Abstract
A glance through Autophagy or any other journal in this field shows that it is very common to block autophagy by RNA interference-based knockdown of ATG mRNAs in mammalian cell lines. Our lab’s experience is that this approach can easily make for failed experiments because good knockdown of even essential autophagy regulators does not necessarily mean you will get good inhibition of autophagy, and, over time, cells can find ways to circumvent the inhibitory effects of the knockdown.
Keywords: :
demonstrates the problem associated with knockdowns. We have shRNAs that work very well to knock down essential autophagy regulators such as ATG5, which is required for LC3-II formation.Citation1 After knockdown of ATG5, autophagosome formation should be blocked and we should be able to see the effects on basal autophagy by monitoring increased LC3-II levels after blocking flux with chloroquine (CQ).Citation2 shows examples from an experiment in which the same batch of HeLa cells were infected with a lentivirus expressing a nonsilencing control or a shRNA that targets ATG5. One day after viral infection, cells were treated with puromycin to kill off any uninfected cells. Two days later, the cells were replated into fresh medium and this population of cells was grown and maintained for one month. The cells were collected at different times from 5 d to 32 d after infection and analyzed for inhibition of basal autophagic flux by treatment with CQ for 6 h. It is clear that although passage 1 cells at 5 d after initial infection already knocked down ATG5 very effectively (as determined by the level of the ATG12–ATG5 protein conjugate), there was little inhibition of basal autophagic flux in these cells as shown by the accumulation of LC3-II with 6 h of CQ treatment. By passages 2 and 3 at 11 and 15 d postinfection, there is much less detectable LC3-II accumulated following CQ treatment; that is, basal autophagy is now blocked to a significant extent. However, by 20 d, we detect LC3-II again following CQ treatment, even though at this stage there is still very good knockdown of the target. Finally, 32 d after infection, the knockdown itself no longer works as well as it once did and basal autophagy occurs in the cells at a similar level to the control cells.
Figure 1. LC3-II formation is blocked only temporarily after ATG5 knockdown. Western blot analysis is shown of HeLa cells treated for 6 h ± 20 µM chloroquine to block basal autophagic flux of PLK0.1-shRNA nonsilencing (control shRNA) compared with shRNA targeting ATG5 cultured postinfection for up to 32 d. Although the knockdown worked well at 5 d postinfection through 26 d, basal autophagy was only blocked effectively in the samples tested at 11 and 15 d after infection.
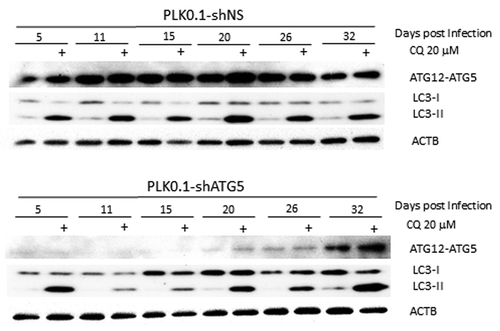
The important points are as follows: First, effective inhibition of autophagic flux is not necessarily coincident with knockdown of the ATG protein. In this case it took longer to see an effect on autophagy than it did to see an effect on the ATG5 protein levels. Second, meaningful inhibition of autophagy is maintained for a shorter period of time than effective knockdown of the autophagy regulator protein. Third, there seems to be selection against the continuous knockdown of the autophagy regulator; eventually the cells re-express the targeted ATG5 protein. Based on similar experiences with other knockdowns we have formulated recommendations that we try to use in our lab for studies using ATG-targeting shRNAs. These recommendations can be summarized as follows:
1. Do not assume that even good knockdown of an autophagy regulator means that autophagy is inhibited.
2. Do not assume that just because the knockdown blocked autophagy last week, the same cells will still be deficient in autophagy this week. Selection against autophagy inhibition may occur, which may be manifested both by re-expression of the protein, or by the cells acquiring the ability to undergo autophagy even with significantly reduced levels of the targeted protein.
3. If possible, test if autophagy has been inhibited for each preparation of cells that is being used for an experiment; obviously, it is better to do this before you spend a lot of time, money and effort on the experiment.
4. After a few weeks, cells expressing ATG-targeting shRNAs should be discarded and a new preparation prepared (or use another aliquot of the original stock) for repeated or follow-up experiments. We think it is usually better to not maintain ATG knockdown cell lines for extended periods of time.
5. Single clones expressing autophagy-inhibiting shRNAs, which will usually take a long time to grow up, may be less useful than mixed clones.
6. LC3 westerns are only suitable for monitoring inhibition of autophagy for some knockdowns. For example, PIK3C3/VPS34 loss does not prevent LC3-II accumulation.Citation3 Indeed LC3-II is elevated in unstimulated PIK3C3-deficient cells due to lack of LC3-II turnover by autophagy. This means that the assay used to test for autophagy inhibition could be different depending upon the knockdown.
7. Because many ATG proteins have other functions in addition to their role in autophagy, it is always prudent to verify the autophagy dependence of a phenotype by knocking down several different ATG genes in accordance with the current guidelines.Citation2 Likewise, if possible, it is also good to reproduce a phenotype using different autophagy-inhibiting shRNAs to the same gene to avoid potential off-target effects.
8. It follows from the above points that if an ATG knockdown is having a consistent effect on the phenotype being studied, but has little effect on autophagy in some passages of the cells, this might actually suggest that the phenotype being affected is not regulated by autophagy.
Ultimately, the problem of selection against ATG knockdown in cell lines reflects the fact that autophagy is critical for the optimal growth and survival of many mammalian cells. Thus, cells that are deficient in autophagy may have a disadvantage that can be selected against. Our experience with multiple cancer cell lines and different shRNAs is that this selection can occur within just a few days. The danger is that time and effort may be wasted doing experiments on cells that do not have autophagy inhibited to a significant extent, even though they maybe did have effective inhibition at one point and may even still have good knockdown of the protein that was targeted. On the one hand, in some cases this issue may manifest itself by an “effect” going away as cells are maintained in culture. On the other hand, if you still see a biological effect of ATG knockdown but are no longer seeing inhibition of autophagy, it would be wise to consider the possibility that in fact whatever you are measuring is not regulated by autophagy (see point 8 above).
Prominent investigators have made recommendations like this before (e.g., see ref. Citation4), however, it has perhaps not been so clear as we see in just how variable even the same ATG knockdown in the same preparation of cells can be at blocking autophagy at different times. We think the moral is: even when you are working with good shRNAs, take the time to remake your knockdown cells at regular intervals. In addition, most importantly, check that the knockdowns really had inhibited autophagy and not just that they reduced the level of the protein that was targeted.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657 - 68; http://dx.doi.org/10.1083/jcb.152.4.657; PMID: 11266458
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Jaber N, Dou Z, Chen J-S, Catanzaro J, Jiang Y-P, Ballou LM, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci USA 2012; 109:2003 - 8; http://dx.doi.org/10.1073/pnas.1112848109; PMID: 22308354
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757