Abstract
As a central controller of cell growth, mechanistic target of rapamycin (MTOR) affects an array of biological processes, in particular protein synthesis, autophagy and cardiac homeostasis. Conflicting findings have been seen with regard to the role of MTOR signaling and autophagy in cardiac and adipocyte function under metabolic syndrome. AKT, an essential insulin-signaling molecule upstream of MTOR, participates in the regulation of glucose homeostasis and cardiac metabolism. Akt2 knockout may rescue against high-fat diet-disrupted autophagy flux, en route to cardioprotection. Thus, inhibition of MTOR may serve as a possible avenue to retard pathological cardiac hypertrophy via rescuing interrupted autophagic flux.
Keywords: :
Uncorrected obesity is a major independent risk factor for human diseases such as cancer, sleep apnea, type 2 diabetes, dyslipidemia, hypertension and cardiovascular diseases. Excessive fat and caloric intake play a pivotal role in the onset and development of obesity. Obesity triggers cardiac hypertrophy and myocardial dysfunction, contributing to the ever-rising cardiac morbidity and mortality rates in obese individuals. Cardiac hypertrophy, as characterized by excess protein synthesis and enlarged cardiomyocytes, promotes arrhythmia and increased risk of heart failure in obesity. Although a number of scenarios including lipid toxicity, sympathetic overactivation and accumulation of reactive oxygen species have been put forward for obesity-related heart diseases, the precise nature behind obesity heart anomalies still remains elusive. Recent evidence has indicated a unique role of disturbed autophagy in the pathogenesis of obesity-related heart diseases although it still remains debatable with regard to the role of autophagy regulation in obesity and obesity-related cardiac geometric and functional anomalies.
Autophagy, a lysosomal-dependent pathway of degradation, is responsible for turnover of long-lived proteins and intracellular structures that are damaged or malfunctioning. The autophagic process is essential to the maintenance of cellular homeostasis. Recent findings have consolidated an important role of autophagy in the pathogenesis of human diseases, implicating the therapeutic value of autophagy as a potential target in their clinical management. In particular, basal levels of autophagy are deemed indispensable for cell survival and serve as a housekeeper for the maintenance of cardiac homeostasis under physiological conditions. In light of the critical role of autophagy in adipogenesis, and glucose and lipid metabolism, recent attention has been drawn toward the role of autophagy regulation in metabolic syndrome, and in particular obesity. For example, hepatic autophagy is defective in obesity and diabetes, whereas its upregulation improves insulin sensitivity and retards complications in diabetes and other metabolic defects.
Nevertheless, controversy exists with regard to the role of autophagy in hypertrophied hearts. In vivo studies depict that inhibition of MTOR, a primary inhibitory regulator of autophagy, attenuates pressure overload-induced cardiac defects. Conversely, suppression of autophagy retards the development of cardiac hypertrophy. Along the same lines, activated autophagy is detrimental for pressure overload-induced heart failure. A role of autophagy in the etiology of myocardial geometric and functional abnormalities has also been unveiled in obesity. Levine and colleagues found modestly suppressed autophagy in hearts following high-fat diet intake (60% fat for 12 weeks). Sadoshima and colleagues also suggested reduced cardiac autophagy in high-fat diet-induced obesity (60% fat for 18 weeks), as evidenced by decreased LC3-II and accumulation of the autophagy adaptor protein SQSTM1/p62. Lerman and colleagues reported suppressed myocardial autophagy in an obese Ossabaw swine model. In obese pigs, diastolic dysfunction and preserved cardiac output as well as ejection fraction were noted associated with compromised myocardial autophagy. These data supported the notion of suppressed cardiac autophagy in obese hearts, although the role of autophagy flux, a dynamic process for autophagosome degradation, remains elusive. Static evaluation of a dynamic autophagy process based merely on several molecular markers remains one major challenge in the field of mammalian autophagy. Not surprisingly, the static assessment of autophagy likely underlies some of the misconceptions in our historical underestimation for the role of autophagy in various diseases.
Recent evidence from our group depicted persevered (in fact activated) myocardial autophagy and disrupted autophagic flux in high-fat diet-induced obese mice. We fed mice 45% high-fat diet for 20 weeks to induce obesity. High-fat diet intake induces overt obesity and cardiac hypertrophy as expected. Echocardiographic examination revealed decreased cardiac output and fractional shortening in obese mice along with compromised cardiomyocyte contractile function and intracellular Ca2+ handling. Interestingly, myocardial protein levels of LC3B-I and LC3B-II are increased in obese mice, favoring the presence of activated autophagy in the heart. Meanwhile, high-fat diet feeding leads to accumulation of SQSTM1 in the heart, favoring the presence of activated autophagy and disrupted autophagosome maturation in obese hearts. To ascertain whether cardiac autophagic flux is disrupted in high-fat diet-induced obese hearts, transmission electron microscopy was used to evaluate formation of autophagosomes and autolysosomes. Our data revealed that the number of double-membrane vacuoles, characteristic of autophagosomes, is much higher in cardiac tissues from high-fat diet-fed mice; however, the number of single-membrane vacuoles, representing autolysosomes, is not significantly increased. The accumulation of double-membrane vacuoles indicates that high-fat diet-induced obesity stimulates autophagy initiation, but prevents autophagosome maturation.
These data suggest that disrupted myocardial autophagic flux contributes to cardiac injuries in high-fat diet-induced obesity. More interestingly, our data revealed that Akt2 knockout rescues the autophagosome maturation deficit and alleviates high-fat diet feeding-induced cardiac geometric and functional anomalies. This is seen by the finding that lysosomal inhibition negates Akt2 knockout-induced beneficial cardiomyocyte mechanical effects. Our data further revealed that high-fat diet significantly suppresses the expression of RAB7, a small GTPase capable of stimulating lysosomal biogenesis and maturation of autophagic vacuoles by promoting their fusion with endosomes and lysosomes, the effect of which is abolished by Akt2 knockout, indicating that RAB7 may play a role in high-fat diet- and Akt2 knockout-induced response of autophagic flux ().
Figure 1. Schematic diagram depicting the role of AKT2 and autophagic flux in high-fat diet-induced change in autophagy and cardiac function (adapted from Xu et al., J Mol Cell Biol 2013; 5:61–3, with modifications).
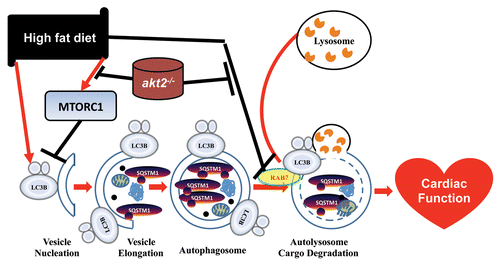
Furthermore, our data revealed that high-fat diet intake overtly increases MTOR phosphorylation, the effect of which is ablated by Akt knockout. As a central controller of cell growth, MTOR signaling may affect a number of biological processes, in particular protein synthesis and autophagy. Conflicting findings have been seen with regard to the role of MTOR signaling and autophagy in adipocyte function under metabolic syndrome. As an important regulator of metabolism and protein synthesis, MTOR regulates cardiac function in response to high-fat diet feeding. Although hypercholesterolemia does not induce obesity in a swine model, activated MTOR signaling is noted with cardiac hypertrophy. In addition, a key role of TOR in regulating cardiomyopathy in high-fat diet-induced obesity was further demonstrated by Birse and colleagues. MTOR can bind with and inhibit EIF4E, which can be reactivated by MTOR-mediated phosphorylation of EIF4EBP1. Besides, MTOR-mediated phosphorylation of RPS6K/p70S6K activates RPS6. The activation of EIF4E and RPS6 may ultimately contribute to protein synthesis and cardiac hypertrophy. Interestingly, our data demonstrated that ablation of Akt2 blocks high-fat diet-induced phosphorylation of MTOR, and thus retards pathological cardiac hypertrophy, rescuing cardiac autophagic flux and cardiac dysfunction following high-fat diet intake. Further studies are needed to uncover the underlying mechanism behind the regulation of MTOR signaling and autophagy flux in both physiological and pathophysiological settings.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.