Abstract
Although suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor, has been used in clinical trials for cancer therapies, its pharmacological effects occur through a poorly understood mechanism. Here, we report that SAHA specifically triggers autophagy and reduces cell viability via promotion of apoptosis in the late phase of glioblastoma stem cells (GSCs). Using a cell line cultured from a glioblastoma biopsy, we investigated the properties and effects of GSCs under SAHA treatment in vitro. In vivo xenograft assays revealed that SAHA effectively caused tumor growth slowdown and the induction of autophagy. SAHA was sufficient to increase formation of intracellular acidic vesicle organelles, recruitment of LC3-II to the autophagosomes, potentiation of BECN1 protein levels and reduced SQSTM1 levels. We determined that SAHA triggered autophagy through the downregulation of AKT-MTOR signaling, a major suppressive cascade of autophagy. Interestingly, upon depletion or pharmacological inhibition of autophagy, SAHA facilitates apoptosis and results in cell death at the early phase, suggesting that SAHA-induced autophagy functions probably act as a prosurvival mechanism. Furthermore, our results also indicated that the inhibition of SAHA-induced autophagy using chloroquine has synergistic effects that further increase apoptosis. Moreover, we found that a reduced dose of SAHA functioned as a potent modulator of differentiation and senescence. Taken together, our results provide a new perspective on the treatment of GSCs, indicating that SAHA is a promising agent for targeting GSCs through the induction of autophagy.
Introduction
Glioblastoma multiforme (GBM, World Health Organization grade IV glioma) is a highly aggressive primary brain tumor. More than 70% of glioblastoma patients die within two years of diagnosis.Citation1 This high mortality rate is due to rapid tumor cell proliferation and invasion and to resistance of these tumor cells to irradiation and chemotherapy.Citation2-Citation4 These malignant characteristics are caused by an abundance of glioblastoma stem cells (GSCs), which possess high potential for DNA repair and resistance to radiation- and drug-induced caspase activation and apoptosis.Citation5 Moreover, the radiation resistance of GSCs leads to enhanced free radical scavenging.Citation6 Therefore, development of novel therapeutics and regimen strategies targeting GSCs is essential for improving GBM treatment efficacy.
Induction of caspase-dependent apoptosis (type I cell death) is a major mechanism by which most chemotherapeutic drugs and radiation kill tumor cells. However, most cancer cells can trigger multiple pathways to escape from apoptosis.Citation7 For example, GBM treatment resistance is strongly correlated with the intrinsic apoptosis resistance of tumor cells, especially that which occurs in diffusely migrating tumor cells.Citation8 More recently, induction of autophagic cell death (type II cell death), one type of nonapoptotic programmed cell death, has been extensively studied as a cancer therapy. Autophagy is a classical cell survival mechanism used during periods of nutrient starvation, which supplies vital components until conditions improve.Citation9 It is a protein degradation system that involves the autophagic/lysosomal compartment. Autophagy is characterized by a cell without nuclear condensation during the process of cellular self-digestion in which cellular constituents are engulfed in double-membrane-containing vesicles called autophagosomes.Citation10 Autophagosomes then fuse with lysosomes and lyse their contents. Microtubule-associated protein 1 light chain 3 (LC3) is required for the elongation of autophagosomes. LC3 has two forms: type I is cytosolic and type II is cleaved, lipidated and membrane-bound. During autophagy, LC3-II increases due to the conversion of LC3-I. LC3-II is localized in both the inner and outer membranes of autophagosomes and is considered the most reliable marker for quantification of the level of autophagy in cells.
Autophagy may facilitate the disposal of unfolded proteins and constitutes a mechanism utilized by tumor cells to survive hypoxic, metabolic, detachment-induced or chemotherapeutic stress.Citation11 However, opposing effects of autophagy on tumor cell survival and death have been documented.Citation12 For instance, enforced overactivation of autophagic flux can lead to autophagic cell death in which massive cellular self-digestion via the autophagosomal pathway occurs beyond the point of cell survival.Citation13 Excessive self-eating through autophagy is likely to contribute to cell death through an as-yet unknown mechanism. Autophagy has been proposed to have a dual role, variously killing and protecting cells, depending on the stage of the disease or the surrounding cellular environment.Citation14 Therefore, assessing the role of autophagy in a context-dependent manner is crucial, particularly when considering if autophagy-targeting can be used during anticancer therapy.
Histone deacetylase (HDAC) overexpression inhibits the expression of tumor suppressor genes. Epigenetic modifications leading to the dysregulation of critical genes that contribute to cell proliferation, differentiation, death and/or senescence improve the prognosis of several cancers.Citation15 Pharmaceutical histone deacetylase inhibitors (HDACi) including SAHA (Vorinostat, ZolinzaTM), are used to treat epilepsy, bipolar disorder and cancers. The first drug of this type has been approved by the US. Food and Drug Administration for the treatment of cutaneous T-cell lymphoma.Citation16 The anticancer effects of SAHA have been linked to the induction of apoptosis, growth arrest, polyploidy and autophagy in cancer cells cultured as a monolayer.Citation17 The aim of the present study was to examine the role of autophagy as mediated by SAHA treatment in GSCs.
Tumor tissue is a precious source of information in assessing the biological profile of the disease in each patient. Therefore, in order to identify cancer stem cells and their roles in GBM progression and therapeutic resistance, our previous studies have shown that two different types of GSCs, including spheroids (PROM1/CD133+) and adherent (PROM1-) GSCs contained stemness features (Fig. S1).Citation4 As a tumor sphere population is heterogeneous and committed progenitor cells can originate from neurospheres, a complementary assay is required to prove that these cells are indeed stem cells. To distinguish the sensitivity of GSCs to SAHA treatment, we exposed these cell types of GSCs to SAHA to evaluate their effects on malignant gliomas.
In this study, we determined that GBM biopsy-derived spheroids and monolayer cells are sensitive to SAHA treatment. The effects of SAHA on GSC growth and autophagy were characterized in vitro and in vivo. We also found that even a reduced dose of SAHA is a potent modulator of differentiation in GSCs. This study is the first to show the overall possible cell types of GSCs.
Results
SAHA, an HDACi, specifically suppresses cell viability in undifferentiated GSCs
First, we determined that the morphologic features of GSCs were distinguishable between spheroid and monolayer cultures. As shown in , scanning electron microscope (SEM) images showed spheroid GSC cultures presenting aggregation and stereoscopic cells, while the monolayer culture presented as flat (). Although the monolayer culture system resulted in stemness loss (PROM1-negative) in GSCs (Fig. S1), the capability of tumorigenesis still remained,Citation4 a phenomenon worthy of further investigation. With this in mind we sought to further examine the behavior of GSCs in the experiments described below. Several reports have proven that HDAC inhibitors induce mitochondria-mediated apoptosis in pituitary adenoma cells and T-leukemia cells.Citation18,Citation19 In the present study, we attempted to investigate the effect of the HDAC inhibitor (SAHA) on GSCs, which may potentially lead to the development of new treatments. Based on light microscopy studies, there was obvious time-dependent cell death in two cellular types, including tumor-spheroid and adherent GSCs after treatment with 5 μM SAHA for 1 to 3 d. There was no observable effect on the numbers of differentiated GSCs (). To confirm the effect of SAHA on cellular toxicity of GSCs, the MTT assay was performed for adherent and differentiated GSCs. As shown in , the cellular viability of GSCs significantly decreased in the group receiving SAHA (5 μM) treatment when compared with the vehicle control group (p < 0.05). The effects of reduced cellular survival increased in a time-dependent manner on the second day after SAHA treatment. The growth curve of GSCs treated with 5 μM SAHA showed a prominent decrease in comparison to that of vehicle control cells. Twenty-four hours after SAHA treatment, the cell number decreased to 91% in comparison to the number of vehicle control cells. At 48 and 72 h after SAHA treatment, these were further decreased to 74% and 57%, respectively. In contrast, differentiated GSCs did not appear to be sensitive to SAHA treatment, even after 7 consecutive days of incubation, as demonstrated by lack of growth inhibition. To further investigate the anticancer activity of SAHA, a clonogenic assay was performed to determine the long-term effect of SAHA-inhibited cellular proliferation on spheroid GSCs and differentiated GSCs in vitro. As shown in and Figure S2, colony formation in the SAHA-treated spheroid GSC group was significantly inhibited when compared with the vehicle control group (p < 0.05). The colonies were counted and colony numbers are shown in . The dose-dependent growth rate of GSCs, not only in terms of colony numbers but also in terms of cell numbers, was significantly lower for cells treated with the highest dose of SAHA than for differentiated GSCs (). These results suggested that SAHA specifically inhibits cellular proliferation of GSCs. However, there were no significant cytotoxic effects on the differentiated GSCs.
Figure 1. HDAC inhibitor SAHA decreased cellular proliferation and colony formation ability of GSCs in vitro. (A) The morphology of GSCs cultured in the gelatin foam present at spheroid formation after 7 d, as observed using SEM analysis. (B) The morphology of GSCs cultured in culture dish as an adherent model system after 7 d, as observed using SEM analysis. Scale bars: 30 μm. (C) Three types of GSCs, including undifferentiated model, spheroids and adherent or differentiated model (Diff.), as described in the text, were treated with 5 μM SAHA for 1 to 3 d. Phase contrast pictures taken at 200 × 200 μm per square. (D) Models of adherent GSCs and differentiated GSCs were treated with 5 μM SAHA for 1 to 7 d and loss of viability was determined by the MTT assay. “*” indicates significant difference from vehicle control of GSCs and p < 0.05 on Student’s t-test. (E) Colony formation was drastically reduced in SAHA-treated GSCs (spheroid model) and differentiated GSCs after 48 h and continued to decrease in a dose-dependent manner. The number of colonies was counted under a dissecting microscope. The number of cells in each colony had to be larger than 50. The data include the relative colony number, and the number of cells without SAHA treatment (as vehicle control) was set at 100%. (F) Spheroid GSCs were treated with 5 μM SAHA for the indicated number of days and then harvested for protein analysis. Cell lysates were resolved in SDS-PAGE and probed with specific antibodies against cleaved PARP, CASP3 and GAPDH. (G) Spheroid GSCs (1 × 106 cells) were treated with 0, 5 and 20 μM of SAHA in the presence of the 10 nM rapamycin for 48 h. Western blot analysis was performed to detect expression of the cleaved PARP and active cleaved CASP3. (H) In some of the above experiments, cell viability of adherent GSCs was determined by MTT assay, as described in Materials and Methods. The data represent the mean and SEM of three independent experiments. *p < 0.05 compared with control using the Student's t-test. “#” indicates significantly different from 20 μM SAHA-alone treatment and p < 0.05 on Student’s t-test.
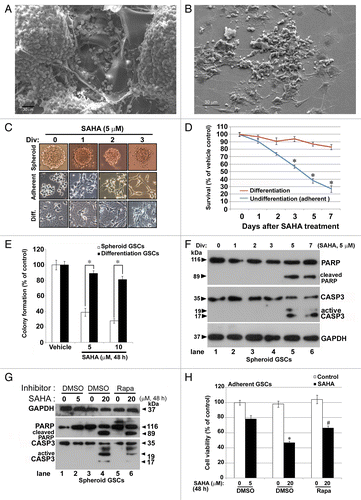
To determine the mechanism by which SAHA induces cell death, we treated GSCs with 5 μM of SAHA and assayed the expression of the apoptosis-related proteins, PARP and CASP3/caspase 3, by western blot analysis. As shown in , time-dependent treatment of GSCs with SAHA resulted in the accumulation of active CASP3 and cleaved PARP in the 5th and 7th days post-treatment, indicating that SAHA simultaneously inhibits cell viability and induces apoptosis at late phase. Under stress conditions, cells always generate some form of resistance in order to survive. To further confirm whether or not SAHA increased apoptosis, we treated spheroid GSCs with various concentrations of SAHA and assayed the expression of cleaved PARP and CASP3 by western blot. As shown in , treatment of GSCs with SAHA increased the accumulation of active CASP3 and cleaved PARP (20 μM of SAHA treatment, lane 4); additional treatment of GSCs with the autophagy activator rapamycin (Rapa) caused a partial decrease inactive CASP3 and cleaved PARP (lane 6) compared with the SAHA-alone treatment (lane 4), indicating that treatment with high concentrations of SAHA induces apoptosis. To further examine the effect of SAHA on cell viability, we treated adherent GSCs with various concentrations of SAHA and assayed using the MTT assay. Consistently, evident inhibited cell viability was only observed in GSCs treated with 20 μM of SAHA for 48 h (), whereas rapamycin reduced the SAHA-induced cytotoxicity in GSCs, indicating that SAHA is capable of inducing apoptosis via highly concentrated or prolonged treatments. Moreover, rapamycin treatment diminished the expression of active CASP3 and cleaved PARP, and increased cell viability under SAHA treatment, suggesting that autophagy is likely to play a critical role in the SAHA-induced cell death process.
SAHA suppresses tumor growth and induces autophagy in a GSC xenograft tumor model
To evaluate the anticancer effect of SAHA, an in vivo antitumor study was performed using a nude mice xenograft model subcutaneously inoculated with spheroid GSCs. The average tumor volume of treatment group (receiving SAHA at 100 mg/kg/mouse weight in saline by intraperitoneal injection, n = 3) was significantly lower than that of the control group (placebo, n = 3) on day 49. The mice were sacrificed on day 56 after treatment. Representative samples of two different tumors from each group (placebo and SAHA-treated, respectively) are shown in . The average tumor volume in untreated mice was 947 ± 152 mm3, whereas in SAHA-treated mice it was 539 ± 104 mm3. This represented a reduction of more than 35% in comparison with the placebo group.
Figure 2. SAHA caused tumor growth slowdown and induction of autophagy in nude mice xenografts of GSCs. (A) The spheroid GSCs (1 × 106 cells) were mixed with 30 μl matrigels and inoculated subcutaneously into nude mice (n = 3 for each group). When the tumors reached 200 mm3 in volume, intraperitoneal injections of SAHA (100 mg/kg body weight) were administered every 24 h for 7 d and tumor volume was measured every 7 d after the cessation of treatment (mean ± SEM). There were highly significant differences at the end (day 49 and 56) of the treatment period (p = 0.031). “*” indicates significantly different from placebo group and p < 0.05 on Student’s t-test. (B) The protein expression levels of LC3 flux, BECN1 and PARP in xenograft tumor were determined by western blotting. GAPDH was a loading control. (C) Top panels: Hematoxylin-eosin staining of a representative section. Xenograft tumor initiated from GSCs exhibited morphologic characteristics of glioma, including marked cellular pleomorphism, large, round nuclei prominent nucleoli, abundant cytoplasm and vasculogenic tubular formation (black arrowheads; placebo, left upper panel), whereas there were significant decreases in tubular structures and increases in differentiated cells in the SAHA treatment (SAHA, right upper panel). Note the presence of vacuolated cytoplasm, fragmented tumor cells, and isolated nuclei of tumor cells characterized by dense eosinophilic cytoplasm and hyperchromatic nuclear fragments (marked by black arrows). Center and bottom panels show the immunochemical staining results for the autophagy BECN1 marker and diagnostic marker of GBM: GFAP. Immunohistochemistry detected BECN1 rarely and, when found, only surrounded the vascular-like channel structures (placebo, center left), whereas the section of SAHA treatment revealed diffuse staining of BECN1 in necrotic parts of the tumor cell cytoplasms, and at the perinuclear region (red arrows), with a relocation of BECN1 around the nucleus (green arrowheads) and at the frontier of the necrotic part of some cells (center right). Bottom panels show the protein expressions of GFAP that served as diagnostic controls. Insert panels show a higher magnification, and each square represents 100 × 100 μm. Scale bars: 500 μm (upper panels) and 50 μm (center and bottom panels).
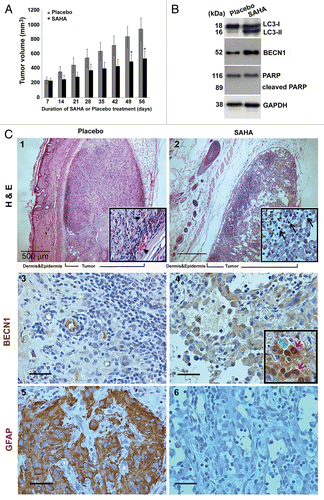
To prove that autophagy was induced by SAHA in xenograft tumors, tumor lysates were analyzed by western blot. As shown in , the protein expressions of LC3-II and BECN1 in tumors of the SAHA group were higher than those in tumors of the placebo group. Although no difference in expression of PARP was observed between the two groups, apoptosis could not be ruled out as a factor behind the tumor growth slowdown following SAHA treatment. In addition, hematoxylin-eosin (H&E) staining showed the presence of apoptotic cells, which exhibited the nuclear condensation typical of apoptosis in SAHA-treated tumors (, part ). Immunohistochemistry sections of autophagic marker demonstrated BECN1 staining in the cytoplasm and the perinuclear region (red arrows), as well as a relocation of BECN1 around the nucleus (green arrowheads), at the frontier of the necrotic part of some cells (, part ), following SAHA treatment. In contrast, BECN1 protein was only present at ductal-like structures in the placebo group (, part ). GFAP is widely expressed in astroglial cells, neural stem cells and astroglial tumors, such as astrocytoma and GBM and increased expression of GFAP represents astroglial activation and gliosis during neurodegeneration.Citation20 The xenografted sections showed high variability in GFAP expression and distribution in the vehicle control. However, the sections treated with SAHA showed rare positive stain for GFAP, indicating that malignant features of GBM are inhibition by SAHA treatment (, parts and ). These data indicated that SAHA caused tumor growth slowdown and induction of autophagy in nude mice xenograft models.
Figure 4. SAHA triggered autophagy flux in GSCs. (A) Conversions of LC3-I to LC3-II and the protein levels of BECN1 and SQSTM1 were determined by western blotting after spheroid GSCs (5 × 105 cells/ 60 mm dish) treated with various concentration (0, 2.5, 5, 10 and 20 μM) of SAHA for 48 h. GAPDH was a loading control. (B) Following above experimental conditions, micrographs showed the appearance of vesicular organelles in the adherent GSCs (phase contrast; blue fluorescence: Hoechst33342 staining as nuclear; B-1, B-4). AVOs induced by 5 μM SAHA were stained with acridine orange (red fluorescence: AVOs; B-5) and 0.05 mM MDC (green fluorescence; B-6). (C) Adherent GSCs were treated with the presence or absence (vehicle control) of the HDAC inhibitor, SAHA, at 5 μM for 48 h, and then stained with LC3 antibody and Hoechst 33342. Immunocytochemical detection of endogenous levels for LC3 (green fluorescence) was visualized on confocal microscope (C-1, C-2). Bottom panels (C-3, C-4) were merged with Hoechst 33342 (blue fluorescence, nuclei) and phase contrast (original magnification, ×1200; scale bars: 20 μm). (D) Time-dependent (0, 6, 12, 24 and 48 h) increase in dephosphorylation of endogenous AKTt and MTOR following SAHA (5 μM) treatment. Each protein was determined on western blot using a specific antibody or phospho-specific antibody as indicated on the left side of each strip. GAPDH protein levels serve as loading controls. Each square represents 100 × 100 μm in (B).
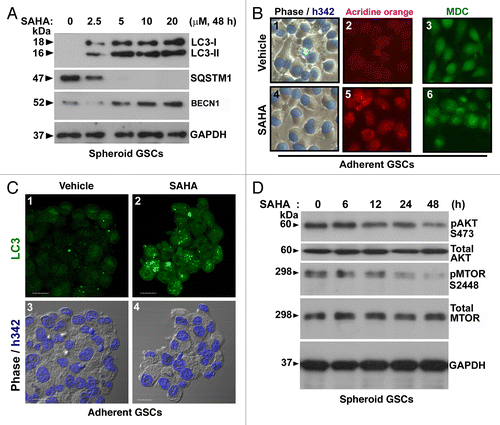
Figure 3. SAHA induced autophagosome formation. Representative ultra-structures using transmission electron micrographs of GSCs treated with either DMSO (control, < 0.1%) or 5 μM SAHA for 48 h. (A) Nuclear and mitochondrial morphologies are normal in control cells (original magnification, ×6000). Control cells also contained a richly granular cytoplasm, relatively more mitochondria. (B) SAHA treatment of GSCs for 48 h resulted in the development of autophagic vacuoles (original magnification, ×6000). The cells exhibited a sparsely granular cytoplasm, few mitochondria, and vacuoles containing membranous and partially degraded granular cytoplasm indicative of autophagic activity. (C) Amplification (original magnification, ×20000) from the square region of (B). Arrowheads denote representative autophagic vacuoles that contain remnant of organelles. N, nucleus; M, mitochondria; L, lysosome. (D–F) are pictures with higher magnification showing detailed autophagosome structure. Some of these vacuoles contained remnants of organelles, including mitochondria (D), (E: arrows) and endoplasmic reticulum (F: arrowhead). The cells with autophagic vacuoles were defined as cells that had five or more autophagic vacuoles. For these treatments, TEM images were randomly chosen, from a field of at least 100 cells. Scale bars: 2 μm; 1 μm (indicated enlargements).
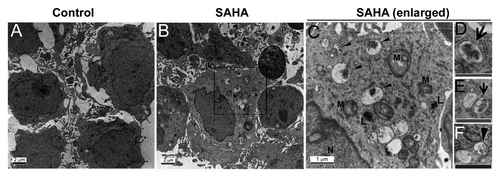
Figure 5. Effects of autophagy inhibitors and knockdown of autophagy-related genes on SAHA-induced autophagy and cytotoxicity in GSCs. (A) Spheroid GSCs (1 × 106 cells/60-mm dish) were treated with 5 μM SAHA alone or pretreated with 5 mM 3-MA or 50 nM wortmannin for 1 h, then treated with SAHA (5 μM) for 24 h, and then harvested for analysis by western blot and detection of LC3, BECN1, SQSTM1, cleaved PARP and CASP3. In some experimental groups, cell viability of adherent GSCs (5 × 103 cells/well of 96-well plate) was analyzed on MTT assay (C). (B) Equal amounts of spheroid GSCs total cell lysates from shLuc, shLC3, shBECN1 and shATG5 GSCs (1 × 106 cells/ 60 mm dish) after 5 μM SAHA treatment for 48 h were analyzed by western blot assays for LC3, BECN1, ATG5 and SQSTM1 to confirm the lack of LC3, BECN1 and ATG5 expression in GSCs. GAPDH was a loading control. In some experimental groups, cell viability of adherent GSCs was analyzed using the MTT assay (D). (E) Spheroid GSCs were treated with 25 nM BafA1 or ATG5 gene knockdown alone or in combination with 5 μM SAHA or 20 μM z-VAD for 48 h and then harvested for western blot analysis. Cell lysates were resolved in SDS-PAGE and probed with indicated antibodies. (F) In some experimental groups, cell viability was determined by the MTT assay after the treatment of adherent GSCs for 24 h. The data represent the mean and SEM of three independent experiments. *p < 0.05 compared with control on Student t-test.
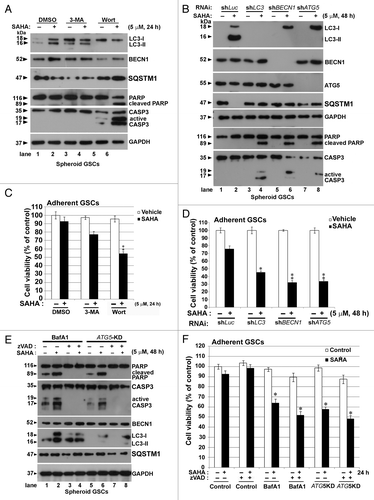
Figure 6. Inhibition of autophagy by CQ augmented SAHA-induced apoptosis in GSCs. (A) PI stain was performed after spheroid GSCs had been exposed to DMSO (vehicle), 5 μM SAHA alone, a combination of 25 μM CQ and 5 μM SAHA or 25 μM CQ alone for 24 h. (B) Spheroid GSCs were treated with SAHA (0 or 5 μM with/without CQ (25 μM) for 24 h. TUNEL assay was performed after the above procedure and finally an analysis was conducted by flow cytometry. **p < 0.01 on Student’s t-test, GSCs cotreated with SAHA and CQ compared with GSCs treated without CQ. #p < 0.05 on Student’s t-test, GSCs cotreatment with NAC, SAHA and CQ compared with GSCs cotreated with SAHA plus CQ. (C) Spheroid GSCs were treated SAHA (S: 5 μM) with/without CQ (25 μM) or NAC (10 mM) for 24 h. western blotting analysis was performed after the above procedure for spheroid GSCs, and to determine the conversions of LC3-I to LC3-II, cleaved CASP3 and PARP. In addition, the proteins of SQSTM1 were extracted by PARP buffer containing 2% (insoluble SQSTM1) or 0.1% SDS (soluble SQSTM1). GAPDH was a loading control. V: vehicle control; S: 5 μM SAHA; S+C: 5 μM SAHA + 25 μM CQ; C: 25 μM CQ, treatment for 24 h.
SAHA-induced autophagy in GSCs is dependent on inhibition of MTOR through inhibition of AKT activation
Several studies have suggested that SAHA could induce autophagy in certain types of cancers, including hepatocellular carcinoma and T-leukemia cells,Citation21,Citation22 and we hypothesized that SAHA causes autophagy in GSCs. Transmission electron microscopy (TEM) is a standard method to reliably detect autophagy. As shown in , the ultrastructure of GSCs treated with 5 μM SAHA or the vehicle control (DMSO; vehicle control, < 0.1%) for 48 h was analyzed by TEM. Numerous obvious autophagic vacuoles seen as membrane-bound vacuoles characteristic of autophagosomes were present in the cytoplasm of SAHA-treated cells (), including features of mitophagy (), whereas control cells exhibited normal nuclei with uniform and finely dispersed chromatin, surrounded by cytoplasm with normal appearing mitochondria, and membrane-bound vacuoles could rarely be found ().
To determine whether SAHA-treatment induces autophagy in GSCs, we monitored the alteration of SAHA-mediated autophagic flux using immunoblot-based LC3 flux assay. The production of the cleaved and lipidated form of LC3 (LC3-II, 16 kDa) in the initial stage of autophagy served as the autophagy marker. The cytosolic form of LC3 (LC3-I, 18 kDa) localizes to the phagophore and the cleaved, lipidated and autophagosome-bound form (LC3-II), together with BECN1, a critical component in the class III PtdIns 3-kinase complex, induce the formation of autophagosomes in mammalian systems. We also monitored the degradation of a known autophagy-specific substrate, sequestosome 1 (SQSTM1/p62).Citation23 As shown in , the production of LC3-II was increased in a dose-dependent manner by SAHA treatment in GSCs. The conversion of LC3-I to LC3-II was augmented by a concentration of 5 μM SAHA. Similar results were obtained for the expression level of BECN1 following SAHA treatment. These results are consistent with multiple studies showing that the protein levels of SQSTM1 are dramatically destabilized in the presence of SAHA at concentrations above 5 μM of SAHA (, lanes 3–5), indicating that the effect of SAHA was not caused by an increase in protein aggregation.Citation21,Citation24,Citation25
To further evaluate if the formation of membrane-bound vacuoles was an autophagic response to SAHA treatment in GSCs, we marked acidic vesicular organelle (AVO) development using acridine orange staining, which is a hallmark of autophagy. As shown in , part , the formation of red fluorescent AVOs in the SAHA-treatment group was significantly induced compared with that of the vehicle control group. The vehicle control group only exhibited slight fluorescence, indicating a lack of AVOs (, part ).
Monodansylcadaverine (MDC) was used to apply a specific marker to autophagic vacuoles. The panel in , part shows higher fluorescent density and more MDC-labeled particles in the SAHA-treated group than in the vehicle control group (, part ), indicating that SAHA increases MDC recruitment to autophagosomes in the cytoplasm of cells.
To investigate LC3 localization and distribution and the recruitment of the cleaved and lipidated microtubule-associated protein 1 light chain 3 (LC3-II) to autophagosomes in response to SAHA treatment, immunocytochemistry was performed on GSCs to detect the production of endogenous LC3. , part shows that SAHA treatment induced a characteristic punctate pattern for LC3-II, indicating autophagosome formation, and produced an increase in fluorescence intensity as compared with control cells. Furthermore, , part shows this using fluorescence confocal microscopy. These results indicate that SAHA increased the expression of markers for autophagy.
Interruption of the MTOR signaling pathway leads to activation of autophagy.Citation26 A previous report has shown that SAHA induces autophagy through interference with the AKT-MTOR pathway in HeLa S3 cells.Citation27 To investigate the molecular mechanisms of SAHA-induced autophagy, MTOR kinase activity, as measured by its phosphorylation, was examined in GSCs. As shown in , time-dependent treatment with 5 μΜ SAHA significantly inhibited the phosphorylation of both AKT (Ser473) and MTOR (Ser2448) compared with the vehicle control. Notably, SAHA-mediated dephosphorylation of both AKT and MTOR was more pronounced when the treatment time was extended to 48 h (). If autophagy is a protective response, downregulation of AKT-MTOR should mitigate SAHA-induced cytotoxicity. These data suggested that SAHA induces autophagic flux through inhibition of AKT-MTOR signaling pathway in GSCs.
Inhibition of SAHA-mediated autophagy by autophagy-inhibiting wortmannin facilitates apoptotic cell death in GSCs
To clarify the role of autophagy in SAHA-induced cell death, GSCs were pretreated with 5 mM 3-methyladenine (3-MA) and 50 nM wortmannin for 1 h, and then co-exposed to 5 μM SAHA for 24 h and analyzed using western blots. 3-MA, a class III PtdIns3K inhibitor, is extensively used as an autophagy inhibitor. Wortmannin has also been widely used to inhibit autophagy.Citation28 We also monitored the degradation of the known autophagy-specific substrate, SQSTM1,Citation23 and the activation of caspase cascade-related substrates. As shown in , expression of LC3-II conversion in the pretreated 3-MA group of GSCs was partially suppressed compared with the SAHA treatment alone (lanes 2 and 4), whereas a significant decrease in LC3-II conversion was observed for spheroid GSCs in the presence of wortmannin. The dynamic process of autophagy, which includes initiation, elongation, maturation and degradation, is also called autophagic flux. An increase of LC3 lipidation may result from increased formation of autophagosomes or decreased degradation of autophagosomes. In our experiments, SAHA decreased SQSTM1 levels in GCSs (lane 2), whereas SQSTM1 levels were increased under wortmannin cotreatment (lanes 5 and 6). Wortmannin, but not 3-MA, significantly decreased the accumulation of LC3-II under SAHA treatment, indicating that wortmannin is likely to be more suitable as a SAHA-induced autophagy inhibitor. Meanwhile, the potential of 3-MA in the autophagy process depends on its differential temporal effects on class I PtdIns3K and class III PtdIns3K; 3-MA blocks class I PtdIns3K persistently, whereas its suppressive effect on class III PtdIns3K is transient.Citation29
Autophagy modulates apoptosis.Citation30 To further clarify the role of SAHA-induced autophagy in the apoptotic process, we detected the protein levels of cleaved PARP and CASP3 after SAHA treatment. When autophagy was inhibited by wortmannin, but not 3-MA, SAHA treatment resulted in the production of cleaved PARP and active CASP3 (, lane 6), suggesting that SAHA probably induces apoptosis under autophagy deficiency.
Inhibition of SAHA-mediated autophagy using knockdown of autophagy-related genes increases SAHA-induced apoptotic cell death in GSCs
To clarify the role of autophagy in SAHA-induced cell death and to rule out the effects of pharmacological agents affecting processes other than autophagy, we performed gene knockdown of autophagy-related gene silencing by performing an LC3, BECN1 and ATG5 silencing experiment. This was performed with a VSV-G pseudotyped lentivirus-shRNA system. The responses of shLuc GSCs were similar to these of parental GSCs after SAHA treatment. In shLuc GSCs, SAHA induced LC3-II conversion (, lane 2), while the induction of LC3-I was not affected by gene silencing, as the responses to the silencing of BECN1 and ATG5 were similar (, lanes 6 and 8). The protein levels of SQSTM1 were destabilized in the presence of SAHA (, lane 2). However, the decrease in SQSTM1 levels was restored by silencing of LC3, BENC1 and ATG5 (, lanes 4, 6 and 8), suggesting that SAHA indeed induces lysosome-mediated degradation of autophagy substrates. That is, it is a functional autophagic flux rather than one via the effect of SAHA on decreased degradation of autophagosomes. Upon treatment of spheroid GSCs with SAHA for 48 h, we noticed a dramatic increase in the protein levels of cleaved PARP and CASP3 in GSCs where LC3, BECN1 and ATG5 had been silenced (, lanes 4, 6 and 8) compared with the shLuc control (, lane 2). Furthermore, we also detected that SAHA-mediated cell death was observed upon depletion or pharmacological inhibition of autophagy using the MTT assay. As shown in , wortmannin facilitated SAHA-induced cell death, while the treatment of an autophagy-related gene using a knockdown system also exacerbated SAHA-induced cell death (), suggesting that the blockage of autophagy exacerbated the cytotoxicity of SAHA treatment. Unexpectedly, 3-MA only slightly increased SAHA-induced cell death (). It appears therefore reasonable that 3-MA-mediated modulation of SAHA-induced autophagy as evidenced by increasing LC3-II conversion and SQSTM1 degradation in the presence of 3-MA (, lanes 3 and 4). Notably, these GSCs with silenced autophagy-related genes exhibited a failure to clear dead cells, and the central regions of spheroids were almost all filled with cellular debris (Fig. S3). Overall, the data suggested that autophagy attenuates the cytotoxicity effect of SAHA treatment to protect GSCs from apoptotic cell death, as autophagy was protective. Inhibition of autophagy using pharmacological agents or a gene knockdown system of autophagy-related genes is likely to facilitate SAHA-triggered apoptotic cell death in GSCs.
Bafilomycin A1 (BafA1) is a vacuolar-type H+-ATPase inhibitor that blocks autophagosome-lysosome fusion to inhibit autophagy. Alternatively, clinical diagnosis reveals that abrogated apoptosis frequently contributes to chemotherapy resistance.Citation31 To test whether SAHA-induced autophagy may cause an increased cell survival rate in the absence of apoptotic cell death, we measured cell survival and caspase activation in GSCs cotreated with SAHA and z-VAD under both genetic and chemical autophagy blockades. As shown in , regardless of whether inhibition of autophagy was accomplished using BafA1 or by ATG5 knockdown, SAHA caused the production of cleaved PARP and CASP3 and the accumulation of SQSTM1 (lanes 2 and 6). However, a decreased cell survival rate was still evident during SAHA and z-VAD cotreatment (), in spite of the cell being treated with z-VAD, which completely abolished caspase activation (, lanes 4 and 8), indicating that upon autophagy-deficiency (BafA1 or ATG5-KD), SAHA exacerbated cellular cytotoxicity under z-VAD treatment (apoptosis-death impairment), compared with the treatment without z-VAD. These results demonstrate that autophagy acts as a protective mechanism to reduce SAHA-induced apoptotic and non-apoptotic cell death.
Inhibition of SAHA-mediated autophagy by CQ cotreatment triggers apoptosis in GSCs
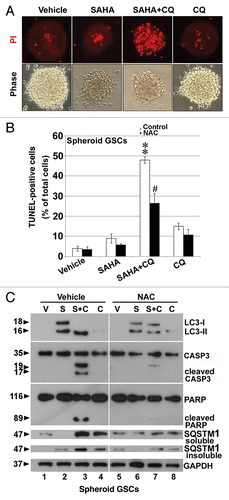
Chloroquine (CQ) is a clinical lysosomotropic autophagy inhibitor that blocks autophagosome-lysosome fusion.Citation32 We were interested to know if, in a condition in which the lysosome system is blocked, the cellular viability and possible regulation of pathways are affected by SAHA treatment. As shown in , cell death of GSCs which were positive stained for PI (red fluorescence) following the combination treatment of SAHA with CQ for 24 h, was significantly increased compared with the vehicle control, SAHA or CQ-alone treatment groups, indicating that the combination treatment of SAHA with CQ rapidly induces the cell death of spheroid GSCs.
Mitochondrial stress and damage are sources of reactive oxygen species (ROS) in autophagy-deficient cells.Citation33 To determine whether ROS contributes to cellular damage upon SAHA and CQ treatment, the stress response without and with the ROS scavenger, N-acetyl cysteine (NAC, 10 mM), a general antioxidant, for 30 min prior to SAHA or CQ exposure, was examined using the TUNEL assay. As shown in , SAHA-induced apoptosis was significantly enhanced in the presence of 25 μM CQ, when compared with that in the presence of SAHA alone (p < 0.001). Similar results were found in differentiated GSCs after serum stimulation under cotreatment with SAHA and CQ (Fig. S4). Upon cotreatment with NAC, SAHA-induced apoptosis resulted from cotreatment with CQ (p < 0.05), suggesting a critical role for basal ROS-mediated oxidative stress in the progression to apoptosis associated with autophagy defects. Autophagy is activated under oxidative stress.Citation34 To determine whether oxidative stress was involved in SAHA-induced autophagy, we pretreated spheroid GSCs with NAC prior to SAHA or CQ exposure, then analyzed with autophagy flux using western blot analysis. As shown in , treatment of GSCs with SAHA and CQ caused an increase of cleaved CASP3 and PARP, while additional pretreatment of cells with NAC resulted in the alleviation of SAHA-induced cellular toxicity and a reduction of the protein levels of cleaved CASP3 and PARP (, lanes 3 and 7). Interestingly, we also examined the levels of LC3-II, and SQSTM1 in a cell lysis buffer containing 2% SDS to evaluate insoluble fraction proteins. Pretreatment of NAC mitigated SAHA, or SAHA plus CQ-induced LC3 lipidation (, lanes 3 and 7). Similar results also were observed under the conditions of 2% (insoluble) or 0.1% SDS (soluble); SAHA decreased the level of SQSTM1, whereas CQ significantly enhanced SQSTM1 levels. NAC reduced the levels of SQSTM1 after cotreatment of SAHA and CQ (, lanes 3 and 7). Cytotoxic effects of SAHA due to defective autophagy were suppressed by the ROS scavenger or by SQSTM1 elimination, indicating that the persistence of SQSTM1 and oxidative stress is likely to cause cellular damage affecting cell survival in GSCs. We also found that both CTSD/cathepsin D, an aspartic protease, and TXN/thioredoxin, related with CTSD substrates, were involved in the induction of apoptotic processes by treatment with SAHA combined with CQ (data not shown). However, proof of these hypotheses will require additional experiments.
SAHA modulates the differentiation of GSCs
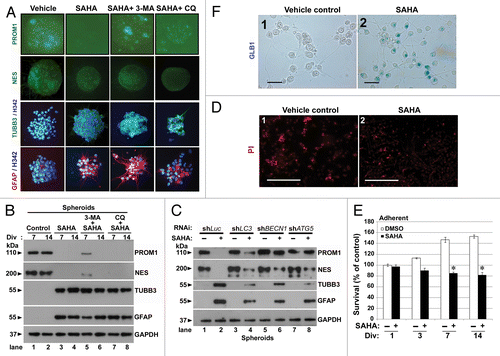
To investigate whether SAHA is a potent modulator of differentiation in GSCs, we assessed the protein expression of neural stem cell (NSC) markers PROM1 and nestin (NES) and the differentiation of markers on TUBB3/β-III-tubulin and GFAP/glial fibrillary acid protein, which are surrogate markers of neuronal and glial lineages, respectively, and are expected to be upregulated upon differentiation.Citation35 As shown in , a reduced dosage of SAHA (2.5 μM) resulted in reduced expression of PROM1 and NES and increased expression of TUBB3 and GFAP in spheroid GSCs. However, the spheroid GSCs cotreated with SAHA and 3-MA continued to express PROM1 but not NES. The addition of autophagy inhibitors (3-MA or CQ) did not prevent the expressions of differentiation markers in GSCs after SAHA treatment including those of TUBB3 and GFAP. To further confirm the differentiation under SAHA treatment, an immunoblotting assay was performed to monitor the expression of stem cell markers (PROM1, NES) or differentiation markers in related neuronal (TUBB3) and glial (GFAP) cells (). After SAHA treatment for 7 or 14 d, GSCs indeed decreased both PROM1 and NES protein levels, and cotreatment with 3-MA or CQ did not prevent the expressions of differentiation markers in GSCs (). In contrast, SAHA induced the expression of differentiation markers (TUBB3 and GFAP) in GSCs. At the same time, to examine whether autophagy deficiency influences the effect of SAHA-induced cell differentiation in GSCs, we used an RNAi system to block autophagy and analyze the protein levels of related differentiated markers. As shown in , the effect of SAHA-induced differentiation was attenuated under autophagy-deficiency (lanes 2 to 4, 6 and 8) no matter what stem cell markers (PROM1 and NES) or differentiation markers (TUBB3 and GFAP) were involved, suggesting that SAHA-induced autophagy affects cell differentiation, given that inhibition of autophagy delays the differentiated phenotype in GSCs. We even examined the expression of senescence-associated GLB1/galactosidase, β 1 to prove that SAHA induces cell senescence in GSCs (). To further determine the effect of reduced dosage of SAHA (2.5 μM) on cell death and cell viability, adherent GSCs were analyzed using PI staining and the MTT assay after SAHA treatment. As shown in , PI positive cells were not significantly induced after SAHA treatment (, part ) as compared with the vehicle control (, part ), indicating that reducing the dosage of SAHA clearly resulted in no significant cytotoxicity in GSCs. Interestingly, cell viability and proliferation were suppressed upon SAHA treatment compared with the control (). These results suggested that epigenetic modification of SAHA determines the differentiation of GSCs, and reduction in the dose of SAHA is a potent modulator of differentiation in GSCs.
Figure 7. Reduced dose of SAHA induced differentiation in GSCs. (A) Immunofluorescence staining was performed after spheroid GSCs (PROM1 and NES, green) and adherent GSCs (TUBB3, green; GFAP, red; nuclear, blue: stained by Hoechst 33324 as nuclear counterstains) had been exposed to DMSO (vehicle), 2.5 μM SAHA alone, a combination of 5 mM 3-MA plus 2.5 μM SAHA or 25 μM CQ plus 2.5 μM SAHA for 7 d. Each square represents 200 × 200 μm. (B) Similar to the above conditions, spheroid GSCs were harvested and immunoblotting assays were performed to detect the protein levels of PROM1, NES, TUBB3 and GFAP for the 7th and 14th day. Div: days in vitro. (C) Equal amounts of spheroid GSC total cell lysates from shLuc, shLC3, shBECN1 and shATG5 GSCs (1 × 106 cells/60-mm dish) after 0 or 2.5 μM SAHA treatment for 7 d were analyzed by western blot to assess the presence of PROM1, NES, TUBB3 and GFAP. GAPDH was a loading control. (D) Adherent GSCs were treated with determination of senescence by colorimetric senescence-associated GLB1 activity assay at the 7th day in the presence or absence of SAHA (2.5 μM) induced differentiation of spheroid GSCs. Original magnification, ×400. Scale bars: 50 μm. (E) Determination of cell death using PI staining at the 7th day in the presence or absence of SAHA (2.5 μM) was not showed significant effect to affect cell death in adherent GSCs. Original magnification, ×40. Scale bars: 500 μm. (F) Using the MTT assays, SAHA-mediated cytotoxicity (2.5 μM treated for indicated time points) was analyzed in adherent GSCs. (1 × 104 cells/ well of 96-well plate). *p < 0.05 compared with control on Student t-test. Data were representative of three independent experiments with similar results. Div, days in vitro.
Discussion
GBM remains one of the most fatal cancers despite optimal therapies. The main reason is the capacity of GSCs to develop therapeutic resistance and that may also be the reason for the high relapse rate. Several studies have proven that a high resistance against classical, caspase-dependent apoptosis is a hallmark of malignant glioma.Citation36,Citation37 Similarly, a defect in autophagy is associated with a malignant phenotype and poor prognosis of GBM.Citation38 Thus, reactivation of autophagy might be an effective strategy for the treatment of GBM. Our results demonstrated that HDAC inhibitor SAHA inhibits cell viability of spheroid and adherent GSCs, but not of differentiated GSCs (IC50 of approximately 5 μM SAHA), and this inhibition is restricted to stemness types of GSCs. SAHA resulted in cell death through late apoptosis in vitro and in the slowdown of tumor growth in vivo. The ultra-structures of GSCs treated with SAHA showed the induction of autophagic vacuoles, autophagosome, and mitophagy. Exposure to SAHA led to the activation of a group of autophagy-related proteins such as BECN1 and LC3 in which there was conversion of LC3-I to LC3-II. SAHA significantly increased endogenous LC3-II accumulation and transduction of cleaved GFP-LC3 puncta/aggregation, a marker of autophagic flux in transfected cells (Fig. S5).Citation39 Further experiments with acridine orange and MDC staining also confirmed SAHA-induced autophagy in GSCs, similar to results of studies on hepatocellular carcinoma and T-leukemia cells.Citation21,Citation22 A possible explanation for the increase in SAHA-mediated autophagy-induction is that SAHA inactivates AKT-MTOR signaling by dephosphorylation of Ser473 and MTOR Ser2448. Depletion of autophagy-related genes or pharmacological inhibition resulted in SAHA-induced apoptosis at the early phase, whereas cotreatment of rapamycin provided a protective effect against the activation of caspase by overdose induction of SAHA treatment, suggesting that SAHA-induced autophagy functions as a prosurvival mechanism to mitigate SAHA-induced apoptotic cell death.Citation25 In fact, we found that rapamycin could rapidly induce LC3 lipidation, and was likely to reduce part of the SAHA-induced cytotoxicity (Fig. S6). Hence, rapamycin offered protection against cell death induced by a high dose of SAHA, whereas wortmannin, BafA1, and CQ exacerbated SAHA-induced cytotoxicity, suggesting that the induction of autophagy provided a potentially protective effect in GSCs. In addition, we attempted to produce a xenograft mouse model of autophagy deficiency using a gene knockdown system. Tumor masses failed to grow from GSCs in the animal models (Fig. S7). In addition, we also showed the induction of autophagic flux depending on the genetic and chemical inhibition of autophagy (), and reduced AKT activation by SAHA treatment may be responsible at least in part for the apoptosis process (), since our studies reveal that AKT activation by repamycin significantly increased cell viability under SAHA treatment in GSCs (). These results strongly support and define that the role of SAHA-induced cell death is in fact a case of late apoptosis.
SAHA is a prototype of a family of hybrid polar compounds, with a longer half-life, lower toxicity, and greater stability than earlier HDACi that induced growth arrest in transformed cells. Manifold effects of SAHA modify a large number of proteins that play a role in the regulation of oncogene pathways. In addition, SAHA can readily cross the blood-brain barrier and can be given orally, which increases the possibility of clinical application.Citation40 In general, SAHA induces differentiation and/or apoptosis in transformed cells in culture.Citation41,Citation42 SAHA has also been shown to alleviate several neurological disorders.Citation43,Citation44 In this study, our results showed that SAHA had specific effects on GSCs: SAHA specifically inhibits the growth of tumor sphere and triggered autophagy functions at the early phase.
Certain reports show that autophagy involved an alternative form of autophagy (or noncanonical autophagy) such as ‘alternative’ macroautophagy, indicating that extreme mitophagy was induced by some of these forms (). It can lead to cellular demise and is regarded as autophagy-induced cell death. Because autophagy is a process that destroys cellular structures in the cytoplasm, depending on the intensity and persistence of the stress, this destruction by SAHA-induced autophagy may pass a threshold in the cell, leading to cell death. Indeed, we observed that caspase activity contributed to this type of cell death after SAHA treatment with high doses or prolonged time frames ().Citation45 Recently, increasing evidence proves that autophagy could exert a protective role toward the activation of apoptosis in cancer cells.Citation25,Citation30 We have provided consistent evidence that specific inhibition of autophagy with RNAi accelerated apoptotic cell death with SAHA treatment (). When autophagy was inhibited by wortmannin, SAHA induced cell death through the activation of CASP3 and PARP (), suggesting that autophagy may reduce SAHA-induced cytotoxicity by buffering metabolic stress. By contrast, PtdIns3K and AKT-MTOR inhibitors (e.g., 3-MA, rapamycin) potently activate autophagy. As these agents often do not produce a durable response in patients, there is interest in increasing their efficacy. Thus, autophagy activation by these agents is potentially counterproductive.
In addition, abrogated apoptosis frequently contributes to chemotherapy resistance. Alterations in key roles are likely to be relevant to autophagy.Citation25 Our results also demonstrated that SAHA induced nonapoptotic cell death in cotreatment with z-VAD () as well as previously reported in cells deficient in apoptotic machinery.Citation25,Citation46 Importantly, we also showed that when apoptosis is pharmacologically blocked, SAHA-induced nonapoptotic cell death can also be potentiated by autophagy inhibition (). Although the nature of this type of nonapoptotic cell death triggered by SAHA remains unresolved, at the very least, these results demonstrate that SAHA has the potential to induce both apoptosis and nonapoptotic cell death depending on the presence or absence of autophagy. Interestingly, knockdown of autophagy-related genes increased cleaved PARP and CASP3 in GSCs, yet the pattern of ANXA5/annexin V staining in spheroid GSCs after silencing of BECN1 and ATG5 was exceedingly low compared with shLuc control (Fig. S3) suggesting that unique mechanisms exist for regulating PS exposure in the three-dimensional tissue models. Taken together, these finding are in agreement with a hypothesis suggesting the existence of a double switch between two principal lethal signaling pathways.Citation47
We observed an increase in LC3 levels induced by SAHA treatment that were mechanistically able to induce autophagy. During persistent autophagy, LC3 protein levels may drop as LC3 is conjugated to autophagosome membranes, which in turn is targeted for degradation upon fusion with the lysosome compartment. Although upregulation of LC3 during autophagy is an important mechanism to avoid the exhaustion of the pathway during prolonged treatment, it is not sufficient to induce autophagy. Indeed, our results indicated that SAHA regulates autophagy by both inducing LC3 expression and inactivating MTOR and AKT. However, it is not clear whether SAHA inactivates MTOR via inhibition of histone deacetylation and thus transcription of certain genes, or via inhibition of deacetylation of a nonhistone protein that is involved in MTOR regulation. We also observed that SAHA not only induced autophagic flux, but also potentiated autolysosome maturation, and SAHA-increased LC3-II conversion through a high dose of SAHA (20 μM) treatment, could not avoid induction of apoptosis (; ), suggesting that the destruction in a dynamic equilibrium of autophagy using chemical agents is likely to induce cellular apoptosis in GSCs. In addition, SAHA increased the protein levels of autophagy-related protein BECN1, but not via a transcriptional-dependent increase (Fig. S8) to indicate that SAHA increased the protein levels of BECN1 is likely to reduce BECN1 protein degradation via protein interactions.Citation48
CQ has been used for symptomatic improvement of malaria, and has a small molecular weight allowing it to easily penetrate the blood-brain barrier. Although early studies indicated that CQ is an autophagy inhibitor, recent reports have shown that CQ induces autophagy and apoptosis.Citation49,Citation50 However, the mechanism of CQ-induced tumor cell death is poorly defined. Our results showed that CQ is less toxic to GSCs, but clearly induces vesicular accumulation. Interestingly, the combination treatment of SAHA with CQ enhanced the cytotoxic effects of SAHA in apoptosis-inducing cells, indicating that SAHA-induced autophagy has a negative effect on apoptosis, since blocking of autophagy by CQ unleashed apoptosis. We showed that CQ-mediated inhibition of SAHA-induced autophagy triggers apoptosis in GSCs, including differentiated GSCs, with an increase of cleaved CASP3 and PARP, and production of apoptotic cells, which was confirmed using TUNEL analysis. In addition, in autophagy-deficient and apoptosis-incompetent tumor cells, metabolic stress leads to the accumulation of SQSTM1, elevated expression of endoplasmic reticulum chaperones, damaged mitochondria and reactive oxygen species,Citation51 resulting in gene instability and leading to tumorigenesis.Citation52 However, our results seem to be consistent with those of previous studies in that the inhibition of autophagy contributed to the cell’s sensitivity to chemo- and radio-resistance.Citation53,Citation54 By inference, the role of autophagy in tumorigenesis and cancer therapy appears to be contextual, functioning as a double-edged sword depending on the status of the tumor cells. In certain cancer treatments, autophagy is required to induce robust cell death in tumor cells as GSCs.Citation55 In addition, in the cancer stem cells that have escaped therapy or at metastatic sites, autophagy-mediated dormancy may provide a period of time to recover and reenter the cycle to cause cancer recurrence.Citation56 Thus, prompt disruption of the autophagy pathway may potentially provide an efficient approach to extend the therapeutic benefits and reduce the incidence of cancer recurrence. Taken together, several pathophysiological mechanisms or their combination may explain the increasing levels of apoptosis, including: (1) altered lysosomal function by CQ, (2) modulation of antiapototic-related proteins by SAHA and/or (3) disruption of the ROS scavenging system (data not shown). Therefore, the combination of CQ with chemotherapy or radiotherapy may improve GBM therapeutic outcomes.Citation57,Citation58
Furthermore, as mentioned in a previous report, autophagy has been recently identified as a new effecter mechanism of senescence,Citation59 and we hypothesize that SAHA preferentially transactivates the regulated differentiation genes of stem cells. The results showed that SAHA reduces the proportion of stem cells positive to markers of GSCs, PROM1 and NES, and induces expression of two differentiation markers, TUBB3 and GFAP. However, autophagy-defective cells using a gene knockdown system seem to affect the protein levels of the SAHA-induced differentiation marker (). This suggests that SAHA induces either selective death or differentiation of GSCs, and autophagy may function as a decision maker to adjust corresponding reactions under damaging stress. Notably, reduced administration of SAHA led to an increase in senescent cells compared with control and caused glial- and neuronal-like differentiation of GBM cells, reducing tumorigenicity.Citation60 It is also important to point out that chronic effects of low doses as metronomic chemotherapy are especially important in view of in vivo studies showing that high micromolar concentrations are very difficult to attain, especially in the central nervous system.Citation61,Citation62 Therefore, SAHA may be a potential therapeutic agent for the induction of terminal differentiation of GSCs.
In conclusion, SAHA inhibits cell viability and induces autophagic flux in GSCs in vitro, and causes tumor growth slowdown in the xenograft tumor model of GSCs. Under the inhibition of autophagy using pharmacological agents or knockdown-related autophagy genes, SAHA facilitates apoptotic cell death and differentiation, suggesting that SAHA is an attractive candidate for tumor therapies, particularly, in cells deficient in apoptotic machinery, such as GBM cells, and that pharmacological targeting of autophagy is probably useful in the management of GBM therapy.
Materials and Methods
Cell culture
The protocol for this study was approved by the Institutional Review Board Committee of Taichung Veterans General Hospital. Confidentiality issues were explained to the participants, and informed consent was obtained. Tumor staging was performed according to the American Joint Committee on Cancer criteria. Detailed isolation procedures of glioblastoma stem cells have been previously described.Citation4 In brief, the obtained tissues were washed and enzymatically dissociated into individual cells. The dissociated cells were cultured in neurosphere conditioned medium using neurobasal media (Invitrogen, 21103-049) containing N2 and B27 supplements (Invitrogen, 17502-048; 0080085SA), plus human recombinant bFGF and EGF (50 ng/ml each; R&D Systems, 233-FB; 236-EG). After 2 to 4 weeks incubation, the serial dilution was performed on surviving GSCs to select a single cell that is able to grow a new sphere. The glioblastoma stem cells (GSCs), spheroid type, were cultured in neurosphere-conditioned medium (NSC medium). The adherent type of GSCs was cultured on a precoated collagen (Upstate, 08115) dish in NSC medium. The differentiated GSCs were cultured in Dulbecco’s modified Eagle medium (GIBCO, 12100-046) supplemented with 10% heat-inactivated fetal bovine serum (GIBCO, 10437), 100 U/ml penicillin, 0.1 mg/ml streptomycin (GIBCO, 15140) for 7 d. To assess the morphology of spheroid GSCs, approximately 5 × 5 × 6 mm of GSCs seeded in gelatin form were then cut from a sheet of gelatin foam dressing material, and after examination it was concluded that this condition did not promote the differentiation of GSCs (). The morphology of cells was observed using a light microscope (Eclipse E600, Nikon Corp.) and scanning electron microscopy (SEM, S-3000N, Hitachi) for observation of survival and proliferation. However, all experiments of spheroid GSCs implied used floating model except . All cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C.
Reagents
Suberoylanilide hydroxamic acid (SAHA, Vorinostat, ZolinzaTM) and z-VAD-FMK were purchased from Cayman (Cayman Chemical, 10009229) and Abcam (Abcam, ab120382) and dissolved in dimethyl sulfoxide (DMSO) (Merck, 1.02931.100). 3-methyladenine (3-MA; Sigma, M9281), an inhibitor of autophagosome formation, bafilomycin A1 (BafA1; Sigma, B1793), chloroquine diphosphate salt (CQ) (Sigma, C6628), N-acetyl cysteine (NAC, Sigma, A7250) acridine orange, rapamycin (Sigma, R0395), acridine orange (Sigma, A6014) and MDC (Sigma, 30432) were purchased from Sigma. Hoech33342 were purchased from Molecular Probes® (H3570).
Antibodies
For the primary antibodies in western blotting assays: LC3 (Abgent, AM18000a), BECN1 (Millipore, MABN16), MTOR/FRAP (Epitomics, 1612), MTOR/FRAP phospho (pS2448) (Epitomics, 2936), AKT (Epitomics, 1080), AKT phospho (p4S73) (Epitomics, 2118), CASP3 (Upstate, 04-439), PARP (Cell Signaling, 9532), SQSTM1 (Novus, NBO1-48320), ATG5 (Cell Signaling, 8540) and GAPDH (Santa Cruz Biotechnology, sc-25778). For the primary antibodies in immunohistochemistry analysis: BECN1 (Millipore, MABN16), glial fibrillary acidic protein (GFAP; Dako Cytomation, Nr. Z 0334).
Cell viability assay
Five × 103 GSCs (undifferentiated or differentiated cells) were seeded onto 96-well plates containing 100 ml of culture medium. After 24 h of incubation, the medium was carefully removed and 100 μl of fresh medium containing various concentration of SAHA or DMSO (< 0.1%, as vehicle control) were added to the wells. The medium was then carefully removed and 100 μl of fresh medium containing 0.5 μg/ml MTT (thiazolyl blue tetrazolium bromide) (Sigma, M2128) was added to the wells. MTT was converted from a yellowish solution to water-insoluble MTT-formazan that is dark blue in color by mitochondrial dehydrogenases of living cells. The blue crystals were solubilized with 100 ml DMSO and the intensity was measured colorimetrically at a wavelength of 570 nm. Absorbance values are presented as the mean ± SE of three independent experiments for each treatment. Cells in controls and compound controls were included. Absorbance of untreated cells was considered to be 100%.
Transmission electron microscopy
The spheroids of GSCs were harvested after drug treatments, washed twice with PBS, and fixed with ice-cold-glutaraldehyde (3% in 0.1 M cacodylate buffer, pH 7.4) overnight. After washing in PBS, the cells were post-fixed in OsO4 and embedded in Resin. 75-nm thin sections were cut, stained with methylene and observed by light microscopy. Representative areas were chosen for ultra-thin sectioning and examined on a JEOL JEM-1230 transmission electron microscope (JEOL USA, Inc.) at 6000× or 20000× magnification, operating at 80 kV.
Western blot analysis
In general, cells were lysed with RIPA buffer containing 0.1% sodium dodecyl sulfate (SDS) protease inhibitor cocktail (Roche, 04693159001) and protein concentrations were assayed with a Bio-Rad Protein Assay Kit (Bio-Rad, 500-0006). For some experiments, RIPA buffer containing 2% SDS was used to evaluate insoluble proteins. Equal amounts of proteins from each sample (50 to 100 μg) were separated by 10–12% SDS PAGE and transferred to polyvinylidene difluoride membrane (Amersham, RPN303F). The membrane was blocked for 1 h in TBS containing 5% nonfat milk and 0.2% Tween 20. For detection with the primary antibodies (described in “Antibodies”), the membranes were incubated with the pertinent antibody at 4°C overnight. Finally, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies and developed using ECL western blot reagents (PerkinElmer, nel104).
Clonogenic assay
The spheroids of GSCs and differentiated GSCs (5 × 105/60 mm dish) were treated with SAHA (0, 5, 10 μM) for 48 h. The cells were dissociated into individual cells and replated at a density of 500 cells/60 mm dish in triplicate, followed by incubation in NSC or differentiated conditions for 10 d. The cells were then fixed with 95% ethanol and stained with a 20% Giemsa solution (Merck, 1.09204.0500).
Visualization and analysis of intracellular vacuoles
Autophagy is the process of sequestrating cytoplasmic proteins into the lytic component and is characterized by the formation and promotion of acidic vesicular organelles. To detect the development of AVOs, we treated GSCs as described above and then performed vital staining with acridine orange as described previously.Citation22 Briefly, the treated adherent GSCs or differentiated GSCs were stained with acridine orange (1 μg/ml) for 15 min. Samples were examined under a fluorescence microscope. The autofluorescent substance MDC, a fluorescent dye known to accumulate in autophagic organelles and capable of staining autophagic vacuoles, which are part of the lysosomal compartment, was applied to the cells at 0.1 mM in PBS at 37°C for 10 min. After incubation, the cells were immediately analyzed using a fluorescence microscope (Olympus IX-71, Inc.).
Indirect immunofluorescent staining
The cell surface markers were analyzed by immunofluorescent staining. The primary antibodies were used to detect specific differentiation surface markers, including PROM1: stem cell marker (Santa Cruz Biotechnology, sc-23797); NES: nerve stem cell marker (Chemicon, MAB5326); TUBB3: early neuronal marker (Sigma, T8660); glial fibrillary acidic protein, glial cell marker (GFAP; DakoCytomation, Nr. Z 0334). The complete protocol has been described in the previous study.Citation4
Autophagosome formation analysis
GSCs were seeded onto coverslips precoated with collagen onto a 24-well plate and incubation at 37°C overnight. After 48 h SAHA treatment, GSCs were fixed and immunocytochemistry assays were performed. The primary antibody used was against the autophagosome marker, LC3 (Novus Biological, R-146-100). The distribution of autophagosomes was visualized on a confocal laser scanning microscope (Olympus FV1000) at 1200× magnification (oil immersion).
VSV-G pseudotyped lentivirus–shRNA system
RNAi reagents were obtained from the National RNAi Core Facility located at the Institute of Molecular Biology/Genomic Research Center, Academia Sinica, which is supported by the National Research Program for Genomic Medicine Grants of the National Science Council (NSC97-3122-B-001-016).
VSV-G pseudotyped lentivirus–shRNA production and infection
Lentiviral infection of GSCs led to stable integration and expression of short hairpin RNA (shRNA) targeting LC3 (91), BECN1 and ATG5 mRNA sequences. The detailed steps of lentivirus production and infection have been described previously.Citation63 Individual clones were identified by their unique TRC number: shLuc TRCN0000072246 for vector control targeted to luciferase, shLC3 (91) (LC3.91) TRCN0000243391 (corresponding sequence: AGC GAG TTG GTC AAG ATC ATC) targeted to LC3 (I and II), shBECN1 (49) TRCN0000033549 (corresponding sequence: CCC GTG GAA TGG AAT GAG ATT) targeted to BECN1, shATG5 (74) TRCN0000151474 (corresponding sequence: CCT TTC ATT CAG AAG CTG TTT) targeted to ATG5.
PI staining and TUNEL assay
Propidium iodide (PI, Sigma, P4170) staining was performed to detect dead cells after drug treatment. After SAHA or combination with CQ treatment, GSCs were added PI buffer solution (1 μg/ml) and the samples were kept in that solution at 37°C for 15 min and protected from light until analysis on the fluorescence microscope. TUNEL assay was performed using the TUNEL enzymatic labeling assay kit (Roche, 11 684 795) according to the manufacturer’s instruction. TUNEL positive cells were examined by flow cytometry (FACS).
Senescence-associated GLB1 assays
GSCs (5 × 104 cells) were plated in duplicate on 24-well plates precoated with collagen and incubated at 37°C O/N. After SAHA treatment, GSCs were stained for senescence-associated GLB1 activity (BioVision, K320-250). Three brightfield micrographs per condition were captured.
In vivo xenograft experiment and histological analysis
All experimental procedures were approved by the Institution Animal Care and Uses Committee of Taichung Veterans General Hospital. For tumorigenesis, spheroids of glioblastoma stem cells (1 × 106 cells, 30 μl) were mixed with 30 μl Matrigels (1:1, v/v; BD Biosciences, 354234) for support of engrafted cells, and inoculated subcutaneously into the 7- to 8-week-old male nude mice (nu+/nu+ BALB-c, n = 3 for each group) which were obtained from the National Laboratory Animal Center, Taipei, Taiwan. Tumor growth was measured every 7 d. Tumor volume was calculated by V = (L × W2)/2, in which L is the length and W is the width in millimeters. Upon the tumor size grow to the certain size (200 mm3), related treatment start to perform. Intraperitoneal injections of SAHA (100 mg/kg body weight) or placebo (PBS, vehicle control) were administered every 24 h for seven consecutive days and tumor volume was measured every 7 d after the cessation of treatment (mean ± SEM). All mice were treated for 56 d and afterwards sacrificed. Although we injected the same number of cells to perform the xenograft assay, we waited until the tumor size was approximately 200 mm3 before we performed the related treatment. The individual variation in tumor growth indeed declined. One tumor was lysed and analyzed by western blotting. Another two tumor slices were cryo-preserved and formalin-fixed (4%) for further analyses, including staining with hematoxylin and eosin (H&E), and immunohistochemistry to visualize tumor cells. The complete protocol has been described in a previous study.Citation4
Statistical analysis
The data were presented as mean ± standard error of the mean. Independent experiments were pooled when the coefficient of variance could be assumed identical. Independent t-tests and one-way analysis of variance (ANOVA) were used to analyze the effects of group (vehicle vs. SAHA) treatment. The results were considered to be of statistical significance with a value of P < 0.05 and P < 0.01.
| Abbreviations: | ||
| 3-MA | = | 3-methyladenine |
| AVOs | = | acidic vesicular organelles |
| BafA1 | = | bafilomycin A1 |
| CQ | = | chloroquine |
| CTSD | = | cathepsin D |
| DMSO | = | dimethyl sulfoxide |
| GBM | = | glioblastoma multiforme |
| GFP | = | green fluorescent protein |
| GSCs | = | glioblastoma stem cells |
| HDACi | = | histone deacetylase inhibitor |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| MDC | = | monodansylcadaverine |
| MTOR | = | mechanistic target of rapamycin |
| NAC | = | N-acetyl cysteine |
| PI | = | propidium iodide |
| PIK3C3 | = | phosphatidylinositol 3-kinase, catalytic subunit type 3 |
| SAHA | = | suberoylanilide hydroxamic acid |
| TEM | = | transmission electron microscopy |
| z-VAD-FMK | = | benzyloxycarbonyl-Val-Ala-Asp(O-methyl)-fluoromethylketone |
Additional material
Download Zip (690 KB)Acknowledgments
We would like to thank Dr Tamotsu Yoshimori and Dr Noboru Mizushima for providing the LC3 cDNA. Flow cytometry and use of microplate reader were performed at the Instrument Center of Taichung Veterans General Hospital. TEM images were performed by a pathologist (S-L Yang) at the Department of Pathology, Taichung Veterans General Hospital. The confocal laser scanning microscope was performed in the Instrument Center, Department of Medical Education, VGHTC. We appreciate Dr Chun-Jung Chen (Molecular Biology Laboratory, Department of Medical Education, VGHTC) and Prof Kuan-Chih Chow (Graduate Institute of Biomedical Sciences, National Chung-Hsing University) for providing valuable suggestions. This study was supported by grants from the VGHUST Joint Research Program (grants TCVGH-994903B, TCVGH-1004902B, TCVGH-1014902B and TCVGH-1014906C), Taiwan.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/25664
Related Research Data
References
- Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 2007; 21:2683 - 710; http://dx.doi.org/10.1101/gad.1596707; PMID: 17974913
- Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010; 468:824 - 8; http://dx.doi.org/10.1038/nature09557; PMID: 21102434
- Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010; 468:829 - 33; http://dx.doi.org/10.1038/nature09624; PMID: 21102433
- Chiao MT, Yang YC, Cheng WY, Shen CC, Ko JL. CD133+ glioblastoma stem-like cells induce vascular mimicry in vivo. Curr Neurovasc Res 2011; 8:210 - 9; http://dx.doi.org/10.2174/156720211796558023; PMID: 21675958
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444:756 - 60; http://dx.doi.org/10.1038/nature05236; PMID: 17051156
- Johannessen TC, Bjerkvig R, Tysnes BB. DNA repair and cancer stem-like cells--potential partners in glioma drug resistance?. Cancer Treat Rev 2008; 34:558 - 67; http://dx.doi.org/10.1016/j.ctrv.2008.03.125; PMID: 18501520
- Coates JM, Galante JM, Bold RJ. Cancer therapy beyond apoptosis: autophagy and anoikis as mechanisms of cell death. J Surg Res 2010; 164:301 - 8; http://dx.doi.org/10.1016/j.jss.2009.07.011; PMID: 20031162
- Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol 2005; 23:2411 - 22; http://dx.doi.org/10.1200/JCO.2005.03.089; PMID: 15800333
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005; 120:237 - 48; http://dx.doi.org/10.1016/j.cell.2004.11.046; PMID: 15680329
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, Carnuccio R, Kroemer G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ 2009; 16:87 - 93; http://dx.doi.org/10.1038/cdd.2008.131; PMID: 18806760
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51 - 64; http://dx.doi.org/10.1016/j.ccr.2006.06.001; PMID: 16843265
- Gozuacik D, Kimchi A. Autophagy and cell death. Curr Top Dev Biol 2007; 78:217 - 45; http://dx.doi.org/10.1016/S0070-2153(06)78006-1; PMID: 17338918
- Hashimoto D, Ohmuraya M, Hirota M, Yamamoto A, Suyama K, Ida S, Okumura Y, Takahashi E, Kido H, Araki K, et al. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J Cell Biol 2008; 181:1065 - 72; http://dx.doi.org/10.1083/jcb.200712156; PMID: 18591426
- Wang L, Zou X, Berger AD, Twiss C, Peng Y, Li Y, Chiu J, Guo H, Satagopan J, Wilton A, et al. Increased expression of histone deacetylaces (HDACs) and inhibition of prostate cancer growth and invasion by HDAC inhibitor SAHA. Am J Transl Res 2009; 1:62 - 71; PMID: 19966939
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007; 109:31 - 9; http://dx.doi.org/10.1182/blood-2006-06-025999; PMID: 16960145
- Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RH, Dunner K Jr., Pal A, Bornmann WG, Chiao PJ, Huang P, et al. Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res 2006; 66:3773 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-05-2961; PMID: 16585204
- Sangeetha SR, Singh N, Vender JR, Dhandapani KM. Suberoylanilide hydroxamic acid (SAHA) induces growth arrest and apoptosis in pituitary adenoma cells. Endocrine 2009; 35:389 - 96; http://dx.doi.org/10.1007/s12020-009-9159-1; PMID: 19291426
- Inoue S, Walewska R, Dyer MJ, Cohen GM. Downregulation of Mcl-1 potentiates HDACi-mediated apoptosis in leukemic cells. Leukemia 2008; 22:819 - 25; http://dx.doi.org/10.1038/leu.2008.1; PMID: 18239621
- Brahmachari S, Fung YK, Pahan K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci 2006; 26:4930 - 9; http://dx.doi.org/10.1523/JNEUROSCI.5480-05.2006; PMID: 16672668
- Liu YL, Yang PM, Shun CT, Wu MS, Weng JR, Chen CC. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010; 6:1057 - 65; http://dx.doi.org/10.4161/auto.6.8.13365; PMID: 20962572
- Li J, Liu R, Lei Y, Wang K, Lau QC, Xie N, Zhou S, Nie C, Chen L, Wei Y, et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells. Autophagy 2010; 6:711 - 24; http://dx.doi.org/10.4161/auto.6.6.12397; PMID: 20543569
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Gammoh N, Marks PA, Jiang X. Curbing autophagy and histone deacetylases to kill cancer cells. Autophagy 2012; 8:1521 - 2; http://dx.doi.org/10.4161/auto.21151; PMID: 22894919
- Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci U S A 2012; 109:6561 - 5; http://dx.doi.org/10.1073/pnas.1204429109; PMID: 22493260
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990 - 5; http://dx.doi.org/10.1126/science.1099993; PMID: 15528435
- Cao Q, Yu C, Xue R, Hsueh W, Pan P, Chen Z, Wang S, McNutt M, Gu J. Autophagy induced by suberoylanilide hydroxamic acid in Hela S3 cells involves inhibition of protein kinase B and up-regulation of Beclin 1. Int J Biochem Cell Biol 2008; 40:272 - 83; http://dx.doi.org/10.1016/j.biocel.2007.07.020; PMID: 17881280
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000; 275:992 - 8; http://dx.doi.org/10.1074/jbc.275.2.992; PMID: 10625637
- Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010; 285:10850 - 61; http://dx.doi.org/10.1074/jbc.M109.080796; PMID: 20123989
- Giansanti V, Torriglia A, Scovassi AI. Conversation between apoptosis and autophagy: “Is it your turn or mine?”. Apoptosis 2011; 16:321 - 33; http://dx.doi.org/10.1007/s10495-011-0589-x; PMID: 21404107
- Basu S, Ganguly A, Chakraborty P, Sen R, Banerjee K, Chatterjee M, Efferth T, Choudhuri SK. Targeting the mitochondrial pathway to induce apoptosis/necrosis through ROS by a newly developed Schiff’s base to overcome MDR in cancer. Biochimie 2012; 94:166 - 83; http://dx.doi.org/10.1016/j.biochi.2011.10.004; PMID: 22037022
- Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, Houghton JA, Huang P, Giles FJ, Cleveland JL. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007; 110:313 - 22; http://dx.doi.org/10.1182/blood-2006-10-050260; PMID: 17363733
- Fariss MW, Chan CB, Patel M, Van Houten B, Orrenius S. Role of mitochondria in toxic oxidative stress. Mol Interv 2005; 5:94 - 111; http://dx.doi.org/10.1124/mi.5.2.7; PMID: 15821158
- Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal 2009; 11:777 - 90; http://dx.doi.org/10.1089/ars.2008.2270; PMID: 18828708
- Dictus C, Tronnier V, Unterberg A, Herold-Mende C. Comparative analysis of in vitro conditions for rat adult neural progenitor cells. J Neurosci Methods 2007; 161:250 - 8; http://dx.doi.org/10.1016/j.jneumeth.2006.11.012; PMID: 17207861
- Ziegler DS, Kung AL, Kieran MW. Anti-apoptosis mechanisms in malignant gliomas. J Clin Oncol 2008; 26:493 - 500; http://dx.doi.org/10.1200/JCO.2007.13.9717; PMID: 18202424
- Bertrand J, Begaud-Grimaud G, Bessette B, Verdier M, Battu S, Jauberteau MO. Cancer stem cells from human glioma cell line are resistant to Fas-induced apoptosis. Int J Oncol 2009; 34:717 - 27; PMID: 19212677
- Zhuang W, Li B, Long L, Chen L, Huang Q, Liang Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int J Cancer 2011; 129:2720 - 31; http://dx.doi.org/10.1002/ijc.25975; PMID: 21384342
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, Moroni F, Chiarugi A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol 2006; 70:1876 - 84; http://dx.doi.org/10.1124/mol.106.027912; PMID: 16946032
- Di Bernardo G, Squillaro T, Dell’Aversana C, Miceli M, Cipollaro M, Cascino A, Altucci L, Galderisi U. Histone deacetylase inhibitors promote apoptosis and senescence in human mesenchymal stem cells. Stem Cells Dev 2009; 18:573 - 81; http://dx.doi.org/10.1089/scd.2008.0172; PMID: 18694296
- Gillenwater AM, Zhong M, Lotan R. Histone deacetylase inhibitor suberoylanilide hydroxamic acid induces apoptosis through both mitochondrial and Fas (Cd95) signaling in head and neck squamous carcinoma cells. Mol Cancer Ther 2007; 6:2967 - 75; http://dx.doi.org/10.1158/1535-7163.MCT-04-0344; PMID: 18025281
- Mielcarek M, Benn CL, Franklin SA, Smith DL, Woodman B, Marks PA, Bates GP. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS One 2011; 6:e27746; http://dx.doi.org/10.1371/journal.pone.0027746; PMID: 22140466
- Benn CL, Butler R, Mariner L, Nixon J, Moffitt H, Mielcarek M, Woodman B, Bates GP. Genetic knock-down of HDAC7 does not ameliorate disease pathogenesis in the R6/2 mouse model of Huntington’s disease. PLoS One 2009; 4:e5747; http://dx.doi.org/10.1371/journal.pone.0005747; PMID: 19484127
- Tormo D, Checińska A, Alonso-Curbelo D, Pérez-Guijarro E, Cañón E, Riveiro-Falkenbach E, Calvo TG, Larribere L, Megías D, Mulero F, et al. Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell 2009; 16:103 - 14; http://dx.doi.org/10.1016/j.ccr.2009.07.004; PMID: 19647221
- Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A 2004; 101:18030 - 5; http://dx.doi.org/10.1073/pnas.0408345102; PMID: 15596714
- Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25:1025 - 40; http://dx.doi.org/10.1128/MCB.25.3.1025-1040.2005; PMID: 15657430
- Ciechomska IA, Goemans GC, Skepper JN, Tolkovsky AM. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene 2009; 28:2128 - 41; http://dx.doi.org/10.1038/onc.2009.60; PMID: 19347031
- Geng Y, Kohli L, Klocke BJ, Roth KA. Chloroquine-induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent. Neuro Oncol 2010; 12:473 - 81; PMID: 20406898
- Kim EL, Wüstenberg R, Rübsam A, Schmitz-Salue C, Warnecke G, Bücker EM, Pettkus N, Speidel D, Rohde V, Schulz-Schaeffer W, et al. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro Oncol 2010; 12:389 - 400; http://dx.doi.org/10.1093/neuonc/nop046; PMID: 20308316
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062 - 75; http://dx.doi.org/10.1016/j.cell.2009.03.048; PMID: 19524509
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961 - 7; http://dx.doi.org/10.1038/nrc2254; PMID: 17972889
- O’Donovan TR, O’Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011; 7:509 - 24; http://dx.doi.org/10.4161/auto.7.5.15066; PMID: 21325880
- Guo XL, Li D, Hu F, Song JR, Zhang SS, Deng WJ, Sun K, Zhao QD, Xie XQ, Song YJ, et al. Targeting autophagy potentiates chemotherapy-induced apoptosis and proliferation inhibition in hepatocarcinoma cells. Cancer Lett 2012; 320:171 - 9; http://dx.doi.org/10.1016/j.canlet.2012.03.002; PMID: 22406827
- Chen N, Debnath J. Autophagy and tumorigenesis. FEBS Lett 2010; 584:1427 - 35; http://dx.doi.org/10.1016/j.febslet.2009.12.034; PMID: 20035753
- Goss P, Allan AL, Rodenhiser DI, Foster PJ, Chambers AF. New clinical and experimental approaches for studying tumor dormancy: does tumor dormancy offer a therapeutic target?. APMIS 2008; 116:552 - 68; PMID: 18834402
- Fan QW, Weiss WA. Autophagy and Akt promote survival in glioma. Autophagy 2011; 7:536 - 8; http://dx.doi.org/10.4161/auto.7.5.14779; PMID: 21266843
- Sundberg AL, Steffen AC. Enhancing the effect of radionuclide tumor targeting, using lysosomotropic weak bases. Int J Radiat Oncol Biol Phys 2007; 67:279 - 87; http://dx.doi.org/10.1016/j.ijrobp.2006.07.1369; PMID: 17189076
- Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavaré S, Arakawa S, Shimizu S, Watt FM, et al. Autophagy mediates the mitotic senescence transition. Genes Dev 2009; 23:798 - 803; http://dx.doi.org/10.1101/gad.519709; PMID: 19279323
- Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett 2009; 280:125 - 33; http://dx.doi.org/10.1016/j.canlet.2009.02.042; PMID: 19359091
- Boocock DJ, Faust GE, Patel KR, Schinas AM, Brown VA, Ducharme MP, Booth TD, Crowell JA, Perloff M, Gescher AJ, et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol Biomarkers Prev 2007; 16:1246 - 52; http://dx.doi.org/10.1158/1055-9965.EPI-07-0022; PMID: 17548692
- Pasquier E, Kavallaris M, André N. Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol 2010; 7:455 - 65; http://dx.doi.org/10.1038/nrclinonc.2010.82; PMID: 20531380
- Hsin IL, Sheu GT, Chen HH, Chiu LY, Wang HD, Chan HW, Hsu CP, Ko JL. N-acetyl cysteine mitigates curcumin-mediated telomerase inhibition through rescuing of Sp1 reduction in A549 cells. Mutat Res 2010; 688:72 - 7; http://dx.doi.org/10.1016/j.mrfmmm.2010.03.011; PMID: 20363232