response into a detrimental outcome.Citation10 Similarly, we propose that the efficiency with which a cell can regenerate mitochondria declines with aging and disease; the ability to balance increased mitochondrial degradation with increased biogenesis may determine the outcome of mitophagy in neurons and other mitochondrially-dependent cells.
Mitophagy and Mitochondrial Quality Control
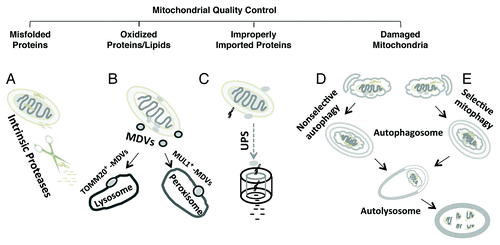
The degradation of misfolded, oxidized or otherwise damaged mitochondrial components plays a key role in mitochondrial quality control (). It has been suggested that the mitochondrion employs its own intrinsic proteases (e.g., LONP1, PARL, and HTRA2/OMI) to degrade unfolded proteins inside the matrix and intermembrane space.Citation11 In response to oxidative stress, mitochondria-derived vesicles (MDVs) expressing either TOMM20/TOM20 or MUL1/MAPL are formed containing cargoes destined for lysosomal or peroxisomal delivery, respectively.Citation12,Citation13 Intriguingly, this process does not require mitochondrial depolarization and is independent of the autophagy proteins ATG5 and microtubule-associated protein 1 light chain 3 (MAP1LC3), key mediators of macroautophagy.Citation13 The proteasome is involved in clearance of damaged or misdirected outer mitochondrial membrane proteins and proteins that fail to be successfully imported.Citation14,Citation15 Mitochondrial dynamics—fission, fusion, transport—also play key roles in regulating mitochondrial function and repair, functioning to segregate or exchange damaged mitochondrial components.Citation16,Citation17 Nevertheless, when mitochondria are subject to severe injury, it is generally accepted that mitophagy is the major mechanism for clearance of the damaged organelles. Programmed or developmental loss of mitochondria, as observed during erythrocyte or lens cell maturation, also occurs through mitophagy. It stands to reason that if mitochondrial degradation is not balanced by a coordinated upregulation of mitochondrial biogenesis, a net loss of mitochondria will occur.
Figure 1. Major clearance pathways engaged in mitochondrial quality control. (A) Intrinsic mitochondrial proteases target and degrade unfolded proteins in the matrix and intermembrane space. (B) Under conditions of oxidative stress, mitochondria-derived vesicles (MDV) deliver oxidized proteins and lipids for lysosomal and peroxisomal degradation. (C) The ubiquitin-proteasome system (UPS) is involved in clearance of damaged outer mitochondrial membrane proteins or improperly targeted nDNA-encoded mitochondrial proteins. Damaged mitochondria may be removed as a consequence of generalized increases in autophagy (D) or through specific cargo targeting mechanisms that result in selective mitophagy (E). Selective mitophagy also functions to sequester unneeded mitochondria under certain physiological conditions (e.g., maturation of reticuloctyes).
Three major types of autophagic pathways have been characterized: (1) macroautophagy, (2) microautophagy, and (3) chaperone-mediated autophagy (CMA). Macroautophagy (referred to hereafter as autophagy) is a tightly regulated degradation pathway involving the generation of a specialized organelle to target cytoplasmic components to the lysosome. In this manner, autophagy acts as a gatekeeper of cellular homeostasis. Increasing evidence has documented that autophagy plays a pivotal role in numerous biological and physiological processes, including mammalian development and differentiation,Citation18,Citation19 aging,Citation20,Citation21 and immunity.Citation22,Citation23 Aberrations in autophagy are described in neurodegenerative,Citation24-Citation26 cardiovascularCitation27 and immunological diseases,Citation28 and in cancer development.Citation29
The process of autophagy involves several steps: induction (nucleation), autophagosome formation (sequestration), maturation to form autolysosomes (fusion), and enzymatic hydrolysis (degradation).Citation30,Citation31 In addition, from a disease perspective, we propose that the impact of autophagy cannot be fully understood without consideration of whether or not the liberated molecules are successfully reutilized in biosynthetic pathways, which may become dysfunctional under many pathological conditions.Citation9,Citation32
Among different regulatory mechanisms, nutrient starvation is linked to changes in AMP-activated protein kinase (PRKAA2/AMPK) and MTOR signaling, which regulate the phosphorylation states of ULK1/2, the mammalian homolog of yeast ATG1. The details of such regulation have been reviewed elsewhere.Citation33,Citation34 In addition, noncanonical BECN1/Beclin 1-independent forms of autophagy or mitophagy have been described in response to the complex I inhibitor MPP+, resveratrol, mutant leucine-rich repeat kinase 2 (LRRK2) or UV irradiation of individual mitochondria.Citation35-Citation38 Involvement of these alternative pathways has been implicated in cell fate decisions.
The yeast Atg8 protein and its mammalian homologs are key mediators of autophagosome formation. Atg8 is a cytosolic protein that, upon induction of autophagy, is covalently conjugated to autophagic membranes as they form from a phagophore-assembly site.Citation39 A portion of Atg8 is recovered from the completed autophagosome by enzymatic cleavage, but a portion remains inside until degraded in the vacuole (the yeast analog of the lysosome).Citation40,Citation41 MAP1LC3, one of the mammalian Atg8 orthologs,Citation42 is similarly regulated through ubiquitin-like conjugation catalyzed by mammalian ATG7 and ATG3, and by delipidation reactions involving mammalian ATG4.Citation43 Thus, lipid-conjugated MAP1LC3 (MAP1LC3-II) is commonly used as a marker of autophagic structures.
MAP1LC3 is regulated by direct phosphorylation of its N-terminal helical region by protein kinase A (PKA), which suppresses its autophagic function.Citation44 Moreover, MAP1LC3 helices regulate selective autophagy through interaction with cargo adaptor proteins such as SQSTM1 (sequestosome 1/p62),Citation45 neighbor of BRCA1 (NBR1),Citation46 the BCL2-related protein BNIP3L/NIX,Citation47 and the autophagy linked FYVE protein (WDFY3/ALFY).Citation48
Role of MAP1LC3-interacting region (LIR) proteins
The BH3 domain-containing BCL2 family members BNIP3 and BNIP3L/NIX were originally identified as intrinsic mitochondrial proapoptotic factors. Later studies suggested both proteins may serve as “receptors” for mitophagy by interacting with key components of the general autophagy machinery. During the course of normal development of the erythroid lineage, the entire cohort of mitochondria is eliminated in mature erythrocytes. BNIP3L is necessary for the sequestration of mitochondria into autophagosomes for selective clearance through a process that is independent of the intrinsic proapoptotic pathway.Citation49,Citation50 Knockout Bnip3l−/− mice develop anemia due to reduced life span of erythrocytes that exhibit mitochondrial retention. However, BNIP3L is not required for engagement of the autophagy machinery, implicating a role primarily in cargo recognition. The signaling mechanism involves BNIP3L-dependent loss of mitochondrial membrane potential (ΔΨm) during erythroid maturation, since the uncoupling agent FCCP can reverse the effects of BNIP3L deficiency.Citation51 BNIP3L-mediated mitophagy is also implicated in the removal of depolarized mitochondria under pathological conditions. In the carbonyl cyanide m-chlorophenyl hydrazone (CCCP) model of mitochondrial injury, the efficient removal of mitochondria is dependent upon two BNIP3L-related events: (1) the reactive oxygen species (ROS)-mediated inhibition of MTOR to induce the general autophagy machinery, and (2) the mitochondrial translocation of PARK2/Parkin and SQSTM1.Citation52 Thus, in contrast to findings in developmental mitophagy, BNIP3L participates in both induction of autophagy and cargo targeting in this pathological model.
The role for BNIP3 in mitophagic regulation was first described in the ceramide-induced autophagic death of malignant glioma cells.Citation53 Another study using an adult myocyte in vitro model showed that BNIP3 can induce dynamin 1-like (DNM1L/Drp1) mitochondrial translocation and mitochondrial fission, which promotes a PARK2-dependent mitophagy.Citation54 Due to the structural similarity of BNIP3 and BNIP3L, it has been proposed that they can regulate autophagy through binding to BCL2 and/or BCL2L1, disrupting the interaction between BCL2 or BCL2L1 and BECN1 to allow the latter to activate the class III phosphatidylinositol 3-kinase PIK3C3/VPS34.Citation55-Citation57
Interestingly, BNIP3L contains a LIR domain that is able to physically interact with MAP1LC3 and GABARAP proteins, thereby recruiting the autophagic machinery.Citation47 Similarly, BNIP3 binds MAP1LC3 to mediate reticulophagy and mitophagy.Citation58 The list of MAP1LC3 interacting proteins that can mediate the selective sequestration of mitochondria within autophagosomes includes other structurally unrelated proteins such as an outer-membrane protein, FUNDC1, which operates in hypoxic fibroblasts.Citation59 Mitochondrial lipids that show altered distribution with injury may also facilitate mitophagy through direct interactions with MAP1LC3 (Chu CT, Bayir H, Kagan V, unpublished data). These studies indicate that several changes to the mitochondrial surface may cooperatively or independently serve to enrich for mitochondrial cargo.
The CCCP-PINK1-PARK2 pathway
A major pathway for injury-induced mitophagy, reported initially by the Youle group,Citation60 has become one of the most widely studied pathways, possibly due to involvement of gene products linked to recessive forms of Parkinson disease. These are PARK2/Parkin, whose mutations represent the most common cause of juvenile onset autosomal recessive parkinsonism,Citation61 and PTEN-induced kinase 1 (PINK1), which is a mitochondrially targeted kinase implicated in the regulation of mitochondrial quality control.Citation62,Citation63
PARK2 is a RING domain containing E3 ubiquitin ligase that can be activated through auto-ubiquitination.Citation64,Citation65 Under conditions of mitochondrial depolarization induced by mitochondrial uncoupling reagents such as CCCP, PARK2 translocates to mitochondria and mediates mitochondrial degradation.Citation60,Citation66 Mitochondrial proteins such as BCL2,Citation67 MFN1/2,Citation68 and VDAC1Citation69 are among the substrates of PARK2; although the importance of these ubiquitination reactions for mitophagy remains controversial. It was reported that PARK2 regulates mitophagy by initially engaging the proteasome,Citation70 whereas other studies suggest that the degradation of mitochondrial outer membrane proteins (TOMM20, TOMM40L, TOMM70A, and SYNJ2BP/OMP25) occurs through a proteasome-dependent, but mitophagy-independent pathway.Citation15 In any case, the E3 ubiquitin ligase activity of PARK2 is essential for depolarization-induced mitochondrial degradation.
PINK1 shows protective effects against mitochondrial injury induced by neurotoxin challenge, while disease-associated PINK1 mutants or loss of function modeled using RNAi knockdown result in ROS-mediated mitochondrial injury.Citation63,Citation71 RNAi studies implicate endogenous PINK1 in the suppression of autophagy,Citation63,Citation72-Citation74 whereas other studies report that dual overexpression of PINK1 and PARK2 promotes mitophagy in the CCCP model.Citation60,Citation75,Citation76 PINK1 overexpression also suppresses 6-OHDA-mediated mitophagy, possibly by reducing fission or enhancing fusion.Citation63 The disparate effects of PINK1 in suppressing or promoting mitophagy may reflect differences in subcellular compartmentalization or post-translational modifications of downstream mediators. Recent studies suggest that PARK2 may be auto-ubiquitinated through a PINK1-dependent self-association mechanism in the cytosol.Citation77,Citation78 These studies disagree, however, on the effect of PARK2 ubiquitination on its participation in mitophagy.
We hypothesize that changes in PINK1 proteolytic processing and/or its subcellular localization regulate whether it serves to stabilize a functional cellular mitochondrial network or regulate its destruction through fission or mitophagy. The effects of PINK1 in promoting autophagy or CCCP-mediated mitophagy require expression of full-length PINK1,Citation79,Citation80 while the predominant form of PINK1 in normal cells is processed.Citation81 Under stressed conditions, mitochondrial membrane depolarization prevents mitochondrial import and processing of PINK1, resulting in accumulation of unprocessed PINK1 on the outer mitochondrial membrane to recruit PARK2.Citation82 This is consistent with studies suggesting that PINK1 acts as an upstream regulator of PARK2,Citation75,Citation83,Citation84 but the ability of PARK2 overexpression to compensate for PINK1 deficiencyCitation63,Citation83 indicates that PINK1 may not be strictly required for the neuroprotective effects of PARK2 overexpression, even those requiring an intact autophagy machinery.Citation63
While the CCCP-PINK1-PARK2 pathway robustly and reproducibly induces nearly complete clearance of mitochondria in transformed cells through proteasomal and lysosomal mechanisms, its biological relevance is less clear. Most studies do not address the viability of the cells either acutely or in longer time frames. In PINK1-deficient cells, mitophagy promotes cell survival, as RNAi knockdown of ATG7 exacerbates cell death.Citation63 However, HeLa cells overexpressing PARK2 are unable to survive in galactose after CCCP-treatment due to mitochondrial loss, while HeLa cells that lack PARK2 are able to survive.Citation60 Interestingly, in some studies, neurons either do not exhibit CCCP-induced mitophagy,Citation85,Citation77 or show delayed, diminished responses compared with HeLa cells.Citation66 These suggest the possibility of cell-type specific modulatory mechanisms.
Mitogen-activated protein kinase/extracellular signal-regulated kinase
The mitogen-activated protein kinases MAPK1/ERK2 and MAPK3/ERK1 play ubiquitous roles in cell proliferation, differentiation, survival, and death. Nonetheless, the requirement for MAPK1/3 activation in selective autophagy has also been demonstrated.Citation35,Citation86,Citation87 We previously found that human brain tissues with Parkinson and other Lewy body diseases display abnormal phospho-MAPK1/3 accumulation in aberrant mitochondria, some of which were inside autophagosomes.Citation88,Citation89 In subsequent in vitro studies, we demonstrated that transfected forms of active, but not kinase-deficient, MAPK1 spontaneously translocate to mitochondria to induce mitophagy, even in the absence of external injury.Citation90 Inhibiting MAPK1/3 activation with U0126 markedly reduces mitophagy and selective mitochondrial protein degradation in response to mitochondrial toxins.Citation35,Citation90 Inhibiting MAPK1/3 activation also restores mitochondrial morphology and interconnectivity, blocks the loss of complex I subunits, and reverses the decrease of aerobic respiratory capacities in a chronic model of MPP+ toxicity.Citation9
In terms of cellular outcomes, mitophagy associated with sustained MAPK1/3 activation tends to promote cell death.Citation35,Citation90 This may reflect noncanonical autophagy induction mechanisms,Citation35,Citation91 or some other factor, such as concurrent suppression of mitochondrial biogenesis.Citation9
Mitochondrial Biogenesis in Health and Disease
Mitochondria consist of over 500 different proteins, with significant variations in composition by organism and tissue type. Most mitochondrial proteins are encoded by nuclear genes and synthesized on cytosolic ribosomes, whereas mtDNA encodes 13 essential respiratory chain complex subunits. Precise coordination and crosstalk between the nuclear and mitochondrial genomes would be expected in mediating appropriate responses to changes in physiological status as well as pathological stresses. However, our understanding of this cross-regulation is largely restricted to how nuclear factors promote the synthesis of proteins required for mitochondrial biogenesis. With the exception of apoptotic signaling, much less is known of mechanisms by which mitochondria communicate back to the nucleus.
Mitochondrial biogenesis is a complex process that includes mtDNA transcription and translation, translation of nDNA-encoded transcripts, membrane recruitment, protein import and assembly of the oxidative phosphorylation complexes.Citation92 Mitochondrial biogenesis is tightly controlled by hormone- or growth factor-initiated intracellular signaling pathways and the resultant activation of nuclear transcription factors.Citation93 Among these, peroxisome proliferator-activated receptor gamma, coactivator 1 α (PPARGC1A/PGC-1α), nuclear respiratory factors (NRF1, NFE2L2/NRF2) and transcription factor A, mitochondrial (TFAM) are the major players. Numerous factors including hormones, second messengers (calcium, endothelial nitric oxide synthase eNOS, cAMP), and kinase pathways (PKA, MAPK, PRKAA2) act to regulate the process at different levels, by regulating PPARGC1A expression, protein localization or post-translational modifications ().
Figure 2. The regulation of mitochondrial biogenesis by the PPARGC1A-NRF1-TFAM pathway. PPARGCA is activated in response to environmental (cold), physiological [exercise or caloric restriction (CR)] or pathological stimuli (oxidative stress). One major pathway involves PKA phosphorylation of CREB that in turn activates PPARGC1A and multiple transcription factors (NRFs, YY1, ERRs, etc.) to result in the synthesis of both nDNA-encoded mitochondrial proteins and mitochondrial transcriptional regulators such as TFAM. TFAM in turn functions to stabilize mtDNA and promote the synthesis of mtDNA-encoded subunits of the electron transport chain (OxPhos/ETC). cAMP-associated phospho-activation and/or deacetylation by SIRT1 increases the activity of PPARGC1A. PPARGC1A can also traffic to mitochondria, where it interacts with TFAM at the mtDNA D-loop. Several major signaling pathways such as MAPK1/3, MYC, MAPK12, and PARK2 are also implicated in modulating mitochondrial biogenesis at different levels. In addition, the PRKAA2, PINK1-PARK2, PKA, and MAPK1/3 pathways also regulate mitophagy, potentially serving to coordinate overall changes in mitochondrial content.
![Figure 2. The regulation of mitochondrial biogenesis by the PPARGC1A-NRF1-TFAM pathway. PPARGCA is activated in response to environmental (cold), physiological [exercise or caloric restriction (CR)] or pathological stimuli (oxidative stress). One major pathway involves PKA phosphorylation of CREB that in turn activates PPARGC1A and multiple transcription factors (NRFs, YY1, ERRs, etc.) to result in the synthesis of both nDNA-encoded mitochondrial proteins and mitochondrial transcriptional regulators such as TFAM. TFAM in turn functions to stabilize mtDNA and promote the synthesis of mtDNA-encoded subunits of the electron transport chain (OxPhos/ETC). cAMP-associated phospho-activation and/or deacetylation by SIRT1 increases the activity of PPARGC1A. PPARGC1A can also traffic to mitochondria, where it interacts with TFAM at the mtDNA D-loop. Several major signaling pathways such as MAPK1/3, MYC, MAPK12, and PARK2 are also implicated in modulating mitochondrial biogenesis at different levels. In addition, the PRKAA2, PINK1-PARK2, PKA, and MAPK1/3 pathways also regulate mitophagy, potentially serving to coordinate overall changes in mitochondrial content.](/cms/asset/95cde86c-58e6-454f-b68b-a7bf6ef37ef5/kaup_a_10924135_f0002.gif)
The regulation of PPARGC1A function
PPARGC1A, a master regulator of mitochondrial biogenesis, belongs to a family of transcriptional coactivators that includes PPARGC1B and the peroxisome proliferator-activated receptor gamma, coactivator-related 1 (PPRC1).Citation94 PPARGC1A is ubiquitously expressed and is capable of driving virtually all aspects of mitochondrial biogenesis, including transcription of respiratory chain and fatty acid oxidation genes, increasing mitochondrial mass, and augmenting mitochondrial respiratory capacity. As an adaptive response to physiological changes such as exercise or caloric restriction, or to pathological stimuli such as oxidative stress, PPARGC1A levels are upregulated, activating transcriptional factors such as nuclear respiratory factors (NRFs), estrogen-related receptors (ERRs), YY1 transcription factor, forkhead box O1 (FOXO1), and myocyte enhancer factors (MEF2/Myef2).Citation95,Citation96
The activity of PPARGC1A is related to its expression level, subcellular localization, and post-transcriptional modifications.Citation97-Citation99 PPARGC1A is predominantly cytoplasmic, but, upon phosphorylation, it is retained in the nucleus to upregulate mitochondrial biogenesis.Citation98 The nuclear accumulation of PPARGC1A can also be initiated by deacetylation.Citation97,Citation98 Thus, there are many points of signaling crosstalk for this master regulator of mitochondrial biogenesis.
Recent studies show that PPARGC1A is also localized in the mitochondrial compartment,Citation9,Citation99,Citation100 where it interacts with TFAM at the mtDNA D-loop.Citation100 In response to exercise, both nuclear and mitochondrial pools of PPARGC1A serve to promote mitochondrial biogenesis,Citation101 while decreased mitochondrial PPARGC1A levels may contribute to impaired mitochondrial biogenesis in response to chronic MPP+ intoxication.Citation9 This transcription factor, therefore, represents one possible mediator of coordinated mtDNA and nDNA transcriptional responses, although its precise role in the mitochondria remains to be elucidated.
There are several major signaling pathways that function upstream to regulate the activation and bioavailability of PPARGC1A. PRKAA2/AMPK functions to upregulate PPARGC1A activityCitation102,Citation103 by sensing changes in the AMP/ATP ratio.Citation104 In skeletal muscle, PRKAA2 phosphorylates PPARGC1A at threonine 177 and serine 538, initiating its gene-regulatory functions.Citation105 SIRT1, an NAD+-dependent deacetylase implicated in longevity and stress responses, also participates in PRKAA2-related pathways.Citation106 PRKAA2 enhances SIRT1 activity by increasing intracellular NAD+ levels.Citation107 SIRT1 deacetylates PPARGC1A to promote its nuclear translocation.Citation97 Alternatively, SIRT1 can activate PRKAA2 via deacetylation, and activation of the PRKAA2 kinase STK11/LKB1, further increasing phosphorylation of PPARGC1A.Citation108 In contrast, acetylation of PPARGC1A by the K(lysine) acetyltransferase 2 (KAT2/GCN5) represses its activity,Citation109 in response to excess calories and the steroid nuclear receptor coactivator 3 (NCOA3/SRC3).Citation110
The cAMP responsive element binding protein (CREB) is another potent inducer of PPARGC1A expression. In response to environmental stimuli, increases in cAMP levels activate PKA. Phosphorylation of CREB activates a downstream cascade of transcriptional complexes involving PPARGC1A, NRF1 and NFE2L2, and TFAM.Citation111 In Huntington disease, mutant HTT/huntingtin inhibits expression of PPARGC1A by interfering with the CREB-TAF4-dependent transcriptional pathway.Citation112 As both PRKAA2 and PKA pathways function to regulate autophagy, these pathways may function to coordinate mitochondrial degradation-biogenesis responses ().
Different branches of the MAPK family show multiple effects on mitochondrial biogenesis. Exercise-induced activation of MAPK12 (p38γ) in mice serves to increase PPARGC1A promoter activity, whereas overexpression of dominant negative MAPK12 attenuates mitochondrial biogenesis.Citation113 Phosphorylation of PPARGC1A by MAPK12 may act by enhancing its ability to promote binding of NRF1 to the cytochrome c promoter and of NFE2L2 to the cytochrome oxidase subunit promoter,Citation114,Citation115 or by disrupting its interaction with MYBBP1A [MYB binding protein (P160) 1a] in myoblasts.Citation116
The MAPK1/3 signaling pathway regulates PPARGC1A activity in a context- or cell-type dependent manner. In Schwann cells, insulin-like growth factor increases mitochondrial DNA replication and mitochondrial biogenesis,Citation117 and acetyl-L-carnitine upregulates PPARGC1A and NRF1 levels in hypoxic hippocampal neurons,Citation118 both through MAPK1/3-dependent mechanisms. However, in other studies, it is the inhibition of MAPK1/3 signaling that enhances PPARGC1A expression, promoting mitochondrial biogenesis in amyloid β injected rats.Citation119 MAPK1/3-dependent decreases in the mitochondrial pool of PPARGC1A level are observed in a chronic model of MPP+ toxicity; inhibiting MAPK1/3 activation restores mitochondrial levels of both PPARGC1A and TFAM, corrects a deficit in mitochondrial protein translation and results in enhanced mitochondrial function.Citation9
While many of the pathways discussed above center on PPARGC1 transcription or post-translational activation, other pathways regulate its bioavailability through degradation. For example, glycogen synthase kinase 3 β (GSK3B) is a kinase, implicated in several models of cell death that negatively regulates PPARGC1A by promoting its proteasomal degradation.Citation97
The NRF transcription factors
The nuclear respiratory factors, NRF1 and NFE2L2/NRF2, were the first nuclear transcription factors implicated in the expression of multiple mitochondrial proteins in vertebrates. NRF1, initially identified through its binding to the cytochrome c promoter, functions as a positive regulator of transcription.Citation120 NRF1 binds to gene promoters encoding respiratory subunits, as well as the mitochondrial transcription factors TFAM and TFBs to regulate mitochondrial transcription and ribosome assembly.Citation121 NFE2L2 was identified based on its specific binding to essential cis-acting elements in the cytochrome oxidase subunit IV (COXIV) promoter,Citation122 and subsequently shown to associate with all 10 nDNA-encoded cytochrome oxidase subunits.Citation123 Inhibition of NFE2L2 by expression of a dominant negative NFE2L2 allele or using siRNA reduces nDNA-encoded COX subunit expression.Citation124 In addition to the effect on COX promoters, NFE2L2, in cooperation with NRF1, regulates mitochondrial transcription through effects on the expression of TFAM, TFB1M, and TFB2M.Citation125
PPARGC1A controls the expression of NRF1 and NFE2L2,Citation126 and their activities are in turn regulated by post-translational modifications. NRF1 exists in a dephosphorylated state in serum-starved cells and is phosphorylated upon addition of serum.Citation127 Serine phosphorylation within its N-terminal domain enhances its DNA bindingCitation127 and trans-activation function.Citation128 In rat hepatoma cells, ROS elicit phosphorylation of NRF1 by AKT1, enhancing its nuclear translocation and increasing TFAM expression.Citation129 In human breast cancer MCF-7 cells treated with sodium butyrate, NRF1 is acetylated by EP300-CREBBP, which enhances DNA binding.Citation130
Mitochondrial transcription factor A
TFAM is a key regulator of mtDNA replication and transcription in vivo, which is essential for mitochondrial biogenesis and embryonic development.Citation131,Citation122 TFAM was first identified as a high mobility group box protein that stimulates transcription through specific binding to recognition sites in mtDNA.Citation132 TFAM protein levels are associated with mtDNA copy number. Heterozygous Tfam+/− knockout mice exhibit reduced mtDNA copy number and respiratory chain deficiencies in heart, while homozygous knockout abolishes oxidative phosphorylation.Citation131 On the other hand, overexpressing human TFAM stimulates mitochondrial biogenesis and ATP synthesis, and prevents MPTP/MPP+-induced neuronal degeneration.Citation9,Citation133,Citation134
In addition to PPARGC1A and NRF proteins, MYC/c-Myc can directly bind to the TFAM promoter, leading to its transcriptional upregulation in lymphocytes.Citation135 Overexpression of MYC leads to increased mitochondrial biogenesis.Citation136
LONP1 is a protease in the eukaryotic mitochondrial matrix that regulates TFAM stability. RNAi knockdown of LONP1 increases TFAM levels and mtDNA copy numbers, while overexpressing LONP1 has the opposite effects;Citation137 RNAi knockdown of LONP1 also partially prevents the MPP+-induced depletion of TFAM.Citation9 Interestingly, recent studies suggest that TFAM may also play a role in the nucleus,Citation138,Citation139 although the relationship to mitochondrial gene transcription remains unknown.
Mitochondrial biogenesis in aged muscle and cardiovascular diseases
Reduced mitochondrial mass and mitochondrial protein quantity are common findings in aging. The decrease in cellular capacity for mitochondrial biogenesis is associated with reduced levels of PRKAA2 and PPARGC1A in skeletal muscle.Citation95,Citation102,Citation140 Exercise training can increase PPARGC1A levels, attenuating age-associated decreases in mitochondrial mass as assayed by measuring citrate synthase or cytochrome c oxidase in young and old rat muscles.Citation141 The effect of exercise on indices of mitochondrial biogenesis has been confirmed in human studies.Citation142,Citation143 Pharmacologically activating PRKAA2 results in improvement of aging-induced cardiomyocyte contractile deficits.Citation144 However, other studies show that exercise training in aged animals fails to increase PPARGC1A levels and does not enhance mitochondrial biogenesis.Citation145 Old rats also show impaired mitochondrial biogenesis in response to chronic activation of PRKAA2 with β-guanidinopropionic acid.Citation102 It is possible that impaired nuclear import, as observed in models of oxidative injury,Citation32 or dysregulation of downstream pathways may limit efficacy of PPARGC1A-targeted therapies in aged or diseased settings.
Treadmill training has been reported to reduce infarct volume and improve post-ischemic recovery of brain function, in association with increased indices of mitochondrial biogenesis.Citation146,Citation147 Other strategies to target mitochondrial biogenesis have also been described in hypoxic/ischemic injury. Selenite treatment increases protein levels of PPARGC1A and NRF1 to restore mitochondrial functional activities, ameliorating cerebral infarct volume.Citation148 In this animal model, selenite pretreatment also attenuates ischemia-induced activation of BECN1 and MAP1LC3-II to inhibit autophagy. Levo-triiodothyronine significantly increases the expression of PPARGC1A and TFAM in the peri-infarct zone, resulting in enhanced markers of mitochondrial biogenesis in a rat myocardial infarction model. The treatment reduces infarct-scar size, and increases angiogenesis and cell survival.Citation149
Mitochondrial biogenesis in cancers
Markers of mitochondrial biogenesis are usually decreased in tumors.Citation150-Citation153 Decreased expression of PPARGC1A and TFAM is found in lung cancers.Citation151 Application of the PPAR agonists bezafibrateCitation150 or resveratrolCitation151 increases the levels of PPARGC1A, TFAM, oxidative phosphorylation components and mtDNA in cultured cervical carcinoma, osteosarcoma and breast cancer cells, decreasing their growth rates and invasive potential.Citation150 Likewise, overexpression of PPARGC1A drives mitochondrial biogenesis while inducing apoptosis in the human intestinal cancer cells.Citation152,Citation153 These observations may relate to the known propensity of cancer cells to downregulate mitochondrial respiration in preference to aerobic glycolysis.
However, in other tumor types such as Bowen diseaseCitation154 or endometrial carcinoma,Citation155,Citation156 increased mitochondrial biogenesis is linked to growth of tumors. An increase in mtDNA content and mitochondrial biogenesis is also associated with activation of PPARGC1A in type I endometrial cancer.Citation155,Citation156 Thus, the potential role for manipulating mitochondrial biogenesis remains unclear in neoplasia.
Mitochondrial biogenesis in neurogenerative diseases
In contrast to cancers, there is more evidence to support a possible therapeutic role for modulating mitochondrial biogenesis and mitochondrial turnover in neurodegenerative diseases such as Huntington, Alzheimer, and Parkinson diseases.Citation112,Citation133,Citation157-Citation162
Decreased mitochondrial function is a common feature for several neurodegenerative diseases. These include significant reductions in the enzymatic activities of mitochondrial oxidative phosphorylation complexes accompanied by decreased expression of PPARGC1A and its downstream targeted proteins in Huntington,Citation163-Citation165 Alzheimer,Citation157,Citation166-Citation168 and Parkinson patients.Citation169,Citation170
Mutant HTT suppresses PPARGC1A expression, whereas crossbreeding of Ppargc1a knockout (KO) mice with HD knockin (KI) mice leads to increased neurodegeneration of striatal neurons and motor abnormalities in the HD mice.Citation112 Overexpressing PPARGC1A or pharmacologically activating PPARGC1A can partially reverse the toxic effects of mutant HTT in cultured striatal neurons, increasing the number of mitochondria, and improving survival in mouse models of HD.Citation112,Citation161 Interestingly, PPARGC1A can also upregulate the autophagy-lysosome pathway through activation of transcription factor EB (TFEB) to promote the clearance of HTT aggregates.Citation171 In Alzheimer disease, overexpression of PPARGC1A improves mitochondrial biogenesis and function in APPswe M17cells,Citation157 protects against amyloidogenic Aβ accumulation and promotes cell survival.Citation168,Citation172,Citation176 Recombinant TFAM restores mitochondrial gene expression in cybrid models of optic neuropathy.Citation173
A meta-analysis of gene expression studies indicates that loss of PPARGC1A is a major factor in Parkinson disease.Citation170 PPARGC1A-deficient mice are much more susceptible to MPTP toxicity,Citation174 while overexpression of TFAM or PPARGC1A can prevent neuronal cell damage induced by MPTP, oxidative stress, mutant SNCA/α-synuclein or the pesticide rotenone.Citation134,Citation170,Citation174,Citation175 However, another study shows that a persistent high level of PPARGC1A expression causes selective loss of dopaminergic neurons, and reverses its protective effects in a model of SNCA overexpression.Citation176 These studies suggest that tight control of PPARGC1A levels in a physiological range is crucial for its neuroprotection.
Proposed Interplay of Mitochondrial Biogenesis and Mitophagy in Determining Cell Fate
Both mitochondrial autophagy and mitochondrial biogenesis are complex, regulated pathways. The balance of mitochondrial biogenesis and degradation responses regulates the content of mitochondria within cells. Under normal situations in which the complete cycle of degradation and regeneration is intact, factors that tip the balance between these processes most likely reflect developmental changes in the demand for mitochondrial function. Under pathological conditions, however, we propose that imbalanced responses in either direction can contribute to cellular degeneration and susceptibility to cell death ().
Figure 3. The balance of mitophagy and mitochondrial biogenesis determines cellular adaptation to physiological and pathological stressors. Developmental or pathological stimuli trigger changes on the mitochondrial surface that signal their elimination by the autophagy machinery (top left). Simultaneously, overlapping triggers (e.g., reactive oxygen species [ROS] or caloric restriction [CR]) or activation of shared regulatory pathways (e.g., PRKAA2 or PARK2) function to regulate mitochondrial biogenesis (top right). Individual kinase pathways (MAPK1/3, MTOR, PKA) may show opposing effects on mitophagy and mitochondrial biogenesis to result in either a net decrease or increase in mitochondrial mass. However, restoration of mitochondrial homeostasis under conditions of increased mitochondrial damage would require simultaneous upregulation of mitophagy and mitochondrial biogenesis. This may occur through cellular integration of multiple competing pathways, or involve localized kinase signaling that triggers mitophagy, while biogenesis is triggered elsewhere in the cell. We propose that balanced mitochondrial recycling, as mediated by appropriately coregulated signals for mitophagy and mitochondrial biogenesis, is required for maintaining mitochondrial homeostasis and normal cellular function and survival. However, overactivation of mitophagy relative to the capacity for mitochondrial biogenesis would lead to a net loss of mitochondria, contributing to “mitophagic” cell death in cell types dependent upon mitochondrial function (lower left). Conversely, impaired mitochondrial clearance or excessive/sustained inputs to drive mitochondrial biogenesis also results in imbalanced responses, resulting in increased oxygen consumption and sensitization to stressors (lower right).
![Figure 3. The balance of mitophagy and mitochondrial biogenesis determines cellular adaptation to physiological and pathological stressors. Developmental or pathological stimuli trigger changes on the mitochondrial surface that signal their elimination by the autophagy machinery (top left). Simultaneously, overlapping triggers (e.g., reactive oxygen species [ROS] or caloric restriction [CR]) or activation of shared regulatory pathways (e.g., PRKAA2 or PARK2) function to regulate mitochondrial biogenesis (top right). Individual kinase pathways (MAPK1/3, MTOR, PKA) may show opposing effects on mitophagy and mitochondrial biogenesis to result in either a net decrease or increase in mitochondrial mass. However, restoration of mitochondrial homeostasis under conditions of increased mitochondrial damage would require simultaneous upregulation of mitophagy and mitochondrial biogenesis. This may occur through cellular integration of multiple competing pathways, or involve localized kinase signaling that triggers mitophagy, while biogenesis is triggered elsewhere in the cell. We propose that balanced mitochondrial recycling, as mediated by appropriately coregulated signals for mitophagy and mitochondrial biogenesis, is required for maintaining mitochondrial homeostasis and normal cellular function and survival. However, overactivation of mitophagy relative to the capacity for mitochondrial biogenesis would lead to a net loss of mitochondria, contributing to “mitophagic” cell death in cell types dependent upon mitochondrial function (lower left). Conversely, impaired mitochondrial clearance or excessive/sustained inputs to drive mitochondrial biogenesis also results in imbalanced responses, resulting in increased oxygen consumption and sensitization to stressors (lower right).](/cms/asset/8a2c3a46-ba9a-4c2d-be1c-18bdae18d8a9/kaup_a_10924135_f0003.gif)
Decreased mitochondrial content or function, often accompanied by reduced levels of PPARGC1A or NRF1, are commonly observed with agingCitation145,Citation177 and in neurodegenerative diseases, cancer, cardiovascular or liver diseases.Citation151,Citation178,Citation179 At the same time, oxidative or other forms of damage related to either genetic or environmental factors serve to promote mitophagic turnover of mitochondria.Citation35,Citation60,Citation63,Citation90 Our recent data suggests that the ability of neuronal cells to undergo mitochondrial biogenesis may dictate whether or not the mitophagic response is successful in compensating for chronic, low level impairment of complex I function.Citation9 We propose that the clearance of marginally functional mitochondria at more rapid rates than regeneration of functional mitochondria is maladaptive in mitochondrially dependent cell types. In this context, blunting or slowing the mitophagic response while enhancing mitochondrial biogenesis may prove effective.
On the other hand, a deficiency in autophagy as observed in other disease models can lead to accumulation of damaged or less efficient mitochondria.Citation180-Citation182 ROS are liberated as a byproduct of oxidative phosphorylation, particularly in damaged or marginally functional mitochondria. Moreover, depolarized mitochondria are unable to effectively buffer cytoplasmic calcium, and may undergo the mitochondrial permeability transition to release cytochrome c and other pro-apoptotic mediators. While loss of mitochondrial function would be expected to trigger compensatory mitochondrial biogenesis, this short-term solution may ultimately lead to larger reservoirs for release of ROS or apoptotic mediators from further accumulation of marginal mitochondria. So how then is feedback between the processes of mitophagy and mitochondrial biogenesis regulated?
Pathways that regulate both mitophagy and mitochondrial biogenesis
Several signaling pathways are implicated in both mitophagy and mitochondrial biogenesis, and are likely to play important roles in coordinating these processes.
PKA is a kinase that functions upstream of both PPARGC1A and autophagy, promoting biogenesis,Citation111 while suppressing autophagy/mitophagy through phosphorylation of MAP1LC3Citation44,Citation74 As PKA phosphorylation of DNM1L/DRP1 inhibits its fission-promoting activity,Citation183 PKA may downregulate mitophagy through that mechanism as well. Whereas PKA is able to promote mammalian mitochondrial mass through more than one mechanism, in yeast, activation of PKA impairs the receptor activity of Tom70 and inhibits the biogenesis of the channel protein Tom40, which affects mitochondrial protein import.Citation184,Citation185 The inappropriate activation of one of the yeast PKA catalytic subunits, Tpk3 promotes the production of dysfunctional, ROS-producing mitochondria and causes cell death.Citation186 It is apparent that the effects of PKA on mitochondrial clearance and biogenesis are context dependent. In mammals, the data suggest that PKA tilts the balance to favor increased mitochondrial mass.
In contrast to PKA, which upregulates mitochondria, the MAPK1/3 pathway serves to downregulate mitochondria through opposing effects on mitophagy and mitochondrial biogenesis. Activation of MAPK1/3 stimulates autophagy/mitophagy in Parkinson models,Citation35,Citation90 potently inhibiting mitochondrial biogenesis,Citation9 and promoting cell death. Interestingly, at high concentrations of MPP+, blocking MAPK1/3 does not improve mitochondrial translation; however, it inhibits mitophagy, modestly reducing cell death.Citation35 In contrast, in a low dose, chronic MPP+ model, inhibiting MAPK1/3 activation not only blunts the autophagy/mitophagy response, but also restores the capacity for mitochondrial biogenesis, resulting in restoration of normal mitochondrial structure and function.Citation9 The protection by U0126 exceeds that induced by ATG7 RNAi, indicating that survival is dependent upon both reducing mitophagy and restoring normal levels of mitochondrial translation.
As discussed above, PINK1 has been implicated in the regulation of mitochondrial dynamics and autophagy/mitophagy, albeit in a complex, context-dependent manner. Loss of PINK1 also results in decreased mtDNA levels as well as decreased mitochondrial ATP synthesis and activity of complex IV of the electron transport chain in human dopaminergic SH-SY5Y cells, suggesting a possible impairment in mitochondrial biogenesis.Citation187 Likewise, mouse PINK1 deficiency affects mtDNA copy number, protein levels of biogenesis factors, fission/fusion proteins and bioenergetics in pink1−/− mouse hearts, affecting post-natal myocardial development and function.Citation188 PINK1 is also markedly reduced in end-stage human heart failure. While a definitive link between PINK1 and mitochondrial biogenesis remains to be established, these findings suggest that manipulating PINK1 expression may provide potential therapeutic strategies for some forms of cardiac disease.
PARK2 not only plays an important role in CCCP-induced mitochondrial degradation, but also is involved in the regulation of mitochondrial biogenesis. PARK2 is associated with TFAM, enhances TFAM-mediated mitochondrial transcription and promotes mitochondrial biogenesis.Citation189 Disease-associated PARK2 mutations abolish its interaction with TFAM and mtDNA.Citation189,Citation190 ZNF746/PARIS, a substrate of PARK2 represses the expression of PPARGC1A and its target gene, NRF1.Citation191 Park2 knockout leads to a ZNF746-related loss of dopaminergic neurons, which is reversed by PPARGC1A overexpression.Citation191 Thus, PARK2 appears to promote mitochondrial health by enhancing both the mitophagy and biogenesis phases of recycling.
Mitochondrial biogenesis and autophagy/mitophagy are also regulated in a coordinated mannerCitation192 by PRKAA2 and PPARGC1A. In response to nutrient starvation or the mitochondrial uncoupler CCCP, the activation of PRKAA2 suppresses the MTOR pathway to elicit autophagy/mitophagy responses.Citation33,Citation193 At the same time, PRKAA2 initiates mitochondrial biogenesis via SIRT1-dependent deacetylation of PPARGC1ACitation107 or upregulation of PPARGC1A expression.Citation102,Citation103 Likewise, PPARGC1A also regulates both biogenesis and degradation through transcriptional mechanisms. In a Huntington disease mouse model, overexpression of PPARGC1A not only enhances mitochondrial biogenesis, but also upregulates autophagy-lysosome function via induction of TFEB to eliminate HTT aggregates.Citation194
Given the central role of PRKAA2 in sensing the bioenergetics status of the cell to modulate both autophagy and PPARGC1A, it is not surprising that either genetic or pharmacological upregulation of PRKAA2 may represent promising therapeutic strategies for several pathological conditions. Injection with AICAR (5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside), an agonist of PRKAA2, induces autophagy and facilitates the elimination of defective mitochondria, which is associated with improvements in a Duchenne muscular dystrophy mouse model.Citation195 Acetylcholine provides cardioprotection against hypoxia/reoxygenation injury in a cellular model through a PRKAA2-PPARGC1A-associated mechanism that increases mitochondrial density, mass, mitochondrial DNA copy number, and ATP synthesis.Citation196
From these examples, it is clear that crosstalk between autophagy/mitophagy and mitochondrial biogenesis is critical for mitochondrial homeostasis. The accumulation of defective mitochondria and lysosomes in aged myocytes suggests that inefficient autophagy is associated with the development of aging-related disorders in the heart,Citation197 while enhancing autophagy gene expression can extend the life span of aging flies.Citation198 Recessive mutations in PINK1 and PARK2 are hypothesized to contribute to defective targeting of damaged mitochondria,Citation80 and enhanced mitophagy is neuroprotective in PINK1 knockdown cells.Citation90 The rapid removal of dysfunctional or damaged mitochondria would serve to prevent accumulation of oxidized lipids, proteins, and DNA, limiting the risk of apoptosis.Citation199 However, overactivation of autophagy/mitophagy without appropriate levels of compensatory mitochondrial biogenesis may also contribute to deficiencies in mitochondrial function and cell death,Citation35,Citation90,Citation200 or to the degeneration of dendrites in neurons.Citation201 Replenishment of new mitochondria through mitochondrial biogenesis accelerates recovery of mitochondrial and cellular functions after oxidative stress.Citation202 Since decreased mitochondrial biogenesis is implicated in aging, cardiovascular diseases, diabetes, and neurodegenerative diseases, therapeutic strategies involving the nonselective stimulation of autophagy may need to consider potential bystander effects on mitochondria.
Conclusions
In summary, mitochondrial quality control includes several tiers: damage prevention, dynamic remodeling and localized repair, mitophagy, and coordinated mitochondrial biogenesis.Citation62 Mitochondrial biogenesis and mitophagy are two key processes, whose balanced induction may be necessary for cellular adaptation and recovery from stress and injury (). We propose that disruption of this balance leads to cellular degeneration or cell death. Understanding the underlying mechanisms that contribute to imbalanced recycling responses and delineating methods to manipulate these two processes are essential for developing new strategies for the treatment of certain human diseases.
Although mitophagy is involved in numerous physiological and pathological conditions, the machineries controlling mitophagy in aging and disease contexts are only beginning to be understood. It appears that the role of mitophagy, like that of autophagy in general, comprises a double-edged sword, emerging as a protective mechanism in response to mild to moderate stress, but leading to cell death when excessively activated. As cell growth, atrophy, and health are dependent upon shifting the balance of anabolic and catabolic processes, we propose that the nebulous concept of “excessive” mitophagy may be defined by the biosynthetic capacities of the aged, injured or diseased cell. In many disease models, promoting mitochondrial biogenesis has been beneficial, although an excess of mitochondrial biogenesis may result in increased oxygen consumption and oxidative stress if not balanced by other quality control mechanisms. Thus, a challenge for the future involves delineating mechanisms that underlie the coordinated regulation of mitochondrial function, autophagy, and biogenesis.
| Abbreviations: | ||
| LRRK2 | = | leucine-rich repeat kinase 2 |
| MDV | = | mitochondria-derived vesicles |
| LIR | = | LC3-interacting region |
| mtDNA | = | mitochondrial DNA |
| nDNA | = | nuclear DNA |
| PKA | = | protein kinase A |
| ROS | = | reactive oxygen species |
| TFAM | = | transcription factor A, mitochondrial |
| UPS | = | ubiquitin-proteasome system |
Acknowledgments
Supported by funding from the National Institutes of Health (R01 AG026389, R01 NS065789). C.T.C. is a Julie Martin Mid-Career Awardee in Aging Research supported in part by The Ellison Medical Foundation and AFAR.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Zorov DB, Krasnikov BF, Kuzminova AE, Vysokikh MYu, Zorova LD. Mitochondria revisited. Alternative functions of mitochondria. Biosci Rep 1997; 17:507 - 20; http://dx.doi.org/10.1023/A:1027304122259; PMID: 9561295
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol 2006; 16:R551 - 60; http://dx.doi.org/10.1016/j.cub.2006.06.054; PMID: 16860735
- Mandal S, Lindgren AG, Srivastava AS, Clark AT, Banerjee U. Mitochondrial function controls proliferation and early differentiation potential of embryonic stem cells. Stem Cells 2011; 29:486 - 95; http://dx.doi.org/10.1002/stem.590; PMID: 21425411
- Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003; 115:629 - 40; http://dx.doi.org/10.1016/S0092-8674(03)00926-7; PMID: 14651853
- Forner F, Foster LJ, Campanaro S, Valle G, Mann M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol Cell Proteomics 2006; 5:608 - 19; http://dx.doi.org/10.1074/mcp.M500298-MCP200; PMID: 16415296
- Perez-Pinzon MA, Stetler RA, Fiskum G. Novel mitochondrial targets for neuroprotection. J Cereb Blood Flow Metab 2012; 32:1362 - 76; http://dx.doi.org/10.1038/jcbfm.2012.32; PMID: 22453628
- Dias N, Bailly C. Drugs targeting mitochondrial functions to control tumor cell growth. Biochem Pharmacol 2005; 70:1 - 12; http://dx.doi.org/10.1016/j.bcp.2005.03.021; PMID: 15907809
- Schapira AH. Targeting mitochondria for neuroprotection in Parkinson’s disease. Antioxid Redox Signal 2012; 16:965 - 73; http://dx.doi.org/10.1089/ars.2011.4419; PMID: 22229791
- Zhu JH, Gusdon AM, Cimen H, Van Houten B, Koc E, Chu CT. Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: dual roles for ERK1/2. Cell Death Dis 2012; 3:e312; http://dx.doi.org/10.1038/cddis.2012.46; PMID: 22622131
- Cherra SJ 3rd, Chu CT. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol 2008; 3:309 - 23; PMID: 18806889
- Matsushima Y, Kaguni LS. Matrix proteases in mitochondrial DNA function. Biochim Biophys Acta 2012; 1819:1080 - 7; http://dx.doi.org/10.1016/j.bbagrm.2011.11.008; PMID: 22172992
- Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, et al. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 2008; 18:102 - 8; http://dx.doi.org/10.1016/j.cub.2007.12.038; PMID: 18207745
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 2012; 22:135 - 41; http://dx.doi.org/10.1016/j.cub.2011.11.057; PMID: 22226745
- Radke S, Chander H, Schäfer P, Meiss G, Krüger R, Schulz JB, et al. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J Biol Chem 2008; 283:12681 - 5; http://dx.doi.org/10.1074/jbc.C800036200; PMID: 18362145
- Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 2011; 286:19630 - 40; http://dx.doi.org/10.1074/jbc.M110.209338; PMID: 21454557
- Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci 2012; 125:2095 - 104; http://dx.doi.org/10.1242/jcs.053850; PMID: 22619228
- Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 2006; 22:79 - 99; http://dx.doi.org/10.1146/annurev.cellbio.22.010305.104638; PMID: 16704336
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823 - 30; http://dx.doi.org/10.1038/ncb0910-823; PMID: 20811354
- Vessoni AT, Muotri AR, Okamoto OK. Autophagy in stem cell maintenance and differentiation. Stem Cells Dev 2012; 21:513 - 20; http://dx.doi.org/10.1089/scd.2011.0526; PMID: 22066548
- Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity?. Nat Cell Biol 2010; 12:842 - 6; http://dx.doi.org/10.1038/ncb0910-842; PMID: 20811357
- Yen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 2008; 23:248 - 62; http://dx.doi.org/10.1152/physiol.00013.2008; PMID: 18927201
- Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev 2012; 249:158 - 75; http://dx.doi.org/10.1111/j.1600-065X.2012.01146.x; PMID: 22889221
- Lu JV, Walsh CM. Programmed necrosis and autophagy in immune function. Immunol Rev 2012; 249:205 - 17; http://dx.doi.org/10.1111/j.1600-065X.2012.01147.x; PMID: 22889224
- Cherra SJ 3rd, Dagda RK, Chu CT. Review: autophagy and neurodegeneration: survival at a cost?. Neuropathol Appl Neurobiol 2010; 36:125 - 32; PMID: 20202120
- Lee JA. Neuronal autophagy: a housekeeper or a fighter in neuronal cell survival?. Exp Neurobiol 2012; 21:1 - 8; http://dx.doi.org/10.5607/en.2012.21.1.1; PMID: 22438673
- Batlevi Y, La Spada AR. Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol Dis 2011; 43:46 - 51; http://dx.doi.org/10.1016/j.nbd.2010.09.009; PMID: 20887789
- Ong SB, Hall AR, Hausenloy DJ. Mitochondrial Dynamics in Cardiovascular Health and Disease. Antioxid Redox Signal 2012; In press http://dx.doi.org/10.1089/ars.2012.4777; PMID: 22793879
- Zhou XJ, Zhang H. Autophagy in immunity: implications in etiology of autoimmune/autoinflammatory diseases. Autophagy 2012; 8:1286 - 99; http://dx.doi.org/10.4161/auto.21212; PMID: 22878595
- Wang SY, Yu QJ, Zhang RD, Liu B. Core signaling pathways of survival/death in autophagy-related cancer networks. Int J Biochem Cell Biol 2011; 43:1263 - 6; http://dx.doi.org/10.1016/j.biocel.2011.05.010; PMID: 21640844
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861 - 73; http://dx.doi.org/10.1101/gad.1599207; PMID: 18006683
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124 - 31; http://dx.doi.org/10.1016/j.ceb.2009.11.014; PMID: 20034776
- Patel VP, Defranco DB, Chu CT. Altered transcription factor trafficking in oxidatively-stressed neuronal cells. Biochim Biophys Acta 2012; 1822:1773 - 82; http://dx.doi.org/10.1016/j.bbadis.2012.08.002; PMID: 22902725
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132 - 41; http://dx.doi.org/10.1038/ncb2152; PMID: 21258367
- Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2 - 11; http://dx.doi.org/10.1128/MCB.06159-11; PMID: 22025673
- Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol 2007; 170:75 - 86; http://dx.doi.org/10.2353/ajpath.2007.060524; PMID: 17200184
- Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ 2008; 15:1318 - 29; http://dx.doi.org/10.1038/cdd.2008.51; PMID: 18421301
- Plowey ED, Cherra SJ 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem 2008; 105:1048 - 56; http://dx.doi.org/10.1111/j.1471-4159.2008.05217.x; PMID: 18182054
- Kim I, Lemasters JJ. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal 2011; 14:1919 - 28; http://dx.doi.org/10.1089/ars.2010.3768; PMID: 21126216
- Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol 2000; 151:263 - 76; http://dx.doi.org/10.1083/jcb.151.2.263; PMID: 11038174
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657 - 68; http://dx.doi.org/10.1083/jcb.152.4.657; PMID: 11266458
- Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 2008; 9:859 - 64; http://dx.doi.org/10.1038/embor.2008.163; PMID: 18704115
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem 2004; 279:47704 - 10; http://dx.doi.org/10.1074/jbc.M407016200; PMID: 15355958
- Cherra SJ 3rd, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, Day BW, et al. Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol 2010; 190:533 - 9; http://dx.doi.org/10.1083/jcb.201002108; PMID: 20713600
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505 - 16; http://dx.doi.org/10.1016/j.molcel.2009.01.020; PMID: 19250911
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010; 11:45 - 51; http://dx.doi.org/10.1038/embor.2009.256; PMID: 20010802
- Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 2010; 38:265 - 79; http://dx.doi.org/10.1016/j.molcel.2010.04.007; PMID: 20417604
- Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A 2007; 104:19500 - 5; http://dx.doi.org/10.1073/pnas.0708818104; PMID: 18048346
- Zhang J, Ney PA. NIX induces mitochondrial autophagy in reticulocytes. Autophagy 2008; 4:354 - 6; PMID: 18623629
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454:232 - 5; http://dx.doi.org/10.1038/nature07006; PMID: 18454133
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 2010; 285:27879 - 90; http://dx.doi.org/10.1074/jbc.M110.119537; PMID: 20573959
- Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res 2004; 64:4286 - 93; http://dx.doi.org/10.1158/0008-5472.CAN-03-3084; PMID: 15205343
- Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 2011; 301:H1924 - 31; http://dx.doi.org/10.1152/ajpheart.00368.2011; PMID: 21890690
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 2009; 29:2570 - 81; http://dx.doi.org/10.1128/MCB.00166-09; PMID: 19273585
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19:5360 - 72; http://dx.doi.org/10.1091/mbc.E08-01-0080; PMID: 18843052
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927 - 39; http://dx.doi.org/10.1016/j.cell.2005.07.002; PMID: 16179260
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 2012; 287:19094 - 104; http://dx.doi.org/10.1074/jbc.M111.322933; PMID: 22505714
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012; 14:177 - 85; http://dx.doi.org/10.1038/ncb2422; PMID: 22267086
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795 - 803; http://dx.doi.org/10.1083/jcb.200809125; PMID: 19029340
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392:605 - 8; http://dx.doi.org/10.1038/33416; PMID: 9560156
- Chu CT. A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for Parkinson disease. Hum Mol Genet 2010; 19:R1 R28 - 37; http://dx.doi.org/10.1093/hmg/ddq143; PMID: 20385539
- Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 2009; 284:13843 - 55; http://dx.doi.org/10.1074/jbc.M808515200; PMID: 19279012
- Shimura H, Hattori N, Kubo Si, Mizuno Y, Asakawa S, Minoshima S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 2000; 25:302 - 5; http://dx.doi.org/10.1038/77060; PMID: 10888878
- Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A 2000; 97:13354 - 9; http://dx.doi.org/10.1073/pnas.240347797; PMID: 11078524
- Cai Q, Zakaria HM, Simone A, Sheng ZH. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol 2012; 22:545 - 52; http://dx.doi.org/10.1016/j.cub.2012.02.005; PMID: 22342752
- Chen D, Gao F, Li B, Wang H, Xu Y, Zhu C, et al. Parkin mono-ubiquitinates Bcl-2 and regulates autophagy. J Biol Chem 2010; 285:38214 - 23; http://dx.doi.org/10.1074/jbc.M110.101469; PMID: 20889974
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 2010; 19:4861 - 70; http://dx.doi.org/10.1093/hmg/ddq419; PMID: 20871098
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119 - 31; http://dx.doi.org/10.1038/ncb2012; PMID: 20098416
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 2010; 191:1367 - 80; http://dx.doi.org/10.1083/jcb.201007013; PMID: 21173115
- Exner N, Treske B, Paquet D, Holmström K, Schiesling C, Gispert S, et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci 2007; 27:12413 - 8; http://dx.doi.org/10.1523/JNEUROSCI.0719-07.2007; PMID: 17989306
- Cui T, Fan C, Gu L, Gao H, Liu Q, Zhang T, et al. Silencing of PINK1 induces mitophagy via mitochondrial permeability transition in dopaminergic MN9D cells. Brain Res 2011; 1394:1 - 13; http://dx.doi.org/10.1016/j.brainres.2011.01.035; PMID: 21262209
- Liu S, Lu B. Reduction of protein translation and activation of autophagy protect against PINK1 pathogenesis in Drosophila melanogaster.. PLoS Genet 2010; 6:e1001237; http://dx.doi.org/10.1371/journal.pgen.1001237; PMID: 21151574
- Dagda RK, Gusdon AM, Pien I, Strack S, Green S, Li C, et al. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ 2011; 18:1914 - 23; http://dx.doi.org/10.1038/cdd.2011.74; PMID: 21637291
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 2010; 107:378 - 83; http://dx.doi.org/10.1073/pnas.0911187107; PMID: 19966284
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 2010; 189:211 - 21; http://dx.doi.org/10.1083/jcb.200910140; PMID: 20404107
- Rakovic A, Shurkewitsch K, Seibler P, Grünewald A, Zanon A, Hagenah J, et al. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem 2013; 288:2223 - 37; http://dx.doi.org/10.1074/jbc.M112.391680; PMID: 23212910
- Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol 2013; 200:163 - 72; http://dx.doi.org/10.1083/jcb.201210111; PMID: 23319602
- Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R, et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ 2010; 17:962 - 74; http://dx.doi.org/10.1038/cdd.2009.200; PMID: 20057503
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8:e1000298; http://dx.doi.org/10.1371/journal.pbio.1000298; PMID: 20126261
- Lin W, Kang UJ. Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem 2008; 106:464 - 74; http://dx.doi.org/10.1111/j.1471-4159.2008.05398.x; PMID: 18397367
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 2012; 13:378 - 85; http://dx.doi.org/10.1038/embor.2012.14; PMID: 22354088
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006; 441:1162 - 6; http://dx.doi.org/10.1038/nature04779; PMID: 16672981
- Sha D, Chin L-S, Li L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum Mol Genet 2010; 19:352 - 63; http://dx.doi.org/10.1093/hmg/ddp501; PMID: 19880420
- Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, Berman SB. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet 2011; 20:927 - 40; http://dx.doi.org/10.1093/hmg/ddq531; PMID: 21147754
- Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J 2010; 277:2 - 21; http://dx.doi.org/10.1111/j.1742-4658.2009.07366.x; PMID: 19843174
- Gao H, Yang WW, Qi ZF, Lu LL, Duan CL, Zhao CL, et al. DJ-1 protects dopaminergic neurons against rotenone-induced apoptosis by enhancing ERK-dependent mitophagy. J Mol Biol 2012; 423:232 - 48; http://dx.doi.org/10.1016/j.jmb.2012.06.034; PMID: 22898350
- Zhu JH, Kulich SM, Oury TD, Chu CT. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am J Pathol 2002; 161:2087 - 98; http://dx.doi.org/10.1016/S0002-9440(10)64487-2; PMID: 12466125
- Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol 2003; 13:473 - 81; http://dx.doi.org/10.1111/j.1750-3639.2003.tb00478.x; PMID: 14655753
- Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson’s disease. Autophagy 2008; 4:770 - 82; PMID: 18594198
- Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy 2007; 3:663 - 6; PMID: 17622797
- Herrmann JM, Longen S, Weckbecker D, Depuydt M. Biogenesis of mitochondrial proteins. Adv Exp Med Biol 2012; 748:41 - 64; http://dx.doi.org/10.1007/978-1-4614-3573-0_3; PMID: 22729854
- Weitzel JM, Iwen KA. Coordination of mitochondrial biogenesis by thyroid hormone. Mol Cell Endocrinol 2011; 342:1 - 7; http://dx.doi.org/10.1016/j.mce.2011.05.009; PMID: 21664416
- Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 2003; 24:78 - 90; http://dx.doi.org/10.1210/er.2002-0012; PMID: 12588810
- López-Lluch G, Irusta PM, Navas P, de Cabo R. Mitochondrial biogenesis and healthy aging. Exp Gerontol 2008; 43:813 - 9; http://dx.doi.org/10.1016/j.exger.2008.06.014; PMID: 18662766
- Jones AW, Yao Z, Vicencio JM, Karkucinska-Wieckowska A, Szabadkai G. PGC-1 family coactivators and cell fate: roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria-nucleus signalling. Mitochondrion 2012; 12:86 - 99; http://dx.doi.org/10.1016/j.mito.2011.09.009; PMID: 21983689
- Anderson RM, Barger JL, Edwards MG, Braun KH, O’Connor CE, Prolla TA, et al. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 2008; 7:101 - 11; http://dx.doi.org/10.1111/j.1474-9726.2007.00357.x; PMID: 18031569
- Chang JS, Huypens P, Zhang Y, Black C, Kralli A, Gettys TW. Regulation of NT-PGC-1alpha subcellular localization and function by protein kinase A-dependent modulation of nuclear export by CRM1. J Biol Chem 2010; 285:18039 - 50; http://dx.doi.org/10.1074/jbc.M109.083121; PMID: 20351112
- Safdar A, Little JP, Stokl AJ, Hettinga BP, Akhtar M, Tarnopolsky MA. Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J Biol Chem 2011; 286:10605 - 17; http://dx.doi.org/10.1074/jbc.M110.211466; PMID: 21245132
- Aquilano K, Vigilanza P, Baldelli S, Pagliei B, Rotilio G, Ciriolo MR. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis. J Biol Chem 2010; 285:21590 - 9; http://dx.doi.org/10.1074/jbc.M109.070169; PMID: 20448046
- Safdar A, Hamadeh MJ, Kaczor JJ, Raha S, Debeer J, Tarnopolsky MA. Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS One 2010; 5:e10778; http://dx.doi.org/10.1371/journal.pone.0010778; PMID: 20520725
- Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab 2007; 5:151 - 6; http://dx.doi.org/10.1016/j.cmet.2007.01.008; PMID: 17276357
- Schulz E, Dopheide J, Schuhmacher S, Thomas SR, Chen K, Daiber A, et al. Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation 2008; 118:1347 - 57; PMID: 18809807
- Schiaffino S, Sandri M, Murgia M. Activity-dependent signaling pathways controlling muscle diversity and plasticity. Physiology (Bethesda) 2007; 22:269 - 78; http://dx.doi.org/10.1152/physiol.00009.2007; PMID: 17699880
- Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A 2007; 104:12017 - 22; http://dx.doi.org/10.1073/pnas.0705070104; PMID: 17609368
- Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010; 464:1313 - 9; http://dx.doi.org/10.1038/nature08991; PMID: 20357764
- Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009; 458:1056 - 60; http://dx.doi.org/10.1038/nature07813; PMID: 19262508
- Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 2012; 15:675 - 90; http://dx.doi.org/10.1016/j.cmet.2012.04.003; PMID: 22560220
- Kelly TJ, Lerin C, Haas W, Gygi SP, Puigserver P. GCN5-mediated transcriptional control of the metabolic coactivator PGC-1beta through lysine acetylation. J Biol Chem 2009; 284:19945 - 52; http://dx.doi.org/10.1074/jbc.M109.015164; PMID: 19491097
- Coste A, Louet JF, Lagouge M, Lerin C, Antal MC, Meziane H, et al. The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1α. Proc Natl Acad Sci U S A 2008; 105:17187 - 92; http://dx.doi.org/10.1073/pnas.0808207105; PMID: 18957541
- De Rasmo D, Signorile A, Papa F, Roca E, Papa S. cAMP/Ca2+ response element-binding protein plays a central role in the biogenesis of respiratory chain proteins in mammalian cells. IUBMB Life 2010; 62:447 - 52; PMID: 20503437
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006; 127:59 - 69; http://dx.doi.org/10.1016/j.cell.2006.09.015; PMID: 17018277
- Pogozelski AR, Geng T, Li P, Yin X, Lira VA, Zhang M, et al. p38gamma mitogen-activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS One 2009; 4:e7934; http://dx.doi.org/10.1371/journal.pone.0007934; PMID: 19936205
- Wright DC, Han DH, Garcia-Roves PM, Geiger PC, Jones TE, Holloszy JO. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1α expression. J Biol Chem 2007; 282:194 - 9; http://dx.doi.org/10.1074/jbc.M606116200; PMID: 17099248
- Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: a potential role in the cardiac metabolic stress response. J Biol Chem 2001; 276:44495 - 501; http://dx.doi.org/10.1074/jbc.M105945200; PMID: 11577087
- Fan M, Rhee J, St-Pierre J, Handschin C, Puigserver P, Lin J, et al. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1α: modulation by p38 MAPK. Genes Dev 2004; 18:278 - 89; http://dx.doi.org/10.1101/gad.1152204; PMID: 14744933
- Echave P, Machado-da-Silva G, Arkell RS, Duchen MR, Jacobson J, Mitter R, et al. Extracellular growth factors and mitogens cooperate to drive mitochondrial biogenesis. J Cell Sci 2009; 122:4516 - 25; http://dx.doi.org/10.1242/jcs.049734; PMID: 19920079
- Hota KB, Hota SK, Chaurasia OP, Singh SB. Acetyl-L-carnitine-mediated neuroprotection during hypoxia is attributed to ERK1/2-Nrf2-regulated mitochondrial biosynthesis. Hippocampus 2012; 22:723 - 36; http://dx.doi.org/10.1002/hipo.20934; PMID: 21542052
- Ashabi G, Ramin M, Azizi P, Taslimi Z, Alamdary SZ, Haghparast A, et al. ERK and p38 inhibitors attenuate memory deficits and increase CREB phosphorylation and PGC-1α levels in Aβ-injected rats. Behav Brain Res 2012; 232:165 - 73; http://dx.doi.org/10.1016/j.bbr.2012.04.006; PMID: 22510382
- Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem 2006; 97:673 - 83; http://dx.doi.org/10.1002/jcb.20743; PMID: 16329141
- Virbasius CA, Virbasius JV, Scarpulla RC. NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev 1993; 7:12A 2431 - 45; http://dx.doi.org/10.1101/gad.7.12a.2431; PMID: 8253388
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 2008; 88:611 - 38; http://dx.doi.org/10.1152/physrev.00025.2007; PMID: 18391175
- Ongwijitwat S, Wong-Riley MT. Is nuclear respiratory factor 2 a master transcriptional coordinator for all ten nuclear-encoded cytochrome c oxidase subunits in neurons?. Gene 2005; 360:65 - 77; http://dx.doi.org/10.1016/j.gene.2005.06.015; PMID: 16126350
- Ongwijitwat S, Liang HL, Graboyes EM, Wong-Riley MT. Nuclear respiratory factor 2 senses changing cellular energy demands and its silencing down-regulates cytochrome oxidase and other target gene mRNAs. Gene 2006; 374:39 - 49; http://dx.doi.org/10.1016/j.gene.2006.01.009; PMID: 16516409
- Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab 2012; 23:459 - 66; http://dx.doi.org/10.1016/j.tem.2012.06.006; PMID: 22817841
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999; 98:115 - 24; http://dx.doi.org/10.1016/S0092-8674(00)80611-X; PMID: 10412986
- Gugneja S, Scarpulla RC. Serine phosphorylation within a concise amino-terminal domain in nuclear respiratory factor 1 enhances DNA binding. J Biol Chem 1997; 272:18732 - 9; http://dx.doi.org/10.1074/jbc.272.30.18732; PMID: 9228045
- Herzig RP, Scacco S, Scarpulla RC. Sequential serum-dependent activation of CREB and NRF-1 leads to enhanced mitochondrial respiration through the induction of cytochrome c. J Biol Chem 2000; 275:13134 - 41; http://dx.doi.org/10.1074/jbc.275.17.13134; PMID: 10777619
- Piantadosi CA, Suliman HB. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J Biol Chem 2006; 281:324 - 33; http://dx.doi.org/10.1074/jbc.M508805200; PMID: 16230352
- Izumi H, Ohta R, Nagatani G, Ise T, Nakayama Y, Nomoto M, et al. p300/CBP-associated factor (P/CAF) interacts with nuclear respiratory factor-1 to regulate the UDP-N-acetyl-alpha-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase-3 gene. Biochem J 2003; 373:713 - 22; http://dx.doi.org/10.1042/BJ20021902; PMID: 12720548
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet 1998; 18:231 - 6; http://dx.doi.org/10.1038/ng0398-231; PMID: 9500544
- Fisher RP, Clayton DA. Purification and characterization of human mitochondrial transcription factor 1. Mol Cell Biol 1988; 8:3496 - 509; PMID: 3211148
- Piao Y, Kim HG, Oh MS, Pak YK. Overexpression of TFAM, NRF-1 and myr-AKT protects the MPP(+)-induced mitochondrial dysfunctions in neuronal cells. Biochim Biophys Acta 2012; 1820:577 - 85; http://dx.doi.org/10.1016/j.bbagen.2011.08.007; PMID: 21856379
- Thomas RR, Khan SM, Portell FR, Smigrodzki RM, Bennett JP Jr.. Recombinant human mitochondrial transcription factor A stimulates mitochondrial biogenesis and ATP synthesis, improves motor function after MPTP, reduces oxidative stress and increases survival after endotoxin. Mitochondrion 2011; 11:108 - 18; http://dx.doi.org/10.1016/j.mito.2010.08.004; PMID: 20727424
- Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, et al. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol 2005; 25:6225 - 34; http://dx.doi.org/10.1128/MCB.25.14.6225-6234.2005; PMID: 15988031
- Dang CV. PKM2 tyrosine phosphorylation and glutamine metabolism signal a different view of the Warburg effect. Sci Signal 2009; 2:pe75; http://dx.doi.org/10.1126/scisignal.297pe75; PMID: 19920249
- Matsushima Y, Goto Y, Kaguni LS. Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM). Proc Natl Acad Sci U S A 2010; 107:18410 - 5; http://dx.doi.org/10.1073/pnas.1008924107; PMID: 20930118
- Watanabe A, Arai M, Koitabashi N, Niwano K, Ohyama Y, Yamada Y, et al. Mitochondrial transcription factors TFAM and TFB2M regulate Serca2 gene transcription. Cardiovasc Res 2011; 90:57 - 67; http://dx.doi.org/10.1093/cvr/cvq374; PMID: 21113058
- Kurita T, Izumi H, Kagami S, Kawagoe T, Toki N, Matsuura Y, et al. Mitochondrial transcription factor A regulates BCL2L1 gene expression and is a prognostic factor in serous ovarian cancer. Cancer Sci 2012; 103:239 - 444; http://dx.doi.org/10.1111/j.1349-7006.2011.02156.x; PMID: 22098591
- Joseph AM, Adhihetty PJ, Buford TW, Wohlgemuth SE, Lees HA, Nguyen LM, et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012; 11:801 - 9; http://dx.doi.org/10.1111/j.1474-9726.2012.00844.x; PMID: 22681576
- Koltai E, Hart N, Taylor AW, Goto S, Ngo JK, Davies KJ, et al. Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am J Physiol Regul Integr Comp Physiol 2012; 303:R127 - 34; http://dx.doi.org/10.1152/ajpregu.00337.2011; PMID: 22573103
- Konopka AR, Douglass MD, Kaminsky LA, Jemiolo B, Trappe TA, Trappe S, et al. Molecular adaptations to aerobic exercise training in skeletal muscle of older women. J Gerontol A Biol Sci Med Sci 2010; 65:1201 - 7; http://dx.doi.org/10.1093/gerona/glq109; PMID: 20566734
- Wang L, Mascher H, Psilander N, Blomstrand E, Sahlin K. Resistance exercise enhances the molecular signaling of mitochondrial biogenesis induced by endurance exercise in human skeletal muscle. J Appl Physiol 2011; 111:1335 - 44; http://dx.doi.org/10.1152/japplphysiol.00086.2011; PMID: 21836044
- Turdi S, Fan X, Li J, Zhao J, Huff AF, Du M, et al. AMP-activated protein kinase deficiency exacerbates aging-induced myocardial contractile dysfunction. Aging Cell 2010; 9:592 - 606; http://dx.doi.org/10.1111/j.1474-9726.2010.00586.x; PMID: 20477759
- Derbre F, Gomez-Cabrera MC, Nascimento AL, Sanchis-Gomar F, Martinez-Bello VE, Tresguerres JA, et al. Age associated low mitochondrial biogenesis may be explained by lack of response of PGC-1α to exercise training. Age (Omaha) 2012; 34:669 - 79; http://dx.doi.org/10.1007/s11357-011-9264-y
- Ploughman M, Attwood Z, White N, Doré JJ, Corbett D. Endurance exercise facilitates relearning of forelimb motor skill after focal ischemia. Eur J Neurosci 2007; 25:3453 - 60; http://dx.doi.org/10.1111/j.1460-9568.2007.05591.x; PMID: 17553014
- Zhang Q, Wu Y, Zhang P, Sha H, Jia J, Hu Y, et al. Exercise induces mitochondrial biogenesis after brain ischemia in rats. Neuroscience 2012; 205:10 - 7; http://dx.doi.org/10.1016/j.neuroscience.2011.12.053; PMID: 22266265
- Mehta SL, Kumari S, Mendelev N, Li PA. Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia. BMC Neurosci 2012; 13:79; http://dx.doi.org/10.1186/1471-2202-13-79; PMID: 22776356
- Forini F, Lionetti V, Ardehali H, Pucci A, Cecchetti F, Ghanefar M, et al. Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodelling in rats. J Cell Mol Med 2011; 15:514 - 24; http://dx.doi.org/10.1111/j.1582-4934.2010.01014.x; PMID: 20100314
- Wang X, Moraes CT. Increases in mitochondrial biogenesis impair carcinogenesis at multiple levels. Mol Oncol 2011; 5:399 - 409; http://dx.doi.org/10.1016/j.molonc.2011.07.008; PMID: 21855427
- Bellance N, Benard G, Furt F, Begueret H, Smolková K, Passerieux E, et al. Bioenergetics of lung tumors: alteration of mitochondrial biogenesis and respiratory capacity. Int J Biochem Cell Biol 2009; 41:2566 - 77; http://dx.doi.org/10.1016/j.biocel.2009.08.012; PMID: 19712747
- D’Errico I, Salvatore L, Murzilli S, Lo Sasso G, Latorre D, Martelli N, et al. Peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α) is a metabolic regulator of intestinal epithelial cell fate. Proc Natl Acad Sci U S A 2011; 108:6603 - 8; http://dx.doi.org/10.1073/pnas.1016354108; PMID: 21467224
- D’Errico I, Lo Sasso G, Salvatore L, Murzilli S, Martelli N, Cristofaro M, et al. Bax is necessary for PGC1α pro-apoptotic effect in colorectal cancer cells. Cell Cycle 2011; 10:2937 - 45; http://dx.doi.org/10.4161/cc.10.17.16791; PMID: 21862870
- Lee CH, Wu SB, Hong CH, Liao WT, Wu CY, Chen GS, et al. Aberrant cell proliferation by enhanced mitochondrial biogenesis via mtTFA in arsenical skin cancers. Am J Pathol 2011; 178:2066 - 76; http://dx.doi.org/10.1016/j.ajpath.2011.01.056; PMID: 21514422
- Guerra F, Kurelac I, Cormio A, Zuntini R, Amato LB, Ceccarelli C, et al. Placing mitochondrial DNA mutations within the progression model of type I endometrial carcinoma. Hum Mol Genet 2011; 20:2394 - 405; http://dx.doi.org/10.1093/hmg/ddr146; PMID: 21470976
- Cormio A, Guerra F, Cormio G, Pesce V, Fracasso F, Loizzi V, et al. Mitochondrial DNA content and mass increase in progression from normal to hyperplastic to cancer endometrium. BMC Res Notes 2012; 5:279; http://dx.doi.org/10.1186/1756-0500-5-279; PMID: 22676897
- Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, et al. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem 2012; 120:419 - 29; http://dx.doi.org/10.1111/j.1471-4159.2011.07581.x; PMID: 22077634
- Trancikova A, Tsika E, Moore DJ. Mitochondrial dysfunction in genetic animal models of Parkinson’s disease. Antioxid Redox Signal 2012; 16:896 - 919; http://dx.doi.org/10.1089/ars.2011.4200; PMID: 21848447
- Patki G, Lau YS. Impact of exercise on mitochondrial transcription factor expression and damage in the striatum of a chronic mouse model of Parkinson’s disease. Neurosci Lett 2011; 505:268 - 72; http://dx.doi.org/10.1016/j.neulet.2011.10.036; PMID: 22040668
- Kim J, Moody JP, Edgerly CK, Bordiuk OL, Cormier K, Smith K, et al. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum Mol Genet 2010; 19:3919 - 35; http://dx.doi.org/10.1093/hmg/ddq306; PMID: 20660112
- Johri A, Calingasan NY, Hennessey TM, Sharma A, Yang L, Wille E, et al. Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington’s disease. Hum Mol Genet 2012; 21:1124 - 37; http://dx.doi.org/10.1093/hmg/ddr541; PMID: 22095692
- Tsunemi T, La Spada AR. PGC-1α at the intersection of bioenergetics regulation and neuron function: from Huntington’s disease to Parkinson’s disease and beyond. Prog Neurobiol 2012; 97:142 - 51; http://dx.doi.org/10.1016/j.pneurobio.2011.10.004; PMID: 22100502
- Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 1997; 41:646 - 53; http://dx.doi.org/10.1002/ana.410410514; PMID: 9153527
- Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol 1996; 39:385 - 9; http://dx.doi.org/10.1002/ana.410390317; PMID: 8602759
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1α in Huntington’s disease neurodegeneration. Cell Metab 2006; 4:349 - 62; http://dx.doi.org/10.1016/j.cmet.2006.10.004; PMID: 17055784
- Kish SJ, Bergeron C, Rajput A, Dozic S, Mastrogiacomo F, Chang LJ, et al. Brain cytochrome oxidase in Alzheimer’s disease. J Neurochem 1992; 59:776 - 9; http://dx.doi.org/10.1111/j.1471-4159.1992.tb09439.x; PMID: 1321237
- Wong-Riley M, Antuono P, Ho KC, Egan R, Hevner R, Liebl W, et al. Cytochrome oxidase in Alzheimer’s disease: biochemical, histochemical, and immunohistochemical analyses of the visual and other systems. Vision Res 1997; 37:3593 - 608; http://dx.doi.org/10.1016/S0042-6989(96)00210-6; PMID: 9425533
- Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, et al. PGC-1α expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol 2009; 66:352 - 61; http://dx.doi.org/10.1001/archneurol.2008.588; PMID: 19273754
- Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, et al. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun 1989; 163:1450 - 5; http://dx.doi.org/10.1016/0006-291X(89)91141-8; PMID: 2551290
- Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, et al, Global PD Gene Expression (GPEX) Consortium. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med 2010; 2:52ra73; http://dx.doi.org/10.1126/scitranslmed.3001059; PMID: 20926834
- La Spada AR. PPARGC1A/PGC-1α, TFEB and enhanced proteostasis in Huntington disease: defining regulatory linkages between energy production and protein-organelle quality control. Autophagy 2012; 8:1845 - 7; http://dx.doi.org/10.4161/auto.21862; PMID: 22932698
- Katsouri L, Parr C, Bogdanovic N, Willem M, Sastre M. PPARγ co-activator-1α (PGC-1α) reduces amyloid-β generation through a PPARγ-dependent mechanism. J Alzheimers Dis 2011; 25:151 - 62; PMID: 21358044
- Iyer S, Thomas RR, Portell FR, Dunham LD, Quigley CK, Bennett JP Jr.. Recombinant mitochondrial transcription factor A with N-terminal mitochondrial transduction domain increases respiration and mitochondrial gene expression. Mitochondrion 2009; 9:196 - 203; http://dx.doi.org/10.1016/j.mito.2009.01.012; PMID: 19460293
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006; 127:397 - 408; http://dx.doi.org/10.1016/j.cell.2006.09.024; PMID: 17055439
- Mudò G, Mäkelä J, Di Liberto V, Tselykh TV, Olivieri M, Piepponen P, et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell Mol Life Sci 2012; 69:1153 - 65; http://dx.doi.org/10.1007/s00018-011-0850-z; PMID: 21984601
- Ciron C, Lengacher S, Dusonchet J, Aebischer P, Schneider BL. Sustained expression of PGC-1α in the rat nigrostriatal system selectively impairs dopaminergic function. Hum Mol Genet 2012; 21:1861 - 76; http://dx.doi.org/10.1093/hmg/ddr618; PMID: 22246294
- Viña J, Gomez-Cabrera MC, Borras C, Froio T, Sanchis-Gomar F, Martinez-Bello VE, et al. Mitochondrial biogenesis in exercise and in ageing. Adv Drug Deliv Rev 2009; 61:1369 - 74; http://dx.doi.org/10.1016/j.addr.2009.06.006; PMID: 19716394
- Arduini A, Serviddio G, Tormos AM, Monsalve M, Sastre J. Mitochondrial dysfunction in cholestatic liver diseases. Front Biosci (Elite Ed) 2012; 4:2233 - 52; PMID: 22202034
- de Moura MB, dos Santos LS, Van Houten B. Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environ Mol Mutagen 2010; 51:391 - 405; PMID: 20544881
- Gusdon AM, Zhu J, Van Houten B, Chu CT. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis 2012; 45:962 - 72; http://dx.doi.org/10.1016/j.nbd.2011.12.015; PMID: 22198378
- Jennings JJ Jr., Zhu JH, Rbaibi Y, Luo X, Chu CT, Kiselyov K. Mitochondrial aberrations in mucolipidosis Type IV. J Biol Chem 2006; 281:39041 - 50; http://dx.doi.org/10.1074/jbc.M607982200; PMID: 17056595
- Suzuki SW, Onodera J, Ohsumi Y. Starvation induced cell death in autophagy-defective yeast mutants is caused by mitochondria dysfunction. PLoS One 2011; 6:e17412; http://dx.doi.org/10.1371/journal.pone.0017412; PMID: 21364763
- Dickey AS, Strack S. PKA/AKAP1 and PP2A/Bβ2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J Neurosci 2011; 31:15716 - 26; http://dx.doi.org/10.1523/JNEUROSCI.3159-11.2011; PMID: 22049414
- Rao S, Schmidt O, Harbauer AB, Schönfisch B, Guiard B, Pfanner N, et al. Biogenesis of the preprotein translocase of the outer mitochondrial membrane: protein kinase A phosphorylates the precursor of Tom40 and impairs its import. Mol Biol Cell 2012; 23:1618 - 27; http://dx.doi.org/10.1091/mbc.E11-11-0933; PMID: 22419819
- Schmidt O, Harbauer AB, Rao S, Eyrich B, Zahedi RP, Stojanovski D, et al. Regulation of mitochondrial protein import by cytosolic kinases. Cell 2011; 144:227 - 39; http://dx.doi.org/10.1016/j.cell.2010.12.015; PMID: 21215441
- Leadsham JE, Gourlay CW. cAMP/PKA signaling balances respiratory activity with mitochondria dependent apoptosis via transcriptional regulation. BMC Cell Biol 2010; 11:92; http://dx.doi.org/10.1186/1471-2121-11-92; PMID: 21108829
- Gegg ME, Cooper JM, Schapira AH, Taanman JW. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS One 2009; 4:e4756; http://dx.doi.org/10.1371/journal.pone.0004756; PMID: 19270741
- Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A 2011; 108:9572 - 7; http://dx.doi.org/10.1073/pnas.1106291108; PMID: 21606348
- Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, et al. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet 2006; 15:883 - 95; http://dx.doi.org/10.1093/hmg/ddl006; PMID: 16449237
- Rothfuss O, Fischer H, Hasegawa T, Maisel M, Leitner P, Miesel F, et al. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet 2009; 18:3832 - 50; http://dx.doi.org/10.1093/hmg/ddp327; PMID: 19617636
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, et al. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 2011; 144:689 - 702; http://dx.doi.org/10.1016/j.cell.2011.02.010; PMID: 21376232
- Gottlieb RA, Carreira RS. Autophagy in health and disease. 5. Mitophagy as a way of life. Am J Physiol Cell Physiol 2010; 299:C203 - 10; http://dx.doi.org/10.1152/ajpcell.00097.2010; PMID: 20357180
- Kwon KY, Viollet B, Yoo OJ. CCCP induces autophagy in an AMPK-independent manner. Biochem Biophys Res Commun 2011; 416:343 - 8; http://dx.doi.org/10.1016/j.bbrc.2011.11.038; PMID: 22119190
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, et al. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med 2012; 4:42ra97; http://dx.doi.org/10.1126/scitranslmed.3003799; PMID: 22786682
- Pauly M, Daussin F, Burelle Y, Li T, Godin R, Fauconnier J, et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol 2012; 181:583 - 92; http://dx.doi.org/10.1016/j.ajpath.2012.04.004; PMID: 22683340
- Sun L, Zhao M, Yu XJ, Wang H, He X, Liu JK, et al. Cardioprotection by acetylcholine: a novel mechanism via mitochondrial biogenesis and function involving the PGC-1α pathway. J Cell Physiol 2013; 228:1238 - 48; http://dx.doi.org/10.1002/jcp.24277; PMID: 23139024
- Terman A, Gustafsson B, Brunk UT. The lysosomal-mitochondrial axis theory of postmitotic aging and cell death. Chem Biol Interact 2006; 163:29 - 37; http://dx.doi.org/10.1016/j.cbi.2006.04.013; PMID: 16737690
- Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008; 4:176 - 84; PMID: 18059160
- Hickson-Bick DL, Jones C, Buja LM. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J Mol Cell Cardiol 2008; 44:411 - 8; http://dx.doi.org/10.1016/j.yjmcc.2007.10.013; PMID: 18062988
- Yan J, Feng Z, Liu J, Shen W, Wang Y, Wertz K, et al. Enhanced autophagy plays a cardinal role in mitochondrial dysfunction in type 2 diabetic Goto-Kakizaki (GK) rats: ameliorating effects of (-)-epigallocatechin-3-gallate. J Nutr Biochem 2012; 23:716 - 24; http://dx.doi.org/10.1016/j.jnutbio.2011.03.014; PMID: 21820301
- Cherra SJ 3rd, Steer E, Gusdon AM, Kiselyov K, Chu CT. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol 2013; 182:474 - 84; http://dx.doi.org/10.1016/j.ajpath.2012.10.027; PMID: 23231918
- Rasbach KA, Schnellmann RG. PGC-1α over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem Biophys Res Commun 2007; 355:734 - 9; http://dx.doi.org/10.1016/j.bbrc.2007.02.023; PMID: 17307137