Abstract
Eukaryotes have two major intracellular protein degradation pathways, namely the ubiquitin-proteasome system (UPS) and autophagy. Inhibition of proteasomal activities has been previously shown to induce autophagy, indicating a coordinated and complementary relationship between these two systems. However, little is known about the regulation of the UPS by autophagy. In this study, we showed for the first time that proteasomes were activated in response to pharmacological inhibition of autophagy as well as disruption of autophagy-related genes by RNA interference under nutrient-deficient conditions in cultured human colon cancer cells. The induction was evidenced by the increased proteasomal activities and the upregulation of proteasomal subunits, including the proteasome β5 subunit, PSMB5. Co-inhibition of the proteasome and autophagy also synergistically increased the accumulation of polyubiquitinated proteins. Collectively, our findings suggest that proteasomes are activated in a compensatory manner for protein degradation upon autophagy inhibition. Our studies unveiled a novel regulatory mechanism between the two protein degradation pathways.
Introduction
The ubiquitin–proteasome system and macroautophagy (hereafter referred to as autophagy) are two major intracellular pathways for protein degradation. Degradation of short-lived proteins through the UPS is initiated by the addition of multiple ubiquitin molecules to target proteins catalyzed by E1-, E2-, and E3-ubiquitinating enzymes. The polyubiquitin-tagged proteins are then recognized and processed by the 26S proteasome, a multicatalytic protease consisting of two major subcomplexes: a 20S core catalytic component that carries multiple proteolytic sites and a 19S regulatory component, which acts as a gate to allow entry of the substrate into the 20S chamber.Citation1 In contrast, autophagy degrades long-lived proteins and damaged organelles. Autophagy, which literally means “to eat oneself,” is characterized by the formation of double-membrane structures known as autophagosomes, which sequester cytosolic proteins and organelles. The cargo-loaded autophagosomes then fuse with lysosomes to form autolysosomes in which the engulfed contents are digested by acidic lysosomal hydrolases.Citation2 Autophagosomes may also selectively sequester polyubiquitinated protein aggregates through the autophagosomal membrane receptors SQSTM1 and NBR1.Citation3,Citation4 To this end, genetic ablation of autophagy results in the accumulation of polyubiquitinated proteins.Citation3
Extensive evidence has shown that there is a connection between the two protein degradation pathways. In this regard, autophagy complements the UPS for the degradation of polyubiquitinated proteins.Citation3–Citation7 Proteasomal inhibition also induces autophagy in various physiological contexts.Citation5–Citation7 For instance, in the central nervous system, autophagy acts as a compensatory protein degradation pathway in response to UPS impairment to retard neurodegeneration.Citation8 The regulation of autophagy by the UPS involves multiple intracellular mediators or cascades, including histone deacetylase 6 (HDAC6), mechanistic target of rapamycin complex 1 (MTORC1), activating transcription factor 4 (ATF4) and the endoplasmic reticulum to nucleus signaling 1 (IRE1)-mitogen-activated protein kinase 8 (MAPK8/JNK1) pathway.Citation2
To date, the connection between the two systems, from autophagy to UPS, remains elusive, as direct evidence for the regulation of the UPS by autophagy is lacking. For instance, in neurons of autophagy-null mice, autophagy deficiency induces the accumulation of polyubiquitinated proteins, but no obvious alteration in proteolytic activities.Citation9,Citation10 In neuroblastoma SHSY56 cells, inhibition of lysosomal activities enhances chaperone-mediated autophagy instead of upregulating proteasomal functions.Citation11 Furthermore, Korolchuk et al. showed that excess SQSTM1 caused by autophagy inhibition could delay the delivery of polyubiquitinated proteins to the proteasome without affecting proteasomal activity and thereby inhibiting their clearance.Citation12 Nevertheless, whether direct regulation of proteasomal activity by autophagy independent of substrate delivery could occur in other cellular contexts remains unclear. Since both the UPS and autophagy are required for protein homeostasis and cell survival,Citation13,Citation14 we speculate that the regulation between the two systems could be reciprocal. In addition to degradation of polyubiquitinated substrates, the proteasome is involved in ubiquitin-independent protein degradation (e.g., CDKN1A/p21, CCND1/cyclin D1) and processing (e.g., cleavage of NFKB1 from p105 to p50).Citation15–Citation17 We therefore hypothesize that autophagy inhibition could increase proteasomal activity and thereby enhance the degradation or processing of ubiquitin-independent proteasomal substrates. We previously demonstrated that proteasomal inhibition induces autophagy in human colon cancer cells,Citation18 suggesting that these cells are sensitive to cellular stress associated with the inhibition of protein degradation. In particular, recent studies report that autophagy is activated in colon cancer cells and confer them tolerance to metabolic stress.Citation19 To test our hypothesis, we sought to determine if inhibition of autophagy could induce proteasomal activity in human colon cancer cells.
Results
Chloroquine prevented autophagy at the late stage in SW1116 and HCT116 colon cancer cells
Chloroquine, a pharmacological inhibitor that interferes with lysosomal function,Citation13 was used to prevent autophagy in two colon cancer cell lines SW1116 and HCT116. To assess the efficacy of chloroquine, the level of LC3B-II was determined in these two cell lines. LC3B-II is an autophagosomal surface protein marker that is ultimately degraded by acidic hydrolases after the formation of autolysosomes.Citation20,Citation21 western blots showed that the LC3B-II level was dramatically increased by chloroquine treatment in serum-deprived SW1116 and HCT116 cells in a dose-dependent manner, indicating lysosomal degradation of autophagosomal LC3B-II was impaired. In contrast, the accumulation of LC3B-II was not evident in serum-replete conditions (). Similarly, the accumulation of SQSTM1, as an indicator of reduced autophagic flux,Citation21 was only observed in serum-deprived but not serum-replete SW1116 and HCT116 cells (). These findings suggest that chloroquine treatment could effectively inhibit autophagy in colon cancer cells in which the autophagic flux was enhanced by serum deprivation as evidenced by the accumulation of LC3B-II and SQSTM1.
Figure 1. Pharmacological inhibition of autophagy by chloroquine in colon cancer cells induced proteasomal activity and subunit expression. (A) Inhibition of autophagy at the late stage was confirmed in serum-deprived SW1116 and HCT116 cells treated with chloroquine (CQ) for 48 h as evidenced by the accumulation of LC3B-II and SQSTM1. Serum-deprived cells were maintained in serum-free medium throughout CQ treatment without prior serum starvation. (B) Treating SW1116 and HCT116 cells with CQ for 48 h in serum-free condition induced the caspase-like (nLPnLD), trypsin-like (LRR), and chymotrypsin-like (LLVY) activities of the proteasome. (C) CQ (48 h) increased the mRNA expression of various proteasomal subunits. (D) The induction of PSMB5 at the protein level by CQ (48 h) was confirmed by western blots. Results were averaged and blots were representative of 3 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.005 significantly different from respective control group.
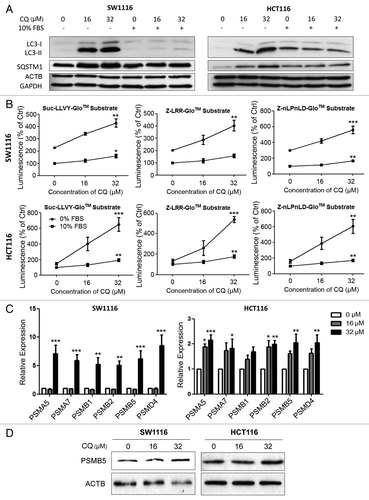
Chloroquine induced proteasomal activity and subunit expression in serum-deprived SW1116 and HCT116 colon cancer cells
The proteasome has three distinct proteolytic activities, namely, caspase-like, trypsin-like and chymotrypsin-like activities, which can be attributed to the β1 (PSMB1), β2 (PSMB2), and β5 (PSMB5) subunits of the 20S core structure of the proteasome in mammalian cells.Citation13 In this study, the caspase-like, trypsin-like and chymotrypsin-like activities of the proteasome were assayed by a chemiluminescence-based method while the expression of proteasomal subunits was measured by real-time PCR. Pharmacological inhibition of autophagy by chloroquine induced all three subtypes of proteasomal proteolytic activities in serum-deprived SW1116 and HCT116 cells (). Increases in proteasomal activities were prominent, approximately 2-fold in SW1116 cells and 4-fold in HCT116. Concordantly, cells cultured in serum-replete conditions showed only a minimal response to chloroquine treatment and had lower proteasomal activity when compared with serum-depleted cells (). Chloroquine also induced the mRNA expression of the genes corresponding to multiple proteasomal subunits, including PSMA5, PSMA7, PSMB1, PSMB2, PSMB5 and PSMD4 in serum-deprived SW1116 and HCT116 cells (). To this end, the upregulation of PSMB5 at the protein level was confirmed by western blot ().
Knockdown of PIK3C3, ATG5 or ATG7 prevented autophagy in the early stage in SW1116 cells
Since lysosomotropic agents may not only inhibit autophagy but also interfere with other lysosome-dependent cellular processes, including endosomal transport and nonautophagic lysosome-dependent cell death, an RNA interference approach that targets nonlysosomal components of autophagy was used to knock down proteins (e.g., PIK3C3, ATG5, ATG7) central to the autophagic machinery.Citation13 As shown in , the knockdown efficacy of PIK3C3-, ATG5-, and ATG7-siRNAs was confirmed by western blot. The efficacy of these siRNAs on autophagy inhibition was also determined by western blots for LC3B-II and SQSTM1. Unlike the blockade of autophagy at the late stage by chloroquine, knockdown of PIK3C3, ATG5, or ATG7 inhibited the conversion of LC3B-I to LC3B-II induced by serum deprivation in SW1116 cells (). Using LC3B-II as a readout of autophagy, our finding is in line with previous reports that the cellular LC3B-II level is decreased when autophagy is intervened at an early stage because of reduced formation of autophagosomes.Citation21 In contrast, the LC3B-II level is increased when autophagy is inhibited at the late stage owing to impaired lysosomal degradation of autophagosomal proteins.Citation22 In this regard, serum deprivation enhanced the autophagic degradation of SQSTM1 in SW1116 cells whereas PIK3C3, ATG5, or ATG7 knockdown prevented the reduction of SQSTM1 caused by serum deprivation (). These findings collectively suggested that knockdown of PIK3C3, ATG5, or ATG7 effectively inhibited the induction of autophagy caused by serum deprivation in SW1116 cells.
Figure 2. Abrogation of autophagy by siRNAs targeting autophagy-related genes in SW1116 colon cancer cells induced proteasomal activity and subunit expression. (A) The knockdown efficacies of PIK3C3-, ATG5-, and ATG7-siRNAs were verified by western blots at 48 h post-transfection. (B) Inhibition of serum deprivation-induced autophagy by PIK3C3-, ATG5-, and ATG7-siRNAs was confirmed in SW1116 cells. Targeting PIK3C3, ATG5, or ATG7 by RNA interference reduced the conversion of LC3B-I to LC3B-II and the accumulation of SQSTM1 caused by serum deprivation at 48 h post-transfection. (C) Inhibition of autophagy by knockdown of PIK3C3, ATG5, or ATG7 in serum-deprived conditions induced the proteasomal activity in SW1116 cells. Proteasomal activity assays were performed 72 h post-transfection. (D) Knockdown of PIK3C3, ATG5 or ATG7 in serum-deprived conditions induced the mRNA expression of proteasomal subunits to various extents in SW1116 cells at 72 h post-transfection. (E) The induction of PSMB5 at the protein level was confirmed by western blots at 72 h post-transfection. Concordant with the real-time PCR results, the induction was more prominent in serum-deprived SW1116 cells transfected with PIK3C3- and ATG7-siRNAs. Results were averaged and blots were representative of 3 independent experiments. The data from (E) are presented in a graph below the western blot panels. *P < 0.05; **P < 0.01, significantly different from respective control group.
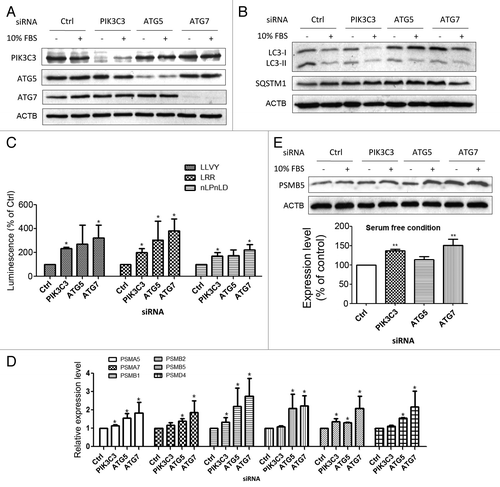
Knockdown of PIK3C3, ATG5, or ATG7 enhanced proteasomal activity and subunit expression in serum-deprived SW1116 cells
To assess if the inhibition of autophagy at the early stage could induce proteasomal activity, caspase-like, trypsin-like and chymotrypsin-like activities of the proteasome were determined in PIK3C3-, ATG5-, and ATG7-depleted SW1116 cells. As shown in , knockdown of PIK3C3, ATG5, or ATG7 upregulated all three types of proteasomal activity. The effect of ATG7 knockdown on the induction of proteasomal activity was more prominent than that of ATG5 or PIK3C3. Similar to the effect of chloroquine, siRNA-mediated inhibition of autophagy increased the mRNA expression of PSMA5, PSMA7, PSMB1, PSMB2, PSMB5, and PSMD4 to various extents in serum-deprived SW1116 cells (). To this end, the induction of PSMB5 at the protein level by knockdown of PIK3C3 and ATG7 in serum-deprived conditions was confirmed by western blot (). A modest but not statistically significant increase in PSMB5 protein level was also observed in ATG5-siRNA-transfected cells. These findings suggest that inhibition of autophagy by knockdown of autophagy-related genes could increase proteasomal activity probably through inducing the expression of multiple proteasomal subunits.
Blockade of proteasomal activity enhanced autophagy inhibition-induced accumulation of polyubiquitin proteins
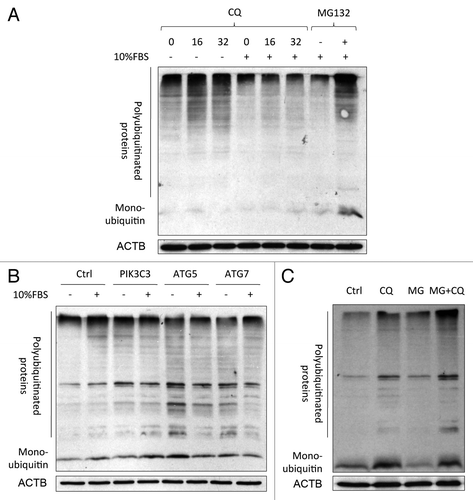
Both the UPS and autophagy have been reported to share common degradation substrates (i.e., polyubiquitinated proteins).Citation4,Citation23,Citation24 Here, we examined if blockade of the proteasome could exaggerate the accumulation of polyubiquitinated proteins upon autophagy inhibition. Autophagy inhibition by chloroquine () or RNA interference () per se substantially increased intracellular levels of polyubiquitinated proteins in serum-deprived SW1116 cells. Concordantly, the accumulation of polyubiquitinated proteins was less severe under serum-replete conditions. Treating cells with the proteasome inhibitor MG132 for 3 h potentiated the accumulation of polyubiquitinated proteins caused by chloroquine, while MG132 by itself had no effect under this condition (). These findings suggest that the induced proteasomal activity secondary to autophagy inhibition was required for efficient removal of polyubiquitinated proteins.
Figure 3. Accumulation of polyubiquitinated proteins caused by co-inhibition of autophagy and the proteasome. (A) Chloroquine (CQ; 48 h) induced the accumulation of polyubiquitinated proteins in SW1116 cells. The accumulation was more prominent in serum-deprived conditions. Cells were treated with MG132 (0.8 μM) for 48 h as a positive control. (B) Knockdown of autophagy-related genes (PIK3C3, ATG5, and ATG7) caused the accumulation of polyubiquitinated proteins at 72 h post-transfection in serum-deprived SW1116 cells. (C) SW1116 cells were treated with or without CQ (16 μM) for 48 h in the absence of presence of MG132 (0.8 μM) for an additional 3 h, and the amount of polyubiquitinated proteins was examined by western blot. Blots were representative of 3 independent experiments.
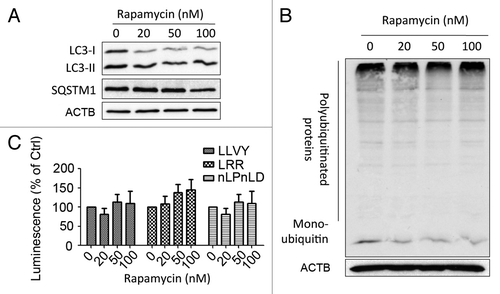
Induction of autophagy by rapamycin did not reduce proteasomal activity
The data presented so far indicated that an unreported regulation exists between the UPS and autophagy in which blockade of autophagy enhanced proteasomal activity. To assess if induction of autophagy could in reverse reduce proteasomal activity, SW1116 cells were treated with increasing concentrations of rapamycin (a known inducer of autophagy). Upon treatment, the LC3-II to LC3-I ratio increased while SQSTM1 decreased, indicating an increased autophagic flux (). Concordantly, the accumulation of polyubiquitinated proteins showed moderate reduction (). Nevertheless, it was found that rapamycin did not show any effect on 3 proteolytic activities of the proteasome ().
Figure 4. Effects of rapamycin-induced autophagy on proteasomal activity in SW1116 cells. (A) The induction of autophagy by rapamycin was evidenced by an increased LC3B-II to LC3B-I ratio and reduction of SQSTM1. Cells were treated with the indicated concentrations of rapamycin for 48 h. (B) Rapamycin (48 h) moderately reduced the accumulation of polyubiquitinated proteins. (C) Rapamycin (48 h) did not alter three proteolytic activities of the proteasome. Results were averaged and blots were representative of 3 independent experiments.
Increased degradation or processing of ubiquitin-independent proteasomal substrates upon autophagy inhibition
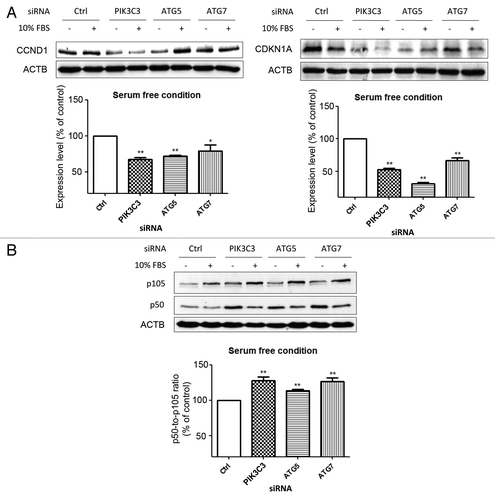
Although our data indicate that autophagy inhibition could induce proteasomal activity, SQSTM1 accumulation caused by autophagy inhibition also impairs the delivery of polyubiquitinated proteins to the proteasome. To demonstrate that there was a real increase in flux through the proteasome, we determined the levels of some proteasomal substrates that have been reported to be degraded or processed in a ubiquitin-independent manner.Citation15–Citation17 To this end, autophagy inhibition led to increased degradation of CCND1 and CDKN1A () as well as enhanced proteolytic cleavage of NFKB from p105 to p50 ().
Figure 5. Enhanced proteolytic degradation or cleavage of ubiquitin-independent proteasomal substrates upon autophagy inhibition in serum-deprived SW1116 cells. (A) The effect of knockdown of autophagy-related genes (PIK3C3, ATG5, and ATG7) on the protein levels of CCND1 and CDKN1A at 72 h post-transfection. (B) siRNA-mediated inhibition of autophagy enhanced proteasomal cleavage of NFKB from p105 to p50. *P < 0.05; **P < 0.01 significantly different from respective control group. Results were averaged and blots were representative of 3 independent experiments.
Discussion
The UPS and autophagy have long been thought of as two separate systems for protein degradation. However, emerging evidence has demonstrated that the UPS and autophagy could serve in a complementary manner to each other to mediate the degradation of polyubiquitinated proteins and the resolution of endoplasmic reticulum stress.Citation2,Citation25,Citation26 Recent studies have also shown that proteasome inhibition may induce autophagy.Citation2 Here we report for the first time that inhibition of autophagy by a pharmacological or RNA interference approach could induce proteasomal activity and the expression of proteasomal subunits, unveiling the unreported regulation between the two systems.
The possible relationship between the UPS and autophagy has been implicated by a previous study. Cuervo et al. reported that proteasomes were probably degraded through the autophagic pathway,Citation27 pointing at the possibility that inhibition of autophagy might enhance the accumulation of proteasomes. This finding is concordant with our postulation that autophagy inhibition enhances proteasomal activity. Their findings together with ours suggest that the regulation of proteasomal activity by autophagy may be mediated through both transcriptional and post-transcriptional mechanisms. However, contradictory evidence also exists in the literature. Qiao and Zhang, and Komatsu et al. reported that autophagy abolition has no effect on neuronal proteasomal activity in vitro and in vivo.Citation9,Citation11 Such discrepancy may be reconciled by the observation that, upon nutritional stress, colon cancer cells switch the major mode of protein degradation to autophagy, in which serum starvation substantially enhances autophagic flux. To this end, autophagy inhibition only induces proteasomal activity when the autophagic flux is high. This is corroborated by our findings that autophagy inhibition had minimal effect on proteasomal activity in nutrient-replete conditions and induction of autophagy by rapamycin did not alter proteasomal activity. These data suggest that the above-mentioned regulation between the two protein degradation systems only occurs in conditions in which autophagy inhibition has caused substantial impairment of the degradation of polyubiquitinated proteins that reached a certain threshold. It is still unclear why neuronal tissue fails to respond to autophagy inhibition by upregulating proteasomal activity in previous studies,Citation9,Citation11 but further experiments using cultured neuronal cells in both serum-replete and -depleted conditions are needed to clarify the potential connection between the two systems. In addition, the regulatory mechanism between autophagy and proteasomal activity has to be elucidated in order to understand how cell type- or tissue-specific transcription factors could potentially interfere with this regulation.
Korolchuk et al. previously demonstrated that SQSTM1 accumulation caused by autophagy inhibition could compromise degradation of ubiquitin-proteasome pathway substrates through delaying their delivery to the proteasome.Citation12 Concordantly, we showed that autophagy inhibition by chloroquine or siRNAs resulted in SQSTM1 accumulation and increased levels of polyubiquitinated proteins. However, our data also revealed that transient blockade of proteasomal activity by MG132, which by itself had no effect, could synergistically increase polyubiquitinated protein levels. This finding indicates that the positive regulation of proteasomal activity by autophagy inhibition may partially offset the impairment of proteasomal flux caused by excess SQSTM1. Importantly, the induction of proteasomal activity by autophagy inhibition is corroborated by the enhanced cellular removal of ubiquitin-independent substrates (i.e., CCND1, CDKN1A) and increased proteolytic cleavage of NFKB, an action that is also independent of ubiquitin.Citation15–Citation17 These findings suggest that autophagy may have crosstalk with other signaling pathways through interplay with the proteasome in both ubiquitin-dependent and -independent manners. However, it is worthwhile to note that knockdown of PIK3C3 by itself reduced CCND1 and CDKN1A in serum-replete conditions, in which autophagy was not substantially inhibited. This finding suggests that the regulation of CCND1 and CDKN1A by PIK3C3 knockdown may be mediated through other cellular processes in addition to autophagy. In this regard, PIK3C3 takes part in intracellular vesicle trafficking independent of autophagy.Citation28
Our data highlight the existence of an intricate regulatory circuit between the two protein degradation systems. The molecular events that mediate such regulation, however, warrant further investigation. Given that autophagy has been implicated in many diseases, including cancer, infection, autoimmunity, inflammatory diseases and neurodegeneration,Citation29,Citation30 our findings not only could have a significant impact on the understanding of the reciprocal regulation of protein degradation pathways in disease development, but also possibly alter the future development of drugs that target the protein degradation systems.
Materials and Methods
Cell culture
SW1116 and HCT116 cells were obtained from American Type Culture Collection (CCL-233 and CCL-247, respectively). The cells were maintained in RPMI 1640 (Invitrogen, 23400062), supplemented with 10% fetal bovine serum (Thermo Scientific, SH30088.03), 100 U/mL penicillin and 100 g/mL streptomycin (liquid penicillin-streptomycin, Invitrogen, 15140122) at 37 °C in a humidified atmosphere of 5% CO2 and 95% air.
RNA interference
The expression of the PtdIns3K PIK3C3, ATG5, and ATG7 was lowered using target-specific siRNA molecules (PIK3C3—Hs_PI3KC3_5, SI00605822; ATG5—Hs_ATG5L_6, SI02655310; ATG7—Hs_ATG7L_5, SI02655373) purchased from Qiagen. Two hundred picomoles of gene-specific or control small interference RNA (siRNA) was transfected into cells at 40–60% confluence using LipofectamineTM 2000 reagent (Invitrogen, 11668-019) according to the manufacturer’s instructions.
Western blot analysis
Equal amounts of protein (40 μg/lane) were resolved by SDS-PAGE, and transferred to Hybond C nitrocellulose membranes (GE Healthcare, RPN203C). The membranes were probed with primary antibodies overnight at 4 °C and incubated for 1 h with secondary peroxidase-conjugated antibodies (anti-rabbit antibodies, Invitrogen, G-21234; anti-mouse antibodies, Invitrogen, 816720). Chemiluminescent signals were then developed with Lumiglo reagent (Cell Signaling Technology, 7003) and detected by the ChemiDoc XRS gel documentation system (Bio-Rad).
Reverse transcription and real-time PCR
Total RNA was isolated from colon cancer cells using TRIZOL Reagent (Life Technologies, 15596-026). Five micrograms of the total RNA was used to generate the first strand of cDNA by the Thermoscript Reverse Transcription System (Life Technologies) in accordance with the manufacturer’s instructions. Real-time PCR was performed with a specific primer set designed by the Primer 3 online tool with ACTB/β-actin as an internal control. Conditions for quantitative PCR were 94 °C for 5 min, 40 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. Quantitative PCR was performed using CyberGreener real-time PCR supermix (Invitrogen, 4309155), and Multicolor Real-Time PCR Detection System (Bio-Rad). The results were analyzed using the comparative threshold cycle (CT) method.
Proteasome activity assay
Proteasome trypsin-like, chymotrypsin-like and caspase-like activities were determined by proteasome-Glo™ assay systems (Promega, Madison, WI). Briefly, cells were harvested in potassium phosphate buffer (pH 7.4) containing 10 mM DTT, 1 mM PMSF, and 0.1 mM EDTA. After sonication for 30 s on ice and centrifugation for 15 min at 12,000 g at 4 °C, the supernatant was collected for proteasome activity assay according to the manufacturer’s instructions. Chemiluminescent signals were measured by the GloMax® microplate Luminometer (Promega). Proteasomal activities were normalized by total protein concentration.
Statistical analysis
Results were expressed as the mean ± SEM. Statistical analysis was performed with an analysis of variance (ANOVA) followed by the Tukey t-test correlation analysis. P values less than 0.05 were considered statistically significant.
| Abbreviations: | ||
| ATG | = | autophagy-related |
| UPS | = | ubiquitin-proteasome system |
Acknowledgments
Work in the authors’ laboratory is supported by Research Fund for the Control of Infectious Diseases/Health and Medical Research Fund (11100082 and 12110332) and CUHK Direct Grant for Research (2041734).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol 2005; 23:4776 - 89; http://dx.doi.org/10.1200/JCO.2005.05.081; PMID: 16034054
- Wu WK, Sakamoto KM, Milani M, Aldana-Masankgay G, Fan D, Wu K, et al. Macroautophagy modulates cellular response to proteasome inhibitors in cancer therapy. Drug Resist Updat 2010; 13:87 - 92; http://dx.doi.org/10.1016/j.drup.2010.04.003; PMID: 20462785
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505 - 16; http://dx.doi.org/10.1016/j.molcel.2009.01.020; PMID: 19250911
- Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, et al. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res 2009; 69:4415 - 23; http://dx.doi.org/10.1158/0008-5472.CAN-08-2839; PMID: 19417138
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007; 447:859 - 63; http://dx.doi.org/10.1038/nature05853; PMID: 17568747
- Wu WK, Cho CH, Lee CW, Wu YC, Yu L, Li ZJ, et al. Macroautophagy and ERK phosphorylation counteract the antiproliferative effect of proteasome inhibitor in gastric cancer cells. Autophagy 2010; 6:228 - 38; http://dx.doi.org/10.4161/auto.6.2.11042; PMID: 20087064
- Pandey UB, Batlevi Y, Baehrecke EH, Taylor JP. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy 2007; 3:643 - 5; PMID: 17912024
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/10.1038/nature04723; PMID: 16625205
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204
- Qiao L, Zhang J. Inhibition of lysosomal functions reduces proteasomal activity. Neurosci Lett 2009; 456:15 - 9; http://dx.doi.org/10.1016/j.neulet.2009.03.085; PMID: 19429125
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517 - 27; http://dx.doi.org/10.1016/j.molcel.2009.01.021; PMID: 19250912
- Wu WK, Coffelt SB, Cho CH, Wang XJ, Lee CW, Chan FK, et al. The autophagic paradox in cancer therapy. Oncogene 2012; 31:939 - 53; http://dx.doi.org/10.1038/onc.2011.295; PMID: 21765470
- Wu WK, Cho CH, Lee CW, Wu K, Fan D, Yu J, et al. Proteasome inhibition: a new therapeutic strategy to cancer treatment. Cancer Lett 2010; 293:15 - 22; http://dx.doi.org/10.1016/j.canlet.2009.12.002; PMID: 20133049
- Newman RM, Mobascher A, Mangold U, Koike C, Diah S, Schmidt M, et al. Antizyme targets cyclin D1 for degradation. A novel mechanism for cell growth repression. J Biol Chem 2004; 279:41504 - 11; http://dx.doi.org/10.1074/jbc.M407349200; PMID: 15277517
- Chen X, Chi Y, Bloecher A, Aebersold R, Clurman BE, Roberts JM. N-acetylation and ubiquitin-independent proteasomal degradation of p21(Cip1). Mol Cell 2004; 16:839 - 47; http://dx.doi.org/10.1016/j.molcel.2004.11.011; PMID: 15574338
- Moorthy AK, Savinova OV, Ho JQ, Wang VY, Vu D, Ghosh G. The 20S proteasome processes NF-kappaB1 p105 into p50 in a translation-independent manner. EMBO J 2006; 25:1945 - 56; http://dx.doi.org/10.1038/sj.emboj.7601081; PMID: 16619030
- Wu WK, Wu YC, Yu L, Li ZJ, Sung JJ, Cho CH. Induction of autophagy by proteasome inhibitor is associated with proliferative arrest in colon cancer cells. Biochem Biophys Res Commun 2008; 374:258 - 63; http://dx.doi.org/10.1016/j.bbrc.2008.07.031; PMID: 18638451
- Sato K, Tsuchihara K, Fujii S, Sugiyama M, Goya T, Atomi Y, et al. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res 2007; 67:9677 - 84; http://dx.doi.org/10.1158/0008-5472.CAN-07-1462; PMID: 17942897
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy 2007; 3:181 - 206; PMID: 17224625
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Wu YC, Wu WK, Li Y, Yu L, Li ZJ, Wong CC, et al. Inhibition of macroautophagy by bafilomycin A1 lowers proliferation and induces apoptosis in colon cancer cells. Biochem Biophys Res Commun 2009; 382:451 - 6; http://dx.doi.org/10.1016/j.bbrc.2009.03.051; PMID: 19289106
- Wenger T, Terawaki S, Camosseto V, Abdelrassoul R, Mies A, Catalan N, et al. Autophagy inhibition promotes defective neosynthesized proteins storage in ALIS, and induces redirection toward proteasome processing and MHCI-restricted presentation. Autophagy 2012; 8:350 - 63; http://dx.doi.org/10.4161/auto.18806; PMID: 22377621
- Engelender S. α-Synuclein fate: proteasome or autophagy?. Autophagy 2012; 8:418 - 20; http://dx.doi.org/10.4161/auto.19085; PMID: 22361583
- Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011; 7:1159 - 72; http://dx.doi.org/10.4161/auto.7.10.16818; PMID: 21691147
- Kardosh A, Golden EB, Pyrko P, Uddin J, Hofman FM, Chen TC, et al. Aggravated endoplasmic reticulum stress as a basis for enhanced glioblastoma cell killing by bortezomib in combination with celecoxib or its non-coxib analogue, 2,5-dimethyl-celecoxib. Cancer Res 2008; 68:843 - 51; http://dx.doi.org/10.1158/0008-5472.CAN-07-5555; PMID: 18245486
- Cuervo AM, Palmer A, Rivett AJ, Knecht E. Degradation of proteasomes by lysosomes in rat liver. Eur J Biochem 1995; 227:792 - 800; http://dx.doi.org/10.1111/j.1432-1033.1995.tb20203.x; PMID: 7867640
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013; 152:290 - 303; http://dx.doi.org/10.1016/j.cell.2012.12.016; PMID: 23332761
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323 - 35; http://dx.doi.org/10.1038/nature09782; PMID: 21248839
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538