Abstract
Mitophagy is an essential process that maintains mitochondrial quality and number, thus limiting cellular degeneration. Along with apoptosis, mitophagy participates in cellular fate decisions by eliminating damaged mitochondria. A variety of mitochondrial parameters, such as structure, membrane potential, and reactive oxygen species, are key determinants in triggering the mitophagic machinery. These parameters are also important regulators of the mitochondrial capacity for calcium (Ca2+) uptake. Rapid Ca2+ accumulation in the mitochondrial matrix allows for prompt stimulation of the organelle. This process requires a close morphofunctional coupling between mitochondria and the main intracellular Ca2+ stores. In mitophagy, the role of Ca2+ remains obscure. What role does mitochondrial Ca2+ play in metabolic sensing or in mitochondrial remodeling? Is endoplasmic reticulum (ER)-Ca2+ crosstalk involved? These are some of the questions that we address in this review.
Introduction
Mitochondria are pivotal organelles within complex endomembrane systems that are capable of transducing a Ca2+ signal into the cell. Currently, it is thought that any mitochondrial dysfunction will inevitably lead to disease. In fact, many pathological conditions are associated with mitochondrial failure, including neurodegenerative diseases, motor neuron disorders, autosomal dominant optic atrophy, ischemia-reperfusion injury, diabetes, aging, and cancer.Citation1 A large body of experimental evidence has unambiguously shown that in addition to their well-established function of producing most cellular ATP, mitochondria trigger cell death, thereby influencing cell fate.Citation2 Mitochondria also have the important role of activating apoptosis by releasing specific mitochondrial pro-apoptotic proteins into the cytoplasm; these proteins activate caspases and lead to apoptotic cell death.Citation3 A variety of extracellular stimuli ranging from the binding of hormones, neurotransmitters, and growth factors to phenomena such as cell–cell interactions, induce a rise in cytosolic Ca2+ concentration ([Ca2+]c) through diverse mechanisms with defined timing, amplitude, and kineticsCitation4,Citation5 that have an impact on mitochondrial activities including ATP production or cell death. Ca2+ plays a key role in the regulation of structural and functional mitochondrial alterations.Citation3 Mitochondrial Ca2+ overload promotes apoptosis by inducing mitochondrial membrane permeabilization, which is characterized by mitochondrial swelling, perturbation of the outer membrane and release of pro-apoptotic factors.Citation2 The execution of this process is connected/accompanied with/by specific mitochondrial dysfunction, including the loss of mitochondrial membrane potential, increased reactive oxygen species (ROS) production and decreased ATP production with the alteration of mitochondrial Ca2+ homeostasis.Citation2
The number of mitochondria, and mitochondrial health, are regulated by mitophagy, a process by which excessive or damaged mitochondria are removed. Mitophagy has a cardinal role in cellular adaptation to stress by acting as a mitochondrial quality control mechanism.Citation6 The maintenance of a healthy mitochondrial population is essential for sustaining cell metabolism in various physiological states. Thus, efficient mitophagic recognition and elimination of dysfunctional mitochondria sets the threshold for mitochondrial viability, thus making the cell able to counteract unwanted degeneration and prevent inflammation and apoptosis.Citation7
As accumulating evidence has indicated, both the induction of autophagy and apoptosis are partially regulated by the same proteins. For example, the anti-apoptotic protein BCL2 regulates autophagy and apoptosis by binding to the pro-autophagic protein BECN1, and the pro-apoptotic protein BAX.Citation8 It is clear that the health of mitochondria and their Ca2+-sensing capacity could determine the difference in cellular outcome between mitophagy and apoptosis. We therefore propose that Ca2+ signaling might be a key regulator of mitophagy and that it maintains a responsive mitochondrial network in the cell. Moreover, several studies in mammalian cells and in yeast models of autophagy have demonstrated a potential role for mitochondrial parameters regulated by Ca2+ signals (mitochondrial ROS production, membrane potential [Ψm] variations and fusion/fission dynamics) in modulating and/or triggering mitophagy.Citation9-Citation15
In this review, we summarize our current knowledge of the mechanisms of mitophagy, focusing our attention on mitochondrial morphology, Ψm and ROS. We also speculate on the possible pathophysiological roles of Ca2+ signaling and associated factors in mitophagy for the maintenance of mitochondrial homeostasis.
Intracellular Ca2+ Circuit
The technological advancements in probe design and detecting systems, allowed the accurate measurement of Ca2+ concentration ([Ca2+]) and distribution in intracellular compartments, and revealed a marked asynchronicity in cell response and a high spatio-temporal complexity of the intracellular Ca2+ signal. We now know that Ca2+ signals can be conveyed as repetitive [Ca2+]c spikes (commonly referred to as Ca2+ oscillations)Citation16 as well as localized [Ca2+]c increases that may either be confined or gradually propagated to the rest of the cell (“Ca2+ waves”).Citation17,Citation18
Cell Ca2+ entry is driven by the presence of a large electrochemical gradient across the plasma membrane. Various entry channels are involved with widely different properties: the voltage-operated channels, receptor-operated channels, second-messenger-operated channels, and store-operated channels or G protein-operated-calcium channels. The interior of the cell is the more negative, yet the [Ca2+]c is less than 10,000th of that in the extracellular milieu. There are also intracellular organelles, such as the ER, the Golgi apparatus, and secretory granules, that contain 1 to 10,000-fold greater [Ca2+] than the cytoplasm. During the “on reaction,” stimuli induce both the entry of external Ca2+ and the formation of second messengers that release internal Ca2+ that is stored within the ER or Golgi apparatus. Ca2+ release from internal store is controlled by Ca2+ itself, or by an expanding group of messengers, such as inositol 1,4,5-triphosphate (IP3), cyclic ADP ribose, nicotinic acid adenine dinucleotide phosphate, and sphingosine-1-phosphate, which either stimulate or modulate the release channels on the internal stores. Most of this Ca2+ is bound to buffers, whereas a small proportion binds to the effectors that activate various cellular processes. During the course of a typical Ca2+ transient wave, the generation of Ca2+ signaling is counteracted by the “off reaction” systems, when various pumps and exchangers remove Ca2+ from the cytoplasm. The pumping mechanisms have important homeostatic functions in which they maintain the resting level of Ca2+ and ensure that internal stores are kept loaded. Four different pumping mechanisms are responsible for the “off reaction”: ATP2B (ATPase, Ca2+ transporting, plasma membrane), SLC8A (Na+/Ca2+ exchanger), ATP2A (ATPase, Ca2+ transporting, cardiac muscle), and MCU (mitochondrial calcium uniporter).Citation4,Citation19,Citation20 SLC8A and ATP2B extrude Ca2+ to the outside, whereas ATP2A pumps Ca2+ back into the ER. Mitochondria also have an active function during the recovery process in which they sequester Ca2+ rapidly through the MCU, and release it more slowly back into the cytosol to be dealt with by ATP2A and ATP2B. Both pumps have lower transport rates but high affinities, which means that they can respond to modest elevations in Ca2+ levels and set basal Ca2+ levels. The SLC8A and MCU have much greater transport rates, and can limit transient Ca2+ changes over a wider dynamic range.Citation21,Citation22
Mitochondrial Morphology and Mitochondrial ER Contact Sites
The mitochondrial morphology of living cells is heterogeneous and is the result of the balance between fusion and fission events. A growing body of evidence indicates that mitochondrial morphology is critical for cell physiology. Changes in mitochondrial shape have been associated with many different processes, such as development, neurodegeneration, Ca2+ signaling, ROS production, cell division, and apoptotic cell death.
Mitochondrial fusion facilitates the exchange of materials between organelles and aids in the repair of defective mitochondria. Mitochondrial fission allows for proper mitochondrial segregation, enhancing distribution along the cytoskeleton or isolation of dysfunctional mitochondria. Impaired mitochondrial fusion results in reduced cell growth, decreased Ψm and defective respiration.Citation23 A similar phenotype is observed when fission is deficient, with additional loss of mitochondrial DNA (mtDNA) and reduced ATP production.Citation24
Studies on the mammalian mitochondrial fusion machinery have shown that MFN1/mitofusin 1 and MFN2/mitofusin 2 are located on the outer mitochondrial membrane (OMM),Citation25 whereas OPA1/optic atrophy 1 (autosomal dominant) is located on the inner mitochondrial membrane.Citation26 Additionally, mitochondrial division is regulated by DNM1L/dynamin 1-like,Citation27 FIS1/fission 1 (mitochondrial outer membrane) homolog (S. cerevisiae)Citation28 and MFF/mitochondrial fission factor.Citation29 DNM1L is a cytosolic GTPase, when its expression is inhibited or downregulated, a highly interconnected mitochondrial network forms.Citation27 The same phenotype results from the downregulation of FIS1 and MFF, two proteins proposed to act as a mitochondrial receptor for DNM1L.Citation28 FIS1 and MFF have been detected by blue native electrophoresis in complexes with different molecular weights, suggesting that at least two different macromolecular complexes participate in mitochondrial fission events. Furthermore, a conditional Fis knockout (KO) had indicated that FIS1 is dispensable for DNM1L-mediated fission, whereas MFF is not ().Citation30
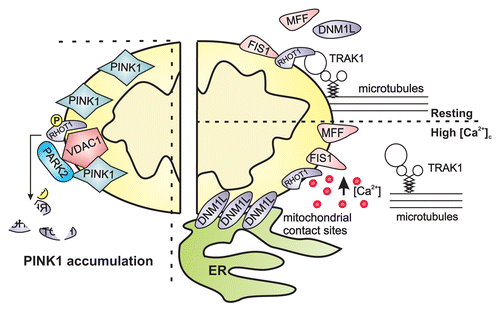
Figure 1. Mitochondrial integrity is a key factor involved in mitophagy, which is regulated by specific fission proteins, including DNM1L, FIS1, and MFF, which are recruited by mitophagy-related inducers. The figure shows the involvement of the mitochondrial Ca2+-sensing protein RHOT1, an OMM protein that under resting conditions interacts with the cytoplasmic adaptor protein TRAK2, mediating mitochondrial sliding along the cytoskeleton. Sustained [Ca2+]c elevations induce the RHOT1-dependent motility block, allowing DNM1L-induced fragmentation. Contact sites have been recently shown to be required for the DNM1L polymerization process that generates a constriction around mitochondria, a process that is necessary for mitochondrial fission. In depolarized neurons, PINK accumulation favors RHOT1 phosphorylation, inducing PINK-RHOT1-PARK2 aggregation. This complex promotes RHOT1 degradation, preventing mitochondrial movement and quarantining the damaged mitochondria.
![Figure 1. Mitochondrial integrity is a key factor involved in mitophagy, which is regulated by specific fission proteins, including DNM1L, FIS1, and MFF, which are recruited by mitophagy-related inducers. The figure shows the involvement of the mitochondrial Ca2+-sensing protein RHOT1, an OMM protein that under resting conditions interacts with the cytoplasmic adaptor protein TRAK2, mediating mitochondrial sliding along the cytoskeleton. Sustained [Ca2+]c elevations induce the RHOT1-dependent motility block, allowing DNM1L-induced fragmentation. Contact sites have been recently shown to be required for the DNM1L polymerization process that generates a constriction around mitochondria, a process that is necessary for mitochondrial fission. In depolarized neurons, PINK accumulation favors RHOT1 phosphorylation, inducing PINK-RHOT1-PARK2 aggregation. This complex promotes RHOT1 degradation, preventing mitochondrial movement and quarantining the damaged mitochondria.](/cms/asset/4b3198c8-6b3f-4905-adfc-2ce0d611eac8/kaup_a_10924795_f0001.gif)
The organization of the mitochondrial network is fundamental for the regulation of Ca2+ homeostasis. In fact, it has been widely demonstrated that mitochondria can uptake Ca2+ due to their close apposition with the ER.Citation31 At these sites, ITPR/inositol 1,4,5-trisphosphate receptor on the ER side is juxtaposed with VDAC1/voltage-dependent anion channel 1 on the OMM via the chaperone activity of HSPA9/heat shock 70 kDa protein 9 (mortalin).Citation32 In this scenario, Ca2+ released through the ITPR generates microdomains at high Ca2+ concentrations, and mitochondria can rapidly uptake Ca2+ through the MCU. Once within the mitochondrial matrix, Ca2+ rapidly diffuses through the whole network and is extruded through the mitochondrial Na+/Ca2+ exchanger or the Ca2+/H+ antiporter.Citation33 Alterations in mitochondrial morphology can clearly impair Ca2+ uptake and its flow through the network. It has been demonstrated that Mfn KO cells, in which the amount of contact with ER is reduced, display impaired Ca2+ uptake.Citation34 Similarly, DNM1L-overexpressing cells, in which mitochondria have normal contacts with the ER but are fragmented, show a consequent loss of matrix connectivity and reduced Ca2+ uptake.
The relationship between mitochondrial shaping and Ca2+ is bidirectional due to Ca2+-dependent regulation of mitochondrial morphology. At least two interconnected mechanisms have to be considered. First, the mitochondrial Rho-GTPase (RHOT1) is an OMM protein with two EF hands for Ca2+ binding that interacts with TRAK1, a cytoplasmic adaptor able to bind the kinesin 1 heavy chain on microtubules. Under resting conditions, RHOT1 mediates mitochondrial sliding along the cytoskeleton, thus favoring mitochondrial fusion by impeding DNM1L activity. Elevation of the [Ca2+]c induces the RHOT1-dependent block in motility and allows DNM1L-induced fragmentation ().Citation35
Second, Scorrano and colleagues showed that elevation of [Ca2+]c induces calcineurin activation and subsequent dephosphorylation at serine 637 of DNM1L that then accumulates in mitochondria.Citation36 Interestingly, another group found that Ca2+ influx through VDACs induces DNM1L phosphorylation at serine 600 via activation of CAMK1/calcium/calmodulin-dependent protein kinase I with the consequent induction of mitochondrial fragmentation.Citation37 These data suggest that different Ca2+ sources can lead to the same result through different pathways.
Mitophagy is also involved in the wide range of physiological pathways that regulate fusion/fission events. Incorporation of mitochondria within an autophagosome, which has a maximum diameter of approximately 1 μm, is sterically impossible if it is not preceded by a fission event. Accordingly, mitochondrial fragmentation has been detected before mitophagy during mitochondrial stress.Citation38 Moreover, OPA1 overexpression decreases mitophagy, whereas blocking mitochondrial fission induces autophagy without mitophagy, and dysfunctional mitochondria accumulate.Citation24,Citation39 It appears clear that fission is relevant for the success of mitophagy; however, this process is not sufficient to induce mitophagy as shown in FIS1-overexpressing cells that display only a weak increase in mitophagy.Citation40 In yeast cells, which are used to model important aspects of autophagy in eukaryotes, the role of mitochondrial fission in promoting autophagy is debated. Genetic screening revealed that deletion of the profission DNM1 gene, the ortholog of mammalian DNM1L, is associated with reduced mitophagy.Citation41 In contrast, in a different study, the KO of fission factor genes, such as DNM1, MDV1, and CAF4, did not alter mitophagy,Citation42 suggesting that Dnm1-dependent fission is not essential for mitophagy in yeast. However, it cannot be excluded that mitochondrial fission may precede mitophagy in yeast through unknown mechanisms and/or upon different types of stimulation. These observations suggest that a fission event is necessary for mitochondrial segregation, but another event, such as Ψm or ROS, is required for mitochondrial engulfment, and will be discussed later.
The work from Wang in 2011 is noteworthy because it describes the first molecular link between mitochondrial morphology and mitochondrial quality control. PINK1 (PTEN-induced putative kinase 1) and the ubiquitin ligase PARK2/Parkin, are two molecular players involved in the recognition of “damaged” mitochondria and their delivery to the autophagosome.Citation43 Wang observed that in mouse and Drosophila depolarized neurons, RHOT1 exists in a molecular complex with PINK1 and PARK2. This interaction induces RHOT1 phosphorylation and degradation that precedes blockage of mitochondrial motility ().Citation44 Unfortunately, the readout of both mitophagy and the Ca2+-dependent activity of RHOT1 after its phosphorylation is not detected in such conditions.
RHOT1 was initially characterized in yeast as a participant in the ER-mitochondria encounter structure (ERMES) that tethers the ER to mitochondria, and this localization was later confirmed in mammalian cells.Citation45 Contact sites have been recently shown to be required for the DNM1L polymerization process that generates a constriction around mitochondria, a process that is necessary for mitochondrial fission.Citation46 In addition, contact sites have just been demonstrated to play a role in autophagosome generation.Citation47 It could thus be speculated that Ca2+ signals at contact sites induce a RHOT1-dependent block in mitochondrial sliding (possibly in front of specific ER subdomains) and DNM1L-dependent fission.
The Mitochondrial Membrane Potential
As mentioned above, mitochondria are the main sites of ATP production. The enzymatic systems involved in this process are the tricarboxylic acid cycle and the mitochondrial electron transport chain complex. The products from glycolysis and fatty acid metabolism are converted to acetyl-CoA and enter the tricarboxylic acid cycle, which generates NADH and FADH2, triggering the electron transport chain. The electrons move along the respiratory chain, and the energy is stored as an electrochemical H+ gradient across the inner membrane, creating the negative mitochondrial membrane potential, which is estimated to be around -180 mV (). This membrane potential provides a huge driving force for Ca2+ entry into the organelle.
Figure 2. The ubiquitination of a subset of OMM proteins including VDAC1, TOMM, and MFNs by the ubiquitin ligase PARK2 promotes the dissipation of Ψm, inducing the recruitment of a series of adaptor proteins (such as SQSTM1) that promote mitochondrial sequestering. Ψm represents a huge driving force for Ca2+-entry into the organelle through the recently identified mitochondrial calcium uniporter MCU. The activity of the channel is regulated by the mitochondrial proteins MICU1/mitochondrial calcium uptake 1 and MCUR1/mitochondrial calcium uniporter regulator 1. The loss of Ψm impairs Ca2+ uptake and mitochondrial Ca2+-buffering causing PINK1 accumulation and stabilization on the OMM. In the inset, Pink1 KO cells show reduced Ψm via the increased mitochondrial Ca2+ uptake and mPTP (mitochondrial permeability transition pore) opening.
Several intracellular signals can induce irreversible mitochondrial damage, destabilizing mitochondrial integrity and favoring the loss of Ψm. This alteration represents an activating factor for the induction of apoptotic cell deathCitation48 and probably for the recruitment of the mitophagic machinery.
It has been demonstrated that the mitochondrial permeability transition (MPT) is an important event for inducing the autophagic process.Citation49 The MPT is a critical event involved in controlling cell fate and results from the opening of the so-called mitochondrial permeability transition pore (mPTP), an aggregate multicomponent protein localized in the mitochondrial compartments that includes factors in the inner and outer mitochondrial membrane.Citation50 Opening of the mPTP can lead to loss of Ψm with subsequent swelling of the mitochondrial matrix and a transient, but strong, release of ROS and Ca2+ ions.
Damaged and depolarized mitochondria are sequestered into autophagic vacuoles identified with the autophagosome marker microtubule-associated protein 1 light chain 3 (MAP1LC3). In particular, the level of mitophagy increases when a subset of mitochondria are experimentally depolarized with ionophores including carbonyl cyanide m-chlorophenylhydrazone (CCCP) or valinomycin that simulate mitochondrial damage.Citation51,Citation52 For these reasons, it appears that Ψm is a critical point for the removal of impaired, aged, and dysfunctional mitochondria.
In aging and degenerative processes, PARK2 selectively translocates to depolarized and disturbed mitochondria and, in turn, promotes their autophagic removal.Citation51 It has been demonstrated that the recruitment of PARK2 to impaired mitochondria is modulated by PINK1 expression and activity.Citation43,Citation53 Loss-of-function mutations in PINK1 have been linked to familial Parkinson disease (PD), where it has been reported that loss of the protein impairs mitochondrial respiratory activity in mouse brains.Citation54 In Pink1 KO cells, the enzymatic activities of the mitochondrial electron transport chain complex are normal, but the Ψm is reduced through increased mitochondrial Ca2+ uptake and mPTP opening ().Citation54
PINK1 contains an N-terminal mitochondrial targeting motif and a highly conserved kinase domain that shows strong homology to the Ca2+/calmodulin-dependent serine/threonine kinase family.Citation55 The kinase domain of PINK1 is oriented toward the cytosol, and it can physically interact with PARK2 and modulate its mitochondrial localization via direct phosphorylation.Citation56 Under normal conditions, PINK1 is localized in the OMM as a 64-kDa full-length protein, which is processed in a cleaved form of 52-kDa and imported into the IMM. This cleaved form of PINK1 is rapidly degraded by a protease; therefore, the endogenous amount of PINK1 is low, and the mitophagy of healthy mitochondria is blocked. When a subset of mitochondria suffer a loss of Ψm (prompted by strong stressors or uncoupling agents), the full-length PINK1 accumulates and stabilizes on the OMM of damaged mitochondria where it recruits PARK2.Citation57 In the mitochondrial compartment, PARK2 ubiquitinates a subset of OMM proteins, such as VDAC1 and MFN.Citation58 This incremental ubiquitination on the mitochondria induces the recruitment of a series of adaptor proteins (such as SQSTM1), which connect the autophagic process to ubiquitinated proteins (). As a result, the damaged mitochondria are recruited by MAP1LC3 and autophagosomal membranes that finally fuse with the lysosome.Citation59 In yeast, selective mitochondrial degradation via micro- and macroautophagy is associated with impairment of Ψm and mitochondrial biogenesis in cells lacking Fmc1, a mitochondrial protein required under heat stress conditions for the proper assembly of the ATP synthase complex.Citation60 Additionally, reduced Ψm, mitochondrial swelling and fragmentation have been described in yeast cells when the mitochondrial K+/H+ exchanger, Mdm38, is depleted, thus leading to increased mitophagy and growth defects on nonfermentable carbon sources.Citation61 However, the reduction in Ψm appears to be insufficient for the induction of mitophagy in yeast. Yeast mutants lacking Cox4 show a similar reduction in Ψm as mdm38Δ cells, but they do not undergo mitophagy.Citation61 In addition, in contrast to mammalian cells, the chemical depolarization of yeast mitochondria via CCCP does not induce mitophagy.Citation62 These lines of evidence suggest that damaged mitochondria are degraded by mitophagy in yeast and a change in the Ψm may only influence, but not trigger, this process. Therefore, yeast and mammalian cells may have different cell parameters and stimuli for inducing and/or regulating mitophagy, although the selective removal of the organelles may be completed through similar mechanisms.
Mitochondrial ROS Production
Oxidative stress is inseparably associated with mitochondrial dysfunction, as mitochondria are both the generators of and targets for reactive oxygen species.Citation63 The mitochondria, especially in brain cells, are a major site of the generation and action of both ROS and reactive nitrogen species (RNS). Specific forms of ROS and RNS include hydrogen peroxide (H2O2), superoxide (O2•−), nitric oxide (NO), and peroxynitrite (ONOO−).Citation64 Both exogenously added H2O2 and mitochondrial ROS activate autophagy. Autophagy is essential for degrading ROS-producing organelles such as mitochondria and peroxisomes.Citation65 However, beyond activating autophagy, it is not clear whether ROS is also involved in the activation of mitophagy. Based on the above findings, very recent papers have proposed a role for ROS in initiating mitophagy. Using a mitochondrial-targeted photosensitizer, mitochondrial KillerRed, three different groups induced an increase in ROS levels in the mitochondrial matrix, resulting in the loss of membrane potential, fragmentation and the subsequent activation of PARK2-dependent mitophagy.Citation66-Citation68 In particular, O2•− was determined to be the specific ROS that damaged mitochondria because the overexpression of SOD2 (superoxide dismutase 2, mitochondrial) or an O2•− scavenger inhibit both mitochondrial fragmentation and the mitophagy induced by mitochondrial KillerRed activation.Citation67 In support of these observations, mammalian cells under mild oxidative stress generate moderate levels of ROS; additionally, a DNM1L-dependent type of mitophagy is triggered, but nonselective autophagy is not detected. These results suggest an earlier temporal role for mitophagy in response to mitochondrial damage that governs the selective removal of dysfunctional mitochondria.Citation69 In yeast, it has been proposed that the physiological role of mitophagy is to remove excess mitochondria that may generate a ROS surplus in cells.Citation70 Under nitrogen starvation, a condition that induces mitophagy, atg32Δ and atg11Δ yeast cells lacking genes essential for the selective recruitment of mitochondria to the autophagosomal machinery, show ROS overproduction associated with impaired mitochondrial function. In these mitophagy-deficient cells, ROS surplus causes mtDNA depletion that ultimately leads cells to a petite phenotype unable to grow on respiratory carbon sources.Citation71 Based on this study, mitophagy appears to be responsible for mitochondrial quality control to sustain cell life when damaged organelles are generated. In the frataxin-deficient yeast cells, yfh1Δ, a model of Friedreich ataxia characterized by mitochondrial iron overload, there is an increase in ROS production and oxidative damage. When these cells are treated with rapamycin, which inhibits the TOR pathway, thus inducing autophagy in yeast and mammals,Citation72 a reduction in both mitochondrial ROS and mass is observed, most likely because of the removal of damaged mitochondria.Citation73
Many proteins involved in mitophagy can be regulated by ROS. For example, SNCA/α-synuclein and PARK2 are S-nitrosylated (the addition of NO to thiol groups). Nitrogen modification of SNCA makes the protein prone to aggregation,Citation74 and S-nitrosylation of PARK2 inactivates it ().Citation75
Figure 3. ROS and RNS are responsible for the decrease in mitochondrial potential, mitochondrial fission and mtDNA mutations. Indeed, ROS and RNS regulate several mitophagic molecular targets through protein modification. ROS, reactive oxygen species; RNS, reactive nitrogen species; mETC, mitochondrial electron transport chain complex.
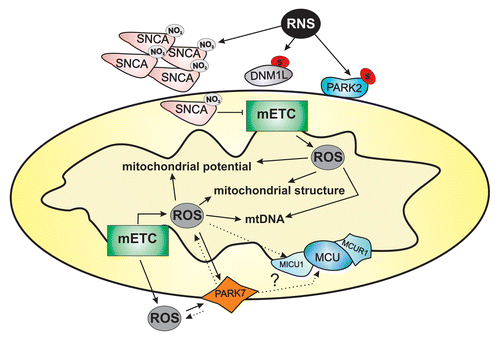
In this scenario, several mitochondrial signals, including impaired ATP production, activation of the MPT and loss of Ψm, appear able to trigger mitophagy, but enhanced ROS generation certainly plays a pivotal role. ROS cooperate with Ca2+ signaling in the control of ER-mitochondrial functions, and their levels are not only mutually regulated but also perfectly mirror the healthy state of a cell.Citation76 What is the role of Ca2+ in the context of oxidative stress-mediated mitophagy? Several observations in studies on PD support the influential activity of Ca2+ in oxidative stress-mediated disease. PD is characterized by mitochondrial dysfunction and oxidative stress as well as an imbalance in protein homeostasis due to an increase in protein misfolding and aggregation and the inability to remove misfolded proteins.Citation77 Among the different proteins implicated in PD, PARK7 plays an important role in the maintenance of mitochondrial integrity. Mutant PARK7 proteins that are associated with PD are stabilized by binding to PARK2, and this interaction is enhanced by oxidative stress. The predominant role of PARK7 is as an antioxidant, and PARK7 knockdown alters Ca2+ homeostasis in skeletal muscle.Citation78 Furthermore, PARK7-deficient dopaminergic neurons exhibit very low levels of mitochondrial uncoupling and correspondingly higher levels of oxidative stress, suggesting that PARK7 might somehow regulate the mitochondrial response to oxidative stress (). Importantly, blocking the L-type Ca2+ channels, which are plasma membrane channels that allow Ca2+ entry into the cytosol, completely rescues the oxidative stress effect.Citation79 The loss of PARK7 function weakens the compensatory mechanisms in mitochondria, making the dopaminergic neuron much more vulnerable to oxidative stress in the substantia nigra. The key finding of this study is that the abolition of Ca2+ accumulation in mitochondria can reverse the intracellular dysregulation resulting from PARK7 mutation. Considering the interplay between PARK2 and PARK7 in mitochondrial quality control,Citation80,Citation81 it will be interesting to investigate the basal level of mitophagy in Park7-null dopaminergic neurons and to determine whether the use of L-type Ca2+ blockers, such as isradipine, might either increase or attenuate the mitophagic process. Interestingly, Ca2+-influx through the channel increases mitochondrial superoxide, NADH production and metabolic activity in quiescent cardiac myocytes, reinforcing the concept that L-type channels could regulate mitochondrial functions.Citation82
As previously mentioned, the state of the mitochondrial network is a critical step in mitophagy because fragmented mitochondria are more readily taken up by autophagosomes due to their size. Mitochondrial fission is a deleterious effect of chronic oxidative stress, especially in neuronal cells,Citation83 and this process is accompanied by a robust decrease in mitochondrial Ca2+ levels. Thus, a mitochondrion that is ready to be engulfed is damaged by ROS, fragmented and, hypothetically, Ca2+ depleted. On the other hand, Ca2+ overload perturbs mitochondrial physiology, causing ROS generation, mPTP opening and the initiation of cell death. In this context, Ca2+ levels might be ideal triggers of mitophagy and could be considered one of the first intracellular mechanisms to avoid apoptosis through the elimination of potentially dangerous elements.
When Does Mitochondrial Ca2+ Intervene in the Mitophagy Process?
During the course of evolution, the mitochondria have developed biological pathways with molecular targets and critical checkpoints to monitor their health. The molecular clarification of involved molecular players and their mechanisms in mitochondrial fate, have highlighted the crosstalk between two important biological processes, mitochondrial-targeted autophagy and mitochondrial-related apoptosis. Both processes have similar molecular targets that trigger and modulate their occurrence. Indeed, they are linked by the Ca2+ ion, since it represents an important modulator of both cellular processes, acting on their common molecular targets.Citation84-Citation87
To date, the role of Ca2+ in mitophagy remains obscure. How does mitochondrial Ca2+ stimulate the selective removal of damaged organelles? Does it reduce the efflux of caspase cofactors? Is Ca2+ crosstalk with the ER involved? We attempt to answer these questions by discussing recent findings.
During apoptosis, by acting in synergy with a variety of cellular stress conditions, the mitochondrial Ca2+ overload is a key “author” of mPTP opening, thus inducing large-scale mitochondrial alterations. If mitochondrial Ca2+ loading is prevented (e.g., by loading an intracellular Ca2+ buffer), then the Ψm and mitochondrial morphology are preserved, and the cells are protected from apoptosis. Mitophagy is generally preceded by the MPT, followed by mitochondrial fission, hence indicating that the regulation of mitochondrial shape is an essential aspect of mitochondrial quality control.Citation39
The Ψm represents a very large driving force for Ca2+ accumulation and permits Ca2+-entry through MCU. Mitochondrial Ca2+ transduction constitutes a fundamental mechanism to regulate cell survival and metabolism, but indirect evidence suggests that it is also pivotal for mitophagy. As previously reported, PINK1 leads to increased mPTP opening, which, in turn, is primarily induced by oxidative stress and/or elevated mitochondrial Ca2+. This observation is consistent with a recent study that showed that loss of PINK1 reduces the activity of the mitochondrial Na+/Ca2+ exchanger.Citation88 Impaired mitochondrial Na+/Ca2+ exchanger activity in Pink1 KO cells may lead to the accumulation of mitochondrial Ca2+, which opens the mPTP. In this context, the mPTP may serve as a Ca2+-activated, Ca2+-releasing channel. These studies support an important function for PINK1 in regulating mitochondrial activity under stress conditions. The loss of Ψm and mitochondrial integrity in a neuronal cell model of PD are partially recovered by treatment with cyclosporin A and fully rescued by treatment with ruthenium red, an inhibitor of MCU, indicating that mitochondrial Ca2+ uptake is once again involved.Citation89 Moreover, recent work has suggested that an increase in mitochondrial Ca2+ content may influence the autophagic pathway.Citation90,Citation91 Pharmacological blocking of MCU activity alters the autophagic level, and the negative regulation of ITPR produces the same effects as the inhibition of the MCU in autophagy.Citation10 In autophagy, Ca2+ may have a dual role, pro- and anti-autophagic, and autophagy is dependent on both ER Ca2+ levels and ITPR activity.Citation10 Although there is no direct evidence, these data suggest that mitophagy could be affected by Ca2+ and its effectors.
The anti-apoptotic proteins BCL2 and BCL2L1/Bcl-xL mediate the decrease in ER Ca2+ levels and Ca2+ release, thus protecting the cell from apoptosis.Citation92 These proteins also inhibit autophagy by lowering ER Ca2+ levelsCitation91 and by interacting with both ITPR and the autophagic protein BECN1. Thus, the proteins have both anti-apoptotic and anti-autophagic roles. Interestingly, BCL2 inhibits autophagy only when localized to the ER membraneCitation93 as indicated by the dynamic interaction between AMBRA1 and BCL2 at the mitochondria,Citation94 where the interaction regulates BECN1-dependent autophagy and apoptotic cell death. The relationship with ER Ca2+ stores points to a role for ER Ca2+ in mitophagy. Ca2+ accumulation in the mitochondrial matrix requires a close morphofunctional coupling between mitochondria and Ca2+ stores such that if the interaction is impaired, the alterations could trigger the mitophagy machinery. For example, an early event in mitophagy is that PARK2 trafficks to the OMM, where it ubiquitinates different mitochondrial targets, such as MFNs,Citation95 DNM1L,Citation96 VDAC1,Citation97 and BCl2,Citation98 that are well-known mitochondrial Ca2+-sensing proteins involved in strategic events that monitor functional interorganelle Ca2+ flux. In addition, PINK1 phosphorylates RHOT1, promoting its proteasomal degradation and triggering PARK2.Citation44 These findings emphasize the pivotal role of crosstalk between the ER and mitochondria in mitophagy because RHOT1 is a Ca2+-sensitive regulator of ER-mitochondria contact sites,Citation45 mitochondrial Ca2+ signaling and motility and fusion-fission dynamics of mitochondria.Citation35 Recent studies have shown that SNCA can associate with mitochondria and that its accumulation increases mitochondrial Ca2+ levels and, as a consequence, oxidative damage, depending on both its expression and intracellular distribution.Citation99,Citation100 Moderate levels of SNCA overexpression enhance mitochondrial Ca2+ homeostasis by augmenting ER-mitochondria contact sites, whereas SNCA loss-of-function impairs mitochondrial Ca2+ levels and enhances the autophagic process.
Specific studies are still required to define the mitophagic scenery and determine how mitochondrial Ca2+ actively participates in triggering and/or modulating mitophagy, thus giving cells the option to follow either the apoptotic or autophagic pathway. Nevertheless, some evidence supports a model where BNIP3 induces selective removal of mitochondria in cardiac myocytes in an independent Ca2+-, ROS generation-, and mPTP opening-dependent manner.Citation101
In this context, yeast cells may represent a suitable model for further identification of proteins and/or cell effectors involved in mitophagy.Citation41 The role of Ca2+ in yeast metabolism and processes controlling cell survival, such as apoptosis and autophagy, still needs to be elucidated. However, there is increasing evidence that changes in ROS production, Ψm and mitochondrial morphology dynamics are either stimuli or consequences of mitophagy in yeast, thus suggesting that a conserved mitophagic mechanism might be shared at least partially in both yeast and mammalian cells.
Future studies will identify this direct link, opening new avenues for investigation and pharmacological intervention, especially for those diseases where the selective removal of mitochondria is one of the critical steps in the pathogenesis of the disorder.
| Abbreviations: | ||
| ATP2A | = | ATPase Ca2+ transporting cardiac muscle |
| ATP2B | = | ATPase, Ca2+ transporting plasma membrane |
| BCL2 | = | B-cell CLL/lymphoma 2 |
| BECN1 | = | Beclin 1, autophagy related |
| DNM1L | = | dynamin 1-like |
| ER | = | endoplasmic reticulum |
| FIS1 | = | fission 1 (mitochondrial outer membrane) homolog (S. cerevisiae) |
| HSPA9 | = | heat shock 70 kDa protein 9 (mortalin) |
| ITPR | = | inositol 1,4,5-trisphosphate receptor |
| MCU | = | mitochondrial calcium uniporter |
| MFF | = | mitochondrial fission factor |
| MPT | = | mitochondrial permeability transition |
| mPTP | = | mitochondrial permeability transition pore |
| OMM | = | outer mitochondrial membrane |
| PINK1 | = | PTEN-induced putative kinase 1 |
| RHOT1 | = | ras homolog family member T1/mitochondrial Rho GTPase 1 |
| ROS | = | reactive oxygen species |
| Ψm | = | mitochondrial potential |
| SLC8A | = | Na+/Ca2+ exchanger |
| VDAC1 | = | voltage-dependent anion channel 1 |
Acknowledgments
AR is financed by the Italian Ministry of Health. SP is supported by a FISM (Fondazione Italiana Sclerosi Multipla) research fellowship (2012/B/11), while SM by FIRC (Fondazione Italiana Ricerca sul Cancro). FML is financed by Telethon (GGP11139). PP is financed by Italian Association for Cancer Research (AIRC), Telethon (GGP09128 and GGP11139B), the Italian Ministry of Education, University and Research and the Italian Ministry of Health.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Giorgi C, Agnoletto C, Bononi A, Bonora M, De Marchi E, Marchi S, et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012; 12:77 - 85; http://dx.doi.org/10.1016/j.mito.2011.07.004; PMID: 21798374
- Rimessi A, Giorgi C, Pinton P, Rizzuto R. The versatility of mitochondrial calcium signals: from stimulation of cell metabolism to induction of cell death. Biochim Biophys Acta 2008; 1777:808 - 16; http://dx.doi.org/10.1016/j.bbabio.2008.05.449; PMID: 18573473
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med 2000; 6:513 - 9; http://dx.doi.org/10.1038/74994; PMID: 10802706
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 2000; 1:11 - 21; http://dx.doi.org/10.1038/35036035; PMID: 11413485
- Clapham DE. Intracellular calcium. Replenishing the stores. Nature 1995; 375:634 - 5; http://dx.doi.org/10.1038/375634a0; PMID: 7791893
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280 - 93; http://dx.doi.org/10.1016/j.molcel.2010.09.023; PMID: 20965422
- Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011; 333:1109 - 12; http://dx.doi.org/10.1126/science.1201940; PMID: 21868666
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571 - 80; http://dx.doi.org/10.1038/cdd.2010.191; PMID: 21311563
- Calì T, Ottolini D, Negro A, Brini M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim Biophys Acta 2013; 1832:495 - 508; http://dx.doi.org/10.1016/j.bbadis.2013.01.004; PMID: 23313576
- Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142:270 - 83; http://dx.doi.org/10.1016/j.cell.2010.06.007; PMID: 20655468
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141:656 - 67; http://dx.doi.org/10.1016/j.cell.2010.04.009; PMID: 20478256
- Scherz-Shouval R, Shvets E, Elazar Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007; 3:371 - 3; PMID: 17438362
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749 - 60; http://dx.doi.org/10.1038/sj.emboj.7601623; PMID: 17347651
- Solesio ME, Saez-Atienzar S, Jordan J, Galindo MF. 3-Nitropropionic acid induces autophagy by forming mitochondrial permeability transition pores rather than activating the mitochondrial fission pathway. Br J Pharmacol 2013; 168:63 - 75; http://dx.doi.org/10.1111/j.1476-5381.2012.01994.x; PMID: 22509855
- Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA, Dreier L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem 2012; 287:40652 - 60; http://dx.doi.org/10.1074/jbc.M112.419721; PMID: 23060438
- Robb-Gaspers LD, Thomas AP. Coordination of Ca2+ signaling by intercellular propagation of Ca2+ waves in the intact liver. J Biol Chem 1995; 270:8102 - 7; http://dx.doi.org/10.1074/jbc.270.14.8102; PMID: 7713913
- Jaffe LF, Créton R. On the conservation of calcium wave speeds. Cell Calcium 1998; 24:1 - 8; http://dx.doi.org/10.1016/S0143-4160(98)90083-5; PMID: 9793683
- Petersen OH, Burdakov D, Tepikin AV. Regulation of store-operated calcium entry: lessons from a polarized cell. Eur J Cell Biol 1999; 78:221 - 3; http://dx.doi.org/10.1016/S0171-9335(99)80054-5; PMID: 10350209
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011; 476:341 - 5; http://dx.doi.org/10.1038/nature10234; PMID: 21685886
- De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011; 476:336 - 40; http://dx.doi.org/10.1038/nature10230; PMID: 21685888
- Collins TJ, Lipp P, Berridge MJ, Bootman MD. Mitochondrial Ca(2+) uptake depends on the spatial and temporal profile of cytosolic Ca(2+) signals. J Biol Chem 2001; 276:26411 - 20; http://dx.doi.org/10.1074/jbc.M101101200; PMID: 11333261
- Colegrove SL, Albrecht MA, Friel DD. Quantitative analysis of mitochondrial Ca2+ uptake and release pathways in sympathetic neurons. Reconstruction of the recovery after depolarization-evoked [Ca2+]i elevations. J Gen Physiol 2000; 115:371 - 88; http://dx.doi.org/10.1085/jgp.115.3.371; PMID: 10694264
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 2005; 280:26185 - 92; http://dx.doi.org/10.1074/jbc.M503062200; PMID: 15899901
- Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, Maechler P, et al. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One 2008; 3:e3257; http://dx.doi.org/10.1371/journal.pone.0003257; PMID: 18806874
- Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci 2001; 114:867 - 74; PMID: 11181170
- Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J 2006; 25:2966 - 77; http://dx.doi.org/10.1038/sj.emboj.7601184; PMID: 16778770
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 2001; 12:2245 - 56; PMID: 11514614
- James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 2003; 278:36373 - 9; http://dx.doi.org/10.1074/jbc.M303758200; PMID: 12783892
- Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 2008; 19:2402 - 12; http://dx.doi.org/10.1091/mbc.E07-12-1287; PMID: 18353969
- Palmer CS, Osellame LD, Stojanovski D, Ryan MT. The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cell Signal 2011; 23:1534 - 45; http://dx.doi.org/10.1016/j.cellsig.2011.05.021; PMID: 21683788
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998; 280:1763 - 6; http://dx.doi.org/10.1126/science.280.5370.1763; PMID: 9624056
- Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 2006; 175:901 - 11; http://dx.doi.org/10.1083/jcb.200608073; PMID: 17178908
- Drago I, Pizzo P, Pozzan T. After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J 2011; 30:4119 - 25; http://dx.doi.org/10.1038/emboj.2011.337; PMID: 21934651
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008; 456:605 - 10; http://dx.doi.org/10.1038/nature07534; PMID: 19052620
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A 2008; 105:20728 - 33; http://dx.doi.org/10.1073/pnas.0808953105; PMID: 19098100
- Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A 2008; 105:15803 - 8; http://dx.doi.org/10.1073/pnas.0808249105; PMID: 18838687
- Han X-J, Lu Y-F, Li S-A, Kaitsuka T, Sato Y, Tomizawa K, et al. CaM kinase I α-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol 2008; 182:573 - 85; http://dx.doi.org/10.1083/jcb.200802164; PMID: 18695047
- Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, et al. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol 2005; 15:2112 - 8; http://dx.doi.org/10.1016/j.cub.2005.10.041; PMID: 16332536
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008; 27:433 - 46; http://dx.doi.org/10.1038/sj.emboj.7601963; PMID: 18200046
- Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta 2008; 1777:860 - 6; http://dx.doi.org/10.1016/j.bbabio.2008.05.442; PMID: 18515060
- Kanki T, Wang K, Baba M, Bartholomew CR, Lynch-Day MA, Du Z, et al. A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol Biol Cell 2009; 20:4730 - 8; http://dx.doi.org/10.1091/mbc.E09-03-0225; PMID: 19793921
- Mendl N, Occhipinti A, Müller M, Wild P, Dikic I, Reichert AS. Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2.. J Cell Sci 2011; 124:1339 - 50; http://dx.doi.org/10.1242/jcs.076406; PMID: 21429936
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8:e1000298; http://dx.doi.org/10.1371/journal.pbio.1000298; PMID: 20126261
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011; 147:893 - 906; http://dx.doi.org/10.1016/j.cell.2011.10.018; PMID: 22078885
- Kornmann B, Osman C, Walter P. The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc Natl Acad Sci U S A 2011; 108:14151 - 6; http://dx.doi.org/10.1073/pnas.1111314108; PMID: 21825164
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science 2011; 334:358 - 62; http://dx.doi.org/10.1126/science.1207385; PMID: 21885730
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013; 495:389 - 93; http://dx.doi.org/10.1038/nature11910; PMID: 23455425
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11:621 - 32; http://dx.doi.org/10.1038/nrm2952; PMID: 20683470
- Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 2001; 15:2286 - 7; PMID: 11511528
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87:99 - 163; http://dx.doi.org/10.1152/physrev.00013.2006; PMID: 17237344
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795 - 803; http://dx.doi.org/10.1083/jcb.200809125; PMID: 19029340
- Rakovic A, Grünewald A, Kottwitz J, Brüggemann N, Pramstaller PP, Lohmann K, et al. Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS One 2011; 6:e16746; http://dx.doi.org/10.1371/journal.pone.0016746; PMID: 21408142
- Vives-Bauza C, de Vries RL, Tocilescu M, Przedborski S. PINK1/Parkin direct mitochondria to autophagy. Autophagy 2010; 6:315 - 6; http://dx.doi.org/10.4161/auto.6.2.11199; PMID: 20200476
- Gautier CA, Giaime E, Caballero E, Núñez L, Song Z, Chan D, et al. Regulation of mitochondrial permeability transition pore by PINK1. Mol Neurodegener 2012; 7:22; http://dx.doi.org/10.1186/1750-1326-7-22; PMID: 22630785
- Mills RD, Sim CH, Mok SS, Mulhern TD, Culvenor JG, Cheng HC. Biochemical aspects of the neuroprotective mechanism of PTEN-induced kinase-1 (PINK1). J Neurochem 2008; 105:18 - 33; http://dx.doi.org/10.1111/j.1471-4159.2008.05249.x; PMID: 18221368
- Springer W, Kahle PJ. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 2011; 7:266 - 78; http://dx.doi.org/10.4161/auto.7.3.14348; PMID: 21187721
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013; 20:31 - 42; http://dx.doi.org/10.1038/cdd.2012.81; PMID: 22743996
- Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol 2011; 23:476 - 82; http://dx.doi.org/10.1016/j.ceb.2011.05.007; PMID: 21705204
- Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci 2012; 125:795 - 9; http://dx.doi.org/10.1242/jcs.093849; PMID: 22448035
- Priault M, Salin B, Schaeffer J, Vallette FM, di Rago JP, Martinou JC. Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ 2005; 12:1613 - 21; http://dx.doi.org/10.1038/sj.cdd.4401697; PMID: 15947785
- Nowikovsky K, Reipert S, Devenish RJ, Schweyen RJ. Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ 2007; 14:1647 - 56; http://dx.doi.org/10.1038/sj.cdd.4402167; PMID: 17541427
- Kissová I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem 2004; 279:39068 - 74; http://dx.doi.org/10.1074/jbc.M406960200; PMID: 15247238
- Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, et al. Mitochondria-ros crosstalk in the control of cell death and aging. J Signal Transduct 2012; 2012:329635; http://dx.doi.org/10.1155/2012/329635; PMID: 22175013
- Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab 2009; 20:332 - 40; http://dx.doi.org/10.1016/j.tem.2009.04.001; PMID: 19733481
- Liu Z, Lenardo MJ. Reactive oxygen species regulate autophagy through redox-sensitive proteases. Dev Cell 2007; 12:484 - 5; http://dx.doi.org/10.1016/j.devcel.2007.03.016; PMID: 17419989
- Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, et al. Mutant A53T α-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 2011; 286:10814 - 24; http://dx.doi.org/10.1074/jbc.M110.132514; PMID: 21252228
- Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012; 8:1462 - 76; http://dx.doi.org/10.4161/auto.21211; PMID: 22889933
- Yang JY, Yang WY. Spatiotemporally controlled initiation of Parkin-mediated mitophagy within single cells. Autophagy 2011; 7:1230 - 8; http://dx.doi.org/10.4161/auto.7.10.16626; PMID: 22011618
- Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, et al. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 2012; 1823:2297 - 310; http://dx.doi.org/10.1016/j.bbamcr.2012.08.007; PMID: 22917578
- Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T, et al. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J Biol Chem 2012; 287:3265 - 72; http://dx.doi.org/10.1074/jbc.M111.280156; PMID: 22157017
- Kondo-Okamoto N, Noda NN, Suzuki SW, Nakatogawa H, Takahashi I, Matsunami M, et al. Autophagy-related protein 32 acts as autophagic degron and directly initiates mitophagy. J Biol Chem 2012; 287:10631 - 8; http://dx.doi.org/10.1074/jbc.M111.299917; PMID: 22308029
- Bhatia-Kiššová I, Camougrand N. Mitophagy in yeast: actors and physiological roles. FEMS Yeast Res 2010; 10:1023 - 34; http://dx.doi.org/10.1111/j.1567-1364.2010.00659.x; PMID: 20629757
- Marobbio CM, Pisano I, Porcelli V, Lasorsa FM, Palmieri L. Rapamycin reduces oxidative stress in frataxin-deficient yeast cells. Mitochondrion 2012; 12:156 - 61; http://dx.doi.org/10.1016/j.mito.2011.07.001; PMID: 21782979
- Takahashi T, Yamashita H, Nakamura T, Nagano Y, Nakamura S. Tyrosine 125 of α-synuclein plays a critical role for dimerization following nitrative stress. Brain Res 2002; 938:73 - 80; http://dx.doi.org/10.1016/S0006-8993(02)02498-8; PMID: 12031537
- Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A 2004; 101:10810 - 4; http://dx.doi.org/10.1073/pnas.0404161101; PMID: 15252205
- Csordás G, Hajnóczky G. SR/ER-mitochondrial local communication: calcium and ROS. Biochim Biophys Acta 2009; 1787:1352 - 62; http://dx.doi.org/10.1016/j.bbabio.2009.06.004; PMID: 19527680
- Santos D, Cardoso SM. Mitochondrial dynamics and neuronal fate in Parkinson’s disease. Mitochondrion 2012; 12:428 - 37; http://dx.doi.org/10.1016/j.mito.2012.05.002; PMID: 22609323
- Shtifman A, Zhong N, Lopez JR, Shen J, Xu J. Altered Ca2+ homeostasis in the skeletal muscle of DJ-1 null mice. Neurobiol Aging 2011; 32:125 - 32; http://dx.doi.org/10.1016/j.neurobiolaging.2009.07.010; PMID: 19683835
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010; 468:696 - 700; http://dx.doi.org/10.1038/nature09536; PMID: 21068725
- Gao H, Yang W, Qi Z, Lu L, Duan C, Zhao C, et al. DJ-1 protects dopaminergic neurons against rotenone-induced apoptosis by enhancing ERK-dependent mitophagy. J Mol Biol 2012; 423:232 - 48; http://dx.doi.org/10.1016/j.jmb.2012.06.034; PMID: 22898350
- Joselin AP, Hewitt SJ, Callaghan SM, Kim RH, Chung YH, Mak TW, et al. ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum Mol Genet 2012; 21:4888 - 903; http://dx.doi.org/10.1093/hmg/dds325; PMID: 22872702
- Viola HM, Hool LC. Cross-talk between L-type Ca2+ channels and mitochondria. Clin Exp Pharmacol Physiol 2010; 37:229 - 35; http://dx.doi.org/10.1111/j.1440-1681.2009.05277.x; PMID: 19671062
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 2008; 9:505 - 18; http://dx.doi.org/10.1038/nrn2417; PMID: 18568013
- Calì T, Ottolini D, Brini M. Mitochondrial Ca(2+) and neurodegeneration. Cell Calcium 2012; 52:73 - 85; http://dx.doi.org/10.1016/j.ceca.2012.04.015; PMID: 22608276
- Decuypere JP, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB. The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta 2011; 1813:1003 - 13; http://dx.doi.org/10.1016/j.bbamcr.2010.11.023; PMID: 21146562
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 2012; 13:566 - 78; http://dx.doi.org/10.1038/nrm3412; PMID: 22850819
- Smaili SS, Pereira GJ, Costa MM, Rocha KK, Rodrigues L, do Carmo LG, et al. The role of calcium stores in apoptosis and autophagy. Curr Mol Med 2013; 13:252 - 65; http://dx.doi.org/10.2174/156652413804810772; PMID: 23228221
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 2009; 33:627 - 38; http://dx.doi.org/10.1016/j.molcel.2009.02.013; PMID: 19285945
- Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, et al. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J Neurochem 2009; 108:1561 - 74; http://dx.doi.org/10.1111/j.1471-4159.2009.05932.x; PMID: 19166511
- Gastaldello A, Callaghan H, Gami P, Campanella M. Ca 2+ -dependent autophagy is enhanced by the pharmacological agent PK11195. Autophagy 2010; 6:607 - 13; http://dx.doi.org/10.4161/auto.6.5.11964; PMID: 20431351
- Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 2009; 16:1006 - 17; http://dx.doi.org/10.1038/cdd.2009.34; PMID: 19325567
- Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, et al. Mitochondrial Ca(2+) and apoptosis. Cell Calcium 2012; 52:36 - 43; http://dx.doi.org/10.1016/j.ceca.2012.02.008; PMID: 22480931
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927 - 39; http://dx.doi.org/10.1016/j.cell.2005.07.002; PMID: 16179260
- Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, et al. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J 2011; 30:1195 - 208; http://dx.doi.org/10.1038/emboj.2011.49; PMID: 21358617
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 2010; 19:4861 - 70; http://dx.doi.org/10.1093/hmg/ddq419; PMID: 20871098
- Wang H, Song P, Du L, Tian W, Yue W, Liu M, et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem 2011; 286:11649 - 58; http://dx.doi.org/10.1074/jbc.M110.144238; PMID: 21292769
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119 - 31; http://dx.doi.org/10.1038/ncb2012; PMID: 20098416
- Chen D, Gao F, Li B, Wang H, Xu Y, Zhu C, et al. Parkin mono-ubiquitinates Bcl-2 and regulates autophagy. J Biol Chem 2010; 285:38214 - 23; http://dx.doi.org/10.1074/jbc.M110.101469; PMID: 20889974
- Calì T, Ottolini D, Negro A, Brini M. α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem 2012; 287:17914 - 29; http://dx.doi.org/10.1074/jbc.M111.302794; PMID: 22453917
- Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int J Biochem Cell Biol 2009; 41:2015 - 24; http://dx.doi.org/10.1016/j.biocel.2009.05.008; PMID: 19460457
- Quinsay MN, Thomas RL, Lee Y, Gustafsson AB. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010; 6:855 - 62; http://dx.doi.org/10.4161/auto.6.7.13005; PMID: 20668412