Abstract
The ARF tumor suppressor, encoded by the CDKN2A gene, has a well-defined role regulating TP53 stability; this activity maps to exon 1β of CDKN2A. In contrast, little is known about the function(s) of exon 2 of ARF, which contains the majority of mutations in human cancer. In addition to controlling TP53 stability, ARF also has a role in the induction of autophagy. However, whether the principal molecule involved is full-length ARF, or a small molecular weight variant called smARF, has been controversial. Additionally, whether tumor-derived mutations in exon 2 of CDKN2A affect ARF’s autophagy function is unknown. Finally, whereas it is known that silencing or inhibiting TP53 induces autophagy, the contribution of ARF to this induction is unknown. In this report we used multiple autophagy assays to map a region located in the highly conserved 5′ end of exon 2 of CDKN2A that is necessary for autophagy induction by both human and murine ARF. We showed that mutations in exon 2 of CDKN2A that affect the coding potential of ARF, but not p16INK4a, all impair the ability of ARF to induce autophagy. We showed that whereas full-length ARF can induce autophagy, our combined data suggest that smARF instead induces mitophagy (selective autophagy of mitochondria), thus potentially resolving some confusion regarding the role of these variants. Finally, we showed that silencing Tp53 induces autophagy in an ARF-dependent manner. Our data indicated that a conserved domain in ARF mediates autophagy, and for the first time they implicate autophagy in ARF’s tumor suppressor function.
Keywords: :
Introduction
The CDKN2A locus produces two independent transcripts controlled by alternative promoters; these transcripts generate the proteins p16INK4a and p14ARF (p14ARF in human and p19ARF in mouse; hereafter referred to as ARF when referring to the protein for both species, and ARF or Arf for the respective human or murine nucleotide sequences). These transcripts have unique first exons, but share exon 2; however, in exon 2 they utilize alternate reading frames, so these two tumor suppressor proteins share no homology.Citation1 Both protein products are bona fide tumor suppressors: p16INK4a regulates the RB1 pathway, and ARF regulates TP53/p53.Citation2 In addition to its rather unusual gene structure, ARF also has unusual protein-coding potential. For example, the murine Arf transcript uses more than one initiating methionine for translation. The full-length murine ARF protein is 169 amino acids and 19 kilodaltons (kDa) in molecular mass. In addition, a small fraction of murine Arf transcript initiates translation at methionine 45, and encodes a protein of 14 kDa; this variant has been denoted smARF (see diagram, Fig. S1A).Citation3 In the case of human ARF, whereas the full-length human ARF coding region has the potential to encode a protein of 173 amino acids, the predominant species is 132 amino acids, encoding a protein of 14 kDa.Citation4
In addition to its well-known role in the control of TP53, the ARF tumor suppressor protein can also induce autophagy. However, there have been conflicting reports regarding this function. Kimchi and colleagues have reported that the smARF variant of this protein traffics preferentially to mitochondria and is the only form of ARF that can induce autophagy.Citation3,Citation5 Subsequently Abida and Gu have reported that full-length ARF, with the downstream methionine at codon 45 mutated to alanine, was capable of inducing autophagy in transfected cells, and conversely that smARF was incapable.Citation6 Similar confusion exists for human ARF: whereas some groups have reported that human ARF can induce autophagy,Citation6 other groups have reported that neither human ARF nor smARF can induce autophagy.Citation7 Some of this confusion may be caused by the use of transient transfection and overexpression of ARF in these studies, along with the use of limited assays used for autophagy. Unfortunately, these conflicting data have led to questions about the domains of ARF required for autophagy, and uncertainty regarding the importance of autophagy for ARF function. What is needed to resolve these discrepancies is a comprehensive analysis of the ability of human and murine ARF to induce autophagy.
In this report we address the role of ARF and smARF in autophagy, and we map the domain(s) of human and mouse ARF that are required. Toward this goal, we created inducible cell lines for each variant in order to avoid transfection artifacts and supraphysiological expression levels. By using low concentrations of doxycycline, we were able to induce ARF protein to levels that are equivalent to the levels seen in cultured tumor cell lines, or in cells with silenced Trp53. Additionally, we use up to four different autophagy assays for each clone analyzed. These studies have led to a clearer picture of the role of ARF and smARF in autophagy; specifically, we showed that full-length ARF can induce autophagy, but that the murine smARF variant instead activates selective autophagy of mitochondria (mitophagy). Using deletion mutants, we have mapped the domains required for autophagy by murine and human ARF, and importantly find that the same highly conserved domain located at the 5′ end of exon 2 is required. Moreover, we showed that tumor-derived mutations of exon 2 of CDKN2A that alter ARF coding potential, but are silent for p16INK4a, impair the ability of ARF to induce autophagy; this finding implicates ARF-mediated autophagy in tumor suppression by this protein. Finally, we confirmed the finding of others that silencing of Trp53 induces autophagy. However, for the first time, we show that ARF is required for this process.
Results
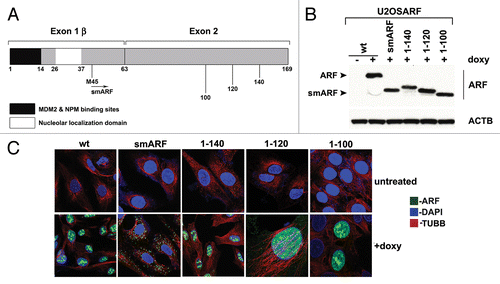
Our group and others previously reported that murine p19ARF (hereafter referred to as ARF) induces autophagy.Citation6,Citation8 We sought to map the domain(s) of ARF required for autophagy induction. Toward this goal we generated several stably transfected inducible versions of murine ARF in the doxycycline-regulatable U2OS cell line. These included inducible cell lines for full-length ARF (amino acids, a.a. 1 to 169), smARF (a.a. 45 to 169) and deletion mutants encoding amino acids 1 to 140, 1 to 120 and 1 to 100 (). For the C-terminal deletion mutants (a.a. 1 to 140, 1 to 120 and 1 to 100) we confirmed that these mutants were properly folded, as they showed comparable ability to bind to MDM2 (data not shown) and stabilize TP53 (see below) compared with wild-type (wt) ARF. We cultured cells containing all deletion mutants in the presence of doxycycline (0.1 μg/ml) for 24 h, and selected clones with comparable levels of ARF for further analysis (). To ensure that the amount of Arf mRNA induced in these cells were within physiologically relevant levels, we compared these levels to that in cells that express endogenous ARF. As ARF mRNA is negligibly detectable in normal, nontransformed adult cells,Citation2 we compared the levels of ARF mRNA induced by doxycycline to that in human tumor cells with mutant TP53, or in murine cells in which Trp53 is silenced (as TP53 transcriptionally represses ARFCitation4). We found that these levels are comparable (Fig. S1B). For all of the analyses described below, we confirmed our results in a minimum of two clones for each deletion mutant. We first analyzed the cellular localization of each ARF variant using immunofluorescence analysis. After doxycycline treatment, we observed that wt ARF was predominantly localized in a punctate pattern consistent with nucleolar staining; a similar staining pattern was observed for the C-terminal deletion mutants 1 to 140, 1 to 120 and 1 to 100 (). In contrast, murine smARF, which lacks the nucleolar localization sequence (a.a. 26 to 37) showed a cytosolic punctate localization consistent with mitochondrial localization (). Consistent with this premise and the findings of others,Citation3,Citation6 we found that murine smARF costained with MitoTracker Red, a marker for mitochondria, although some nucleo-cytoplasmic staining of smARF was also evident in some experiments (Fig. S1C).
Figure 1. Generation of inducible cell lines for deletion mutants of mouse ARF. (A) Schematic of the coding region of Arf. Exon 1β contains the binding sites for MDM2 and nucleophosmin/B23 (NPM). Exon 2, shared in an alternative reading frame with the sequence for the p16INK4a protein, is the site for the majority of tumor-derived mutations in CDKN2A. Internal initiation of translation at methionine M45 generates a short form of ARF, denoted smARF. The nucleolar localization sequence is denoted. (B) Western blot analysis of murine ARF deletion mutants in stably transfected clones of U2OS-ARF cell lines untreated or treated with 0.1 µg/mL doxycycline (doxy) for 24 h. Actin (ACTB) is included as a loading control. Bands for ARF and smARF are denoted; note that a small amount of smARF is generated in the wt ARF-induced cells (lane 2). (C) Immunofluorescence analysis of U2OS-ARF cells treated with 0.1 µg/mL doxycycline (+doxy) for 48 h, immunostained with antisera to murine ARF (green) and β-tubulin (TUBB, red), and stained with DAPI (blue).
The mitochondrial localization domain maps to amino acids 45 to 100
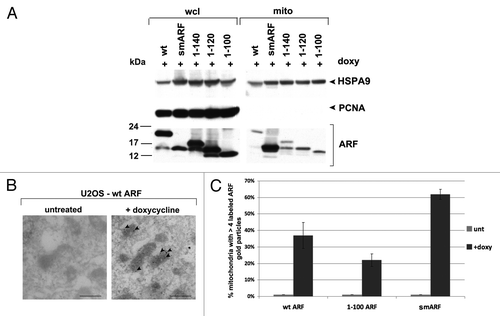
We next used our cell lines containing ARF deletion mutants in order to map the domain required for mitochondrial localization of ARF. Toward this end we cultured U2OS-ARF cells in the presence of doxycycline for 24 h, and used a mannitol gradient fractionation procedure to isolate highly purified mitochondria.Citation9 The purity of these mitochondrial fractions was monitored by western analysis for HSPA9 (also called GRP75) to monitor mitochondrial purity, and PCNA, to control for contamination from the nuclear fraction. Western blot analysis of whole cell vs. mitochondrial lysates revealed that wild type ARF, smARF and all of the ARF deletion mutants copurified with mitochondria (). Notably however, there was greatly enhanced copurification of smARF with mitochondria, compared with wt ARF and the C-terminal deletion mutants (). These data are consistent with the findings of Kimchi, who reports enhanced mitochondrial localization of this internal translation variant.Citation3 We next confirmed these results using immuno-electron microscopy with ARF antisera and Protein-G Gold. For these studies, we chose to focus on three variants: wt ARF, smARF, and the 1 to 100 mutant. Immuno-electron microscopy for these variants confirmed that all three versions localize to mitochondria, but that smARF shows enhanced localization (). The combined data indicate that the mitochondrial localization sequence of murine ARF maps to amino acids 45 to 100; notably this region overlaps with the region of human ARF (a.a. 41 to 64) found to function as a mitochondrial localization sequence when fused to GFP.Citation7
Figure 2. The mitochondrial localization sequence of murine p19ARF maps to amino acids 45 to 100; murine smARF shows enhanced localization to mitochondria. (A) Western blot analysis of whole cell lysate (wcl) vs. lysate from purified mitochondria (mito) isolated from U2OS-ARF cells following treatment with doxycycline for 24 h (doxy, 0.1 μg/ml). Lysates were probed for the mitochondrial protein HSPA9 (GRP75) and the nuclear/cytosolic protein PCNA as an assessment of purity. The bottom lanes depict ARF protein level in these fractions. The data depicted are representative of three independent experiments, for multiple clones for each mutant. (B) Immuno-electron microscopy using ARF antisera followed by protein G-Gold in U2OS-ARF cells following 24 h treatment with doxycycline. The arrowheads depict gold particles colocalizing with mitochondria. Scale bar: 500 nm. (C) Graphic analysis of the percent of mitochondria immunostained with ARF antisera in (B). Mitochondria with four or greater gold particles were considered positive, and at least 200 mitochondria were counted for each sample.
The ARF autophagy domain maps to amino acids 100 to 120
We next chose to define the domain(s) of ARF required for autophagy induction. To do this, we performed time course analyses in ARF-inducible cells for the level of SQSTM1/p62 (hereafter SQSTM1), which is an adaptor protein that is degraded by autophagy.Citation10 We also used western blot analysis to monitor the level of MAP1LC3A protein (hereafter LC3-I and -II); LC3-I is converted into the faster-migrating, lipid-conjugated form, LC3-II, when autophagy is induced. Western analysis for SQSTM1 revealed that wt ARF, and the 1 to 140 and 1 to 120 mutants, were capable of inducing SQSTM1 degradation as early as 24 h after ARF induction (); these mutants were also capable of causing LC3-II accumulation (). In contrast, the 1 to 100 and smARF mutants both failed to induce SQSTM1 degradation, and induced minor amounts of LC3-II (). Interestingly, the 1 to 100 and smARF mutants led to an accumulation, instead of decrease, of SQSTM1; in contrast, doxycycline treatment of parental cells did not cause an accumulation of SQSTM1 (Fig. S1D), raising the possibility that these ARF proteins function in a dominant-negative manner. We did not pursue this latter observation further.
Figure 3. The murine ARF autophagy domain maps to amino acids 100 to 120. (A) Western blot analysis of U2OS-ARF cells containing the deletion mutants indicated, untreated or treated with doxycycline (doxy, 0.1 μg/ml) for the indicated time points (hours). Degradation of SQSTM1/p62 (SQSTM1) and accumulation of LC3-II (bottom LC3 band) are indicators of autophagy induction. Actin (ACTB) is included as a loading control. The data depicted are representative of three independent experiments, for multiple clones of each mutant. (B) Western analysis of the level of SQSTM1 and LC3-I and -II following cessation of autophagic flux with 10 mM NH4Cl. Note that whereas wt ARF induces increased LC3-II, above that which accumulates following NH4Cl (compare lanes 4 and 3), there is no increase in LC3-II in smARF-induced cells (compare lanes 8 and 7). (C) Electron microscopy of U2OS-ARF cells containing wild-type ARF (wt) or the deletion mutants indicated, untreated (unt) or treated with doxycycline (0.1 µg/mL) for 48 h. The arrowheads point to autophagic vesicles that appear following ARF induction. The average area of autophagosomes, calculated with ImageJ software, is indicated below each panel. The data depicted are representative of three independent experiments, in multiple independent clones of each mutant. Scale bar: 500 nm. (D) Western blot analysis of U2OS-ARF cells containing wild type ARF (wt) or the valine-to-alanine mutant at codon 113 (V113A), untreated or treated with doxycycline (doxy, 0.1 μg/ml) for 24 h. Degradation of SQSTM1 and accumulation of LC3-II are indicators of autophagy induction. Actin (ACTB) is included as a loading control. The data depicted are representative of three independent experiments.
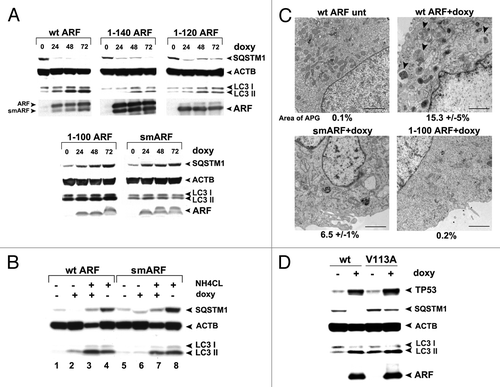
Because analysis of LC3-II levels to assess autophagy induction are most accurately measured when autophagic flux is halted, we next repeated measurements of LC3-II in the presence of NH4Cl, which inhibits lysosome function and halts autophagic flux.Citation10 This analysis revealed that wt ARF led to increased LC3-II even when autophagic flux is halted; in contrast, smARF did not induce LC3-II to levels above that induced by NH4Cl alone ( compare lanes 4 to 3, and 8 to 7). These data suggest that smARF is unable to induce autophagy to an appreciable extent. To confirm and extend these findings, we performed transmission electron microscopy (EM) to analyze autophagosome formation. As expected, numerous autophagosomes were observed in cells expressing wt ARF following doxycycline treatment, which were not observed in untreated cells (). We quantitated the area of autophagosomes and found that this area increased from 0.1% to 15.3% when ARF was induced. Conversely, smARF had markedly reduced ability to induce autophagosomes, and multiple clones of the 1 to 100 mutant failed to induce autophagosomes (). To ensure that the 1 to 100 protein was folded correctly, we analyzed the ability of this protein to stabilize TP53, and found that it has comparable, if not increased, ability to stabilize TP53 compared with wt ARF (Fig. S1E). The combined data indicate that the smARF variant is greatly impaired for autophagosome formation compared with wt ARF, and that a region comprising amino acids 100 to 120 in exon 2 is required for ARF-mediated autophagy.
In an effort to further confirm the relevance of amino acids 100 to 120 to autophagy induction by murine ARF, we cloned amino acids 90 to 120 of ARF downstream of a hemagluttinin tag (HA-90 to 120). To determine whether this protein could function autonomously to induce autophagy, we enforced the expression of the HA-90 to 120 construct in transfected cells, and assayed for SQSTM1 degradation and LC3-II accumulation. Unfortunately, we discovered that this protein is very poorly expressed in transfected cells, and likely is misfolded (A. Budina-Kolomets, data not shown). Therefore, in an additional attempt to assess the importance of this region to autophagy induction by ARF, we next generated several point mutants in this region, selected at random. One of these mutants, which converts valine at amino acid 113 to alanine (V113A) maintains the ability to stabilize TP53, but consistently has impaired ability to induce autophagy, as assessed by SQSTM1 degradation and LC3-II accumulation (). This point mutant supports the relevance of amino acids 100 to 120 to autophagy induction by murine ARF. Notably, the V113A mutant is also the first point mutant that seperates the autophagy-inducing activity of ARF from its ability to stabilize TP53.
smARF induces mitophagy
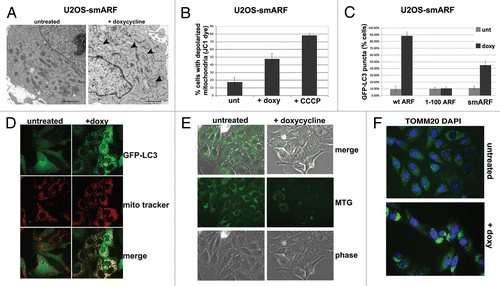
Published reports indicate that the smARF variant can induce autophagy; in these reports, the authors used assays for GFP-LC3 vacuoles as well as loss of mitochondrial transmembrane potential.Citation3 In contrast, our data indicated that smARF fails to induce SQSTM1 degradation, or to induce LC3-II when autophagic flux is halted. Additionally, we found that multiple independent inducible smARF clones showed significantly reduced ability to induce autophagosomes (see for example ). Interestingly, our EM data revealed that the majority of mitochondria in smARF-induced cells had grossly abnormal morphology (). Therefore one reconciliation of these combined data might be that smARF alters mitochondrial integrity, leading to mitophagy (selective autophagy of mitochondria). We therefore sought to test this hypothesis. We first confirmed in our cell lines that, as described by Kimchi,Citation3 smARF induction leads to a loss of mitochondrial transmembrane potential () and an increase in GFP-positive vacuoles in GFP-LC3 transfected cells (). We next costained smARF-induced cells with GFP-LC3 (autophagic vacuoles) along with MitoTracker Red (mitochondria). This experiment revealed that the overwhelming majority of GFP-LC3 vacuoles colocalized with MitoTracker Red in smARF-induced cells (). To further support the premise that smARF might induce mitophagy, we assessed the change in mitochondrial mass in smARF-induced cells using MitoTracker Green staining. We also analyzed the immunostaining of TOMM20, a mitochondrial protein that has been shown to cluster at perinuclear spaces when mitophagy is induced.Citation11 MitoTracker Green staining revealed a marked decrease in mitochondrial mass 48 h after smARF induction (). These experiments also revealed marked perinuclear clustering of TOMM20 in smARF-induced cells after 24 h, consistent with induction of mitophagy (). As a final test of the possibility that smARF might induce mitophagy, we next analyzed smARF- and ARF-induced cells for the mobilization of SQSTM1 to purified mitochondria, which is another indicator of mitophagy.Citation12 As a control for this experiment, we first showed that the mitochondrial uncoupler CCCP, which induces mitophagy, causes SQSTM1 to copurify with mitochondria in both U2OS-ARF and –smARF clones when uninduced, in the absence of doxycycline (Fig. S1F). Notably, we found that SQSTM1 copurified with mitochondria only when smARF, but not ARF, was induced with doxycycline (Fig. S1G). The data from these six assays best support a model whereby smARF induces preferential autophagy of mitochondria, or mitophagy, but not non-selective (macro-) autophagy; this function is consistent with smARF’s greatly enhanced mitochondrial localization.
Figure 4. smARF induces morphological indicators of mitophagy. (A) Electron microscopy of U2OS-smARF cells untreated or treated with doxycycline for 48 h (0.1 µg/mL). The arrowheads indicate swollen mitochondria evident after smARF induction. The data depicted are representative of several independent experiments, and two different subclones of U2OS-smARF cells. Scale bar: 500 nm. (B) U2OS-smARF cells were treated with either doxycycline (doxy, 0.1 µg/mL) or a positive control (the mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone, CCCP, 10 μM) for 48 h or left untreated (unt). The mitochondrial transmembrane potential was measured using the flow cytometry-based MitoPotential assay from Millipore (Guava EasyCyte). Note that induction of smARF increases the percentage of cells with depolarized mitochondria. The graph depicts data from three independent experiments, with standard error. (C) U2OS-ARF cells were transiently transfected with GFP-LC3 plasmid for 24 h, treated with doxycycline for 48 h (0.1 µg/mL) and examined by confocal microscopy. The chart shows the percentage of cells containing more than four GFP-LC3 vacuoles compared with untreated controls. The results are the average plus standard error of three independent experiments in which at least 200 cells were counted for each sample. (D) Immunofluorescence analysis of U2OS-smARF cells transiently transfected with GFP-LC3 plasmid for 24 h, then untreated or treated with doxycycline for 48 h (0.1 µg/mL) and stained with MitoTracker Red. The bottom panels depict the colocalization of GFP-LC3 vacuoles with MitoTracker stain following smARF induction. (E) U2OS cells with inducible smARF were left untreated or incubated with 0.1 µg/mL doxycycline for 48 h, and mitochondrial mass was assessed following incubation with MitoTracker Green (MTG). The data depicted are representative of three independent experiments. (F) U2OS cells with inducible smARF were left untreated or incubated with 0.1 µg/mL doxycycline for 24 h, and then analyzed by immunofluorescence for TOMM20. The data depicted are representative of three independent experiments.
A conserved C-terminal region is required for autophagy by human ARF
Although several reports have shown that most of ARF's tumor supressor functions are conserved in mice and humans, the majority of the studies analyzing autophagy have utilized murine ARF. Therefore, we sought to confirm that human ARF could induce autophagy as well, and to see if homologous domain(s) of human ARF are required. To accomplish this, we first compared the coding sequences for the generation of human and murine ARF proteins. This alignment revealed that exon 1β of human ARF has the potential to encode an extra 41 amino acids at the N-terminus (starting at amino acids MGRG, see Fig. S1A). To our knowledge, no group has analyzed the function of this cDNA. Using primers specific for this extra N-terminal region, we were able to show that mRNA transcripts containing this region exist in several human tumor cell lines, including Capan2 and PC3 (Fig. S2A), and we were able to clone this full-length cDNA from RNA isolated from Capan2 cells. However, upon transfection of this cDNA in human tumor cell lines, or following generation of tetracycline-inducible clones in U2OS cells using this cDNA, we found little evidence for initiation of translation from the upstream methionine; instead, the predominant species of ARF generated from this construct is clearly the 14 kDa variant, which initiates at sequence MVRR in the amino acid 1 to 132 species of human ARF; western blotting using antisera specific for the higher molecular weight species confirms this assessment (data not shown). Therefore, whereas we opted to use this ‘full-length’ ARF cDNA as the template for our deletion mutants, our data indicate that translation from the +1 MVRR sequence occurs predominantly; because of this, and in order to remain consistent with the numbering system of human ARF used by others, we have opted to denote this version of ARF −41/+132, again with the consideration that translation from this construct occurs predominantly from the methionine designated +1 in this construct (see for example ).
Figure 5. A conserved region in the C terminus of ARF is required for autophagy in both human and mouse. (A) Schematic representation of deletion mutants of human ARF p14ARF (ARF) that were cloned into the U2OS/Tet-On-inducible cell line (U2OS-TREx). To keep the numbering of human ARF consistent with other studies, the extended transcript is denoted −41/+132. (B) Protein levels of ARF deletion mutants, and level of TP53 induced by each mutant, in inducible cells following 24 h in doxycycline (doxy, 0.1 µg/mL). Note that the monoclonal antibody that is used to detect ARF in the left two samples (−41/+132 and 1–132) does not recognize the −41/+99 and −41/+59 deletion mutants; therefore, for the right two samples (−41/+99 and −41/+59), a different antibody, which maps to the N-terminus of ARF, was used (Abcam, Ab14930). (C) Western blot analysis of SQSTM1 in U2OS cells following induction of full-length ARF (−41/+132), the −41/+59 mutant, and the 1 to 132 protein. Actin (ACTB) is indicated as a loading control. Note that the −41/+59 mutant lacks the epitope detected by the Sigma ARF antibody. (D) Electron microscopy of U2OS-ARF cells untreated or treated with doxycycline (0.1 µg/mL) for 48 h. The average and standard deviation of the area of autophagosomes (APG), calculated with ImageJ software, is indicated on the bottom. The results were consistent in three independent experiments, using two clones for each mutant. Scale bar: 500 nm. (E) U2OS-ARF cells were transiently transfected with GFP-LC3 plasmid for 24 h and untreated or treated with doxycycline for 48 h (0.1 µg/mL), and then analyzed by confocal microscopy. Cytoplasmic GFP-LC3 vesicles were detectable in full-length (−41/+132) ARF cells and for the 1 to 132 version, but not in cells expressing the −41/+99 deletion mutant. (F) The percentage of cells with four or more GFP-LC3 vacuoles in (E) was quantified in three independent experiments; error bars mark standard error.
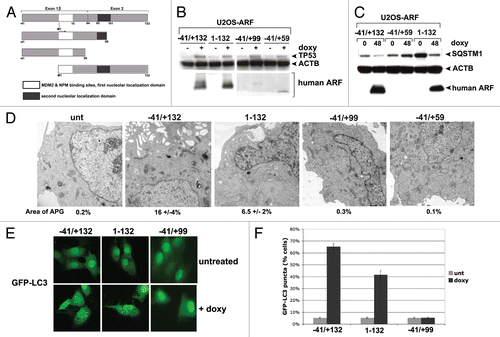
From the human ARF cDNA (−41/+132) we generated the following deletion mutants: a cDNA initiating exclusively from the +1 methionine (1 to 132) and two different C-terminal truncation mutants (−41/+99 and −41/+59, see diagram ); the −41/+99 deletion mutant represents a deletion of the 20 amino acids required for autophagy induction by murine ARF (see map, Fig. S1A). These constructs were cloned into a doxycycline-responsive promoter and used to create stable transfectants in U2OS-TRex cells; as in the case of our murine ARF-inducible clones, we confirmed that the level of ARF protein induced in these clones was physiologically-relevant (that is, comparable to that in human tumor lines mutant or null for TP53, Fig. S1B). We also confirmed by real-time RT-PCR that the level of transcripts induced for each mutant following doxycycline treatment was comparable (Fig. S2B). Because of the lack of an antibody that could recognize all four human ARF proteins, we were unable to directly compare the levels of ARF protein induced in each clone; however, we were able to show that ARF protein was induced in all clones, and further that each mutant had comparable ability to stabilize TP53, suggesting that similar levels of deletion mutants were generated (). Further, we repeated all experiments in multiple clones of each mutant (data not shown).
Analysis of the level of SQSTM1 in our human ARF clones indicated that the −41/+132 version of ARF, and the 1 to 132 version, both caused decreased SQSTM1 after induction with doxycycline (). In contrast, the −41/+99 and −41/+59 mutants showed no change in SQSTM1 level ( and data not shown). We next assessed autophagy induction in our U2OS-ARF clones using GFP-LC3 analysis as well as EM. We found that the −41/+132 and 1 to 132 human ARF clones were consistently able to induce autophagosomes () and GFP-LC3 vacuoles (), but the −41/+99 and −41/+59 mutants were not ( and data not shown). To ensure that TP53 was not involved in any of these functions of human ARF, we also created inducible cell lines for human ARF in the human H1299 (TP53-null adenocarcinoma) doxycycline-inducible cell line. Analysis of GFP-LC3 vacuoles and autophagosomes by electron microscopy confirmed that human ARF induces autophagy in TP53-null cells (Fig. S3A–S3C), confirming the lack of a requirement for TP53 in autophagy induced by ARF. The combined data indicate that the autophagy function of ARF is conserved between human and mouse, and further that an overlapping region in exon 2 of CDKN2A is required in both species.
Tumor-derived mutants of ARF fail to induce autophagy
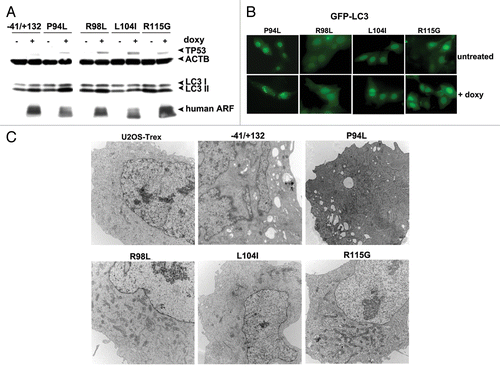
The conserved region of ARF that is required for autophagy lies at the 5′ end of exon 2, where the majority of mutations in CDK2NA occur in human cancer (Fig. S1A). Interestingly, an estimated 7% of mutations in this region of exon 2 of CDKN2A are predicted to alter the ARF protein coding region, but are silent in the p16INK4a gene product.Citation13 Because autophagy is known to be suppressive to tumor initiation,Citation14 we chose to test the hypothesis that some of the tumor-derived mutations in exon 2 of CDKN2A that alter ARF but not p16INK4a protein coding region might have altered autophagy function. Toward this end, we generated cell lines containing doxycycline-inducible mutants encoding R98L (arginine at amino acid 98 to leucine), L104I (leucine at amino acid 104 to isoleucine) and R115G in ARF (arginine at amino acid 115 to glycine).Citation13 As a negative control, we generated a point mutant in this region that is known to alter the coding region for both the ARF protein as well as the p16INK4a protein, but which is known to impair the ability of p16INK4a to inhibit CDK4; this mutant is P94L (proline at amino acid 94 to leucine).Citation15 Western analysis for ARF and TP53 level 24 h after induction of these mutants revealed that all expressed equal levels of ARF, and each had comparable ability to stabilize TP53 protein (). Notably, whereas wt ARF and the P94L mutant showed comparable ability to induce LC3-II, the three tumor-derived mutants that affect only ARF coding region had reduced ability (particularly L104I and R115G; ). We next transfected these cells for 24 h with GFP-LC3, and monitored GFP-LC3 vesicle formation 24 h after doxycycline addition. This experiment revealed that the P94L mutant was effective at inducing GFP-LC3 vesicles, but that the R98L, L104I and R115G mutants were completely incapable (). To confirm the premise that these tumor-derived mutants of ARF fail to induce autophagy, we next performed electron microscopy for autophagosomes. Both wt and the P94L mutant of ARF showed consistent ability to induce autophagosomes following ARF induction; however, the R98L, L104I and R115G mutants were unable (). All five cell lines, however, showed identical induction of autophagy by HBSS, indicating these clones were not defective in autophagy induction in response to other stimuli (A. Budina-Kolomets, data not shown). In sum, these findings show that mutations in exon 2 of CDKN2A that affect ARF coding region but not p16INK4a all impair the autophagy function of this protein.
Figure 6. Tumor-derived mutants of ARF are impaired for autophagy. (A) Western blot analysis of U2OS-ARF cells containing wild-type ARF (−41/+132) or the mutants indicated, untreated or treated with doxycycline (doxy, 0.1 μg/ml) for 24 h. Stabilization of TP53 and accumulation of LC3-II are indicated. Actin (ACTB) is included as a loading control. The data depicted are representative of three independent experiments, for multiple clones of each mutant. (B) U2OS-ARF cells containing the mutants indicated were transiently transfected with GFP-LC3 plasmid for 24 h and untreated or treated with doxycycline for 24 h (0.1 µg/mL), and then analyzed by confocal microscopy for GFP-LC3 vesicles. Cytoplasmic GFP-LC3 vesicles were detectable in the P94L mutant, but not in the R98L, L104I and R115G mutants. (C) Electron microscopy of autophagosomes in U2OS-ARF cells containing the ARF mutants indicated, or parental cells (U2OS-TRex), untreated or treated with doxycycline (0.1 µg/mL) for 48 h. The results depicted were consistent in three independent experiments, using two clones for each mutant. Scale bar: 500 nm.
TP53 negatively regulates ARF-induced autophagy
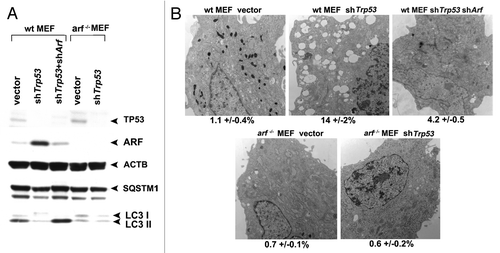
We next sought to analyze a physiologically-relevant scenario whereby ARF-mediated autophagy is induced. The role of TP53 in autophagy is complex. TP53 is required for autophagy induced by certain chemotherapeutic compounds,Citation16 but TP53 also inhibits basal autophagy.Citation17-Citation19 Consistent with an inhibitory role for TP53 in autophagy, we previously reported that silencing Trp53 in mouse embryo fibroblasts (MEFs) is sufficient to induce ARF and autophagy.Citation8 We therefore chose to address the possibility that the autophagy induced by TP53 inhibition relies in part on ARF upregulation. In normal nontransformed cells the level of Arf mRNA is extremely low, in part because TP53 negatively regulates the expression of this gene.Citation20,Citation21 To test the contribution of ARF to autophagy induced by Trp53 silencing, we isolated mouse embryonic fibroblasts from wild-type and Arf knockout mice, and infected these with a retrovirus expressing a short hairpin to Trp53 (shTrp53). As expected, silencing of the Trp53 gene in wt MEFs led to induction of ARF and degradation of SQSTM1; notably, however, silencing of Trp53 in arf−/− MEFs did not lead to SQSTM1 degradation, despite the fact that Trp53 was efficiently silenced (). These data were supported by electron microscopy, which revealed that autophagosomes were induced in wt MEFs infected with shTrp53, but not in arf−/− MEFs infected with this short hairpin (). To further confirm this finding, we compared the level of SQSTM1 and autophagosomes in wt MEFs infected with shTrp53, to that in wt MEFs infected with both shTrp53 and a short hairpin to ARF (shArf). These experiments revealed that each of the autophagy markers induced by silencing Trp53 (SQSTM1 degradation and autophagosome formation) was effectively eradicated when cells were simultaneously infected with a short hairpin to Arf (). Similar findings were made when analyzing LC3-II in the presence of NH4Cl to freeze autophagic flux (Fig. S3D). These data show for the first time that autophagy induced by silencing of Trp53 is dependent upon the resultant upregulation of ARF, at least in MEFs.
Figure 7. Autophagy induced by Trp53 silencing requires ARF. (A) Primary mouse embryo fibroblasts (wt MEFs) and MEFs from the Arf knockout mouse (arf−/− MEFs) were infected with parental MLP retrovirus (vector), retroviral short hairpin to Trp53 (shTrp53), or retroviral short hairpin to Arf (shArf). Whole cell lysates following infection and selection were subjected to western blot analysis using the antibodies indicated. The level of autophagy induced is assessed by the degradation of SQSTM1; the lower molecular weight SQSTM1 band is thought to be a degradation product. Note that silencing of Trp53 in arf−/− MEFs, and silencing of both Trp53 and Arf in wt MEFs, does not induce appreciable autophagy. The data presented are representative of three independent experiments, in two independent batches of MEFs from each genotype. (B) Electron microscopy of wild-type and arf−/− MEFs following infection with the retroviruses indicated. The average area of autophagosomes calculated with ImageJ software, with standard error, is indicated on the bottom of each panel. Scale bar: 500 nm.
Discussion
It is presently thought that the chief role of the ARF tumor suppressor protein is to suppress hyperproliferative signals in precancerous cells, in part by inhibiting MDM2 and stabilizing the TP53 tumor suppressor protein.Citation2,Citation22 Additionally, ARF possesses TP53-independent tumor suppressor function. For example, overexpression of ARF can suppress the proliferation of Trp53-null cells,Citation23-Citation25 and triple knockout mice null for Trp53, Mdm2 and Arf show increased tumor spectrum and aggressiveness compared with double-knockout mice null for only Trp53 and Mdm2.Citation26 The TP53-independent tumor suppressor function of ARF has multiple potential pathways, including inhibition of ribosome biogenesis,Citation22,Citation27 upregulation of TIMP3,Citation28 maintenance of chromosomal stability,Citation29,Citation30 activation of ATM-ATR-CHEK pathways,Citation2 and nucleolar sequestration of cancer-relevant transcription factors, including MYC and E2F1,Citation22,Citation31 among others. In addition to these functions, several groups have shown that ARF also plays a role in autophagy. However, this literature has been fraught with contradictory findings. Kimchi and colleagues have been the first to report that murine smARF, but not full-length ARF, could traffic to mitochondria and induce autophagy.Citation3 Conversely, Abida and Gu report that full-length murine ARF (in which the internal methionine at amino acid 45 is mutated to alanine) can induce autophagy, and further that smARF was unable to do so.Citation6 The reasons for these conflicting data have been largely unclear. The end-result of ARF-mediated autophagy was likewise subject to controversy: whereas Kimchi reports that ARF-mediated autophagy was cytotoxic to tumor cells, our group has reported that silencing Arf in established tumors could inhibit autophagy and impede tumor development.Citation32 Finally, whether human smARF existed, and/or could traffic to mitochondria, was also subject to debate. Whereas one group finds no evidence for full-length human ARF trafficking to mitochondria,Citation6 another group has published that full-length human ARF (1 to 132), but not smARF, could localize to mitochondria.Citation7
Several factors likely contribute to the abundance of discrepant results in this field: the first is that many of these experiments were performed on transiently-transfected cells, where ARF levels and/or localization may be nonphysiological. Second, many of these studies relied only upon two autophagy assays: the conversion of LC3-I to LC3-II, and the presence of GFP-LC3 vacuoles. However, these studies did not analyze LC3-II levels after halting autophagic flux, and this is now known to be important for accurately assessing autophagy.Citation33 Importantly, neither of these assays can distinguish between autophagy and mitophagy. To circumvent these constraints we created cell lines containing inducible versions of ARF. We used extremely low doses of doxycycline, such that physiologically-relevant levels of ARF are achieved. Further, we performed four assays for autophagy, including electron microscopy for autophagosomes, accumulation of LC3-II after halting autophagic flux, GFP-LC3 vacuoles and analysis of SQSTM1 degradation. Our data indicate that, for murine ARF, only the full-length version of ARF can induce autophagy. In addition, our experiments support the premise that smARF may instead induce selective autophagy of mitochondria, or mitophagy. This latter premise is supported by decreased autophagosomes after murine smARF induction (even at multiple timepoints), along with the presence of mitochondria with altered morphology, a loss of mitochondrial transmembrane potential, a clustering of TOMM20 staining and a loss of mitochondrial mass. This premise is also supported by the ability of murine smARF to induce SQSTM1 recruitment to mitochondria. The concept that murine smARF may induce mitophagy instead of autophagy is certainly supported by the enhanced localization of this variant at mitochondria, seen by our group and others.Citation3,Citation6 In sum, our data are more supportive of a role for smARF in mitophagy, and for ARF in autophagy. One question arising from this finding is whether smARF induces mitophagy solely because of its preferential localization to mitochondria, or whether this protein has unique functions at the mitochondria. To address this issue, we analyzed a full-length murine ARF construct that we previously created and characterized.Citation8,Citation32 This mutant protein, OTC-ARF, is predominantly directed to mitochondria by virtue of a mitochondrial leader peptide from ornithine transcarbamylase. We find that transfection of OTC-ARF into cells is sufficient to induce mitophagy, as assessed by MitoTracker Green staining (Fig. S4). These data suggest that it is likely the preferential localization of smARF to mitochondria that drives its mitophagy function.
We find that an overlapping, conserved domain of murine and human ARF is required for autophagy induction. Specifically, we show that amino acids 100 to 120 of murine ARF (and amino acids 99 to 132 of human ARF) are required for autophagy induction. This region is one of only two well-conserved domains of human and murine ARF, and is also where the majority of tumor-derived mutations exist in CDKN2A. Our data indicate that three point mutants in this region, which in CDKN2A alter p14ARF coding potential but not p16INK4a, are all impaired for autophagy induction. These data firmly support the premise suggested by othersCitation13 that exon 2 mutations in CDKN2A can alter ARF function, and they implicate ARF’s autophagy function in tumor suppression by this protein. Because autophagy suppresses tumor initiationCitation14 it is not surprising that these mutations impair the ability of ARF to induce autophagy. Interestingly, we also identified a point mutation by random mutagenesis, V113A in murine ARF, which has impaired autophagy induction. Further analysis of this residue revealed that this mutation is actually identical to a tumor-derived mutation that occurs in human ARF in cancer, and affects the coding region of both ARF and p16INK4a; importantly, however, this mutation in CDKN2A has no effect on p16INK4a function,Citation15 suggesting that it might impair ARF function. Therefore, of the four different mutations in ARF that do not impact p16INK4a coding region or function, all of them impair ARF’s autophagy function. These combined data support the premise that some tumor-derived mutations in exon 2 of CDKN2A are selected for to impair the autophagy function of ARF, instead of the cell cycle inhibitory function of p16INK4a. It will now be of interest to identify interacting proteins that bind to this domain of ARF; to date, this region is not known to be required for association with any ARF-interacting proteins. Along these lines, we find that neither BCL2 nor BCL2L1,Citation8 which are implicated in autophagy, nor the mitochondrial protein C1QBP (also called p32), are impaired for interaction with our ARF tumor-derived mutants (Budina-Kolomets A, unpublished observations). We are currently analyzing these tumor-derived mutants, compared with wt ARF, for potential protein-protein interactions in this region.
One unresolved question from these studies involves the significance of the larger −41/+132 transcript of human ARF. Whereas our RT-PCR studies clearly indicate this transcript exists in human cells, western blot analyses using antisera that can specifically detect this protein indicates that it is low to undetectably expressed in normal and transformed cells (Budina-Kolomets A, unpublished observations). This finding is consistent with the findings of other groups, which suggests that the 1 to 132 version of ARF is the predominant species in human tumor cells. This then begs the question, why is the 1 to 132 protein the predominant species in human tumor cells? Possibly a unique structure at the 5′ UTR of full-length human ARF causes preferential translation of the downstream methionine. Alternatively, it is possible that this full-length protein is generated in cells but is largely unstable; this latter premise is supported by our finding that mutation of the internal methionine of human ARF (−41/+132, M1A) renders this protein extremely unstable (Budina-Kolomets A, unpublished data). One interesting alternate possibility is that the translation of human ARF may be regulated, such as by the MTOR pathway. MTOR has been shown to regulate the translation of ARF; specifically, hyperactive MTOR drives the recruitment of ARF mRNA to polysomes, thus increasing ARF translation.Citation34 It has never been tested if this pathway also controls the selection of the initiating methionine. Understanding the controls that regulate the level of ARF translation in the cell, and the impact of the MTOR pathway on this process, remains an important next question to be addressed.
Materials and Methods
Cell lines, drug treatments, transfections, retroviral infections
The U2OS/Tet-On/p19ARF-inducible cell line (U2OS-ARF) and U2OS-Trex cells were provided by Pradip Raychaudhuri (University of Illinois, Chicago). Murine smARF, 1 to 140 ARF, 1 to 120 ARF and 1 to 100 ARF mutants were amplified from the nucleotide sequence coding for the p19ARF protein using the following primers:
One to 140 Arf: 5′-GGGACATCAA GACATTGAGC GATATTTGCG-3′ and 5′-CGCAAATATC GCTCAATGTC TTGATGTCCC-3′.
1 to 120 Arf: 5′-CGGCTGGATG TTAGCGATGC CTGGGGTCG-3′ and 5′-CGACCCCAGG CATCGCTAAC ATCCAGCCG-3′.
1 to 100 Arf: 5′-GCACGACGCA GCTAGGGAAG GCTTCCTGG-3′ and 5′-CCAGGAAGCC TTCCCTAGCT GCGTCGTGC-3′.
SmArf: 5′-GCAGCGGGAG CGCGGGTCGC AGG-3′ and 5′-CCTGCGACCC GCGCTCCCGC TGC-3′.
Human full-length ARF −41/+132, 1 to 132 ARF, −41/+99 ARF, −41/+59 ARF mutants were amplified using the following primers:
−41/+132 full-length ARF: 5′-GGTCCCAGTC TGCAGTTAAG GGGTCAGGAG TGGCGCTGTT CACCTCTGGT-3′ and 5′- ATCTTGCTCG AGTCAGCCAG GTCCACGGGC AGAC-3′.
1 to 132 ARF: 5′-ATCTTGAAGC TTATGGTGCG CAGGTTCTTG GTGAC-3′ and 5′- ATCTTGCTCG AGTCAGCCAG GTCCACGGGC AGAC-3′.
−41/+59 ARF: 5′-ATCTTTAAGC TTATGGGTAG GGGGCGGTGC GTGGGT-3′ and 5′-ATCTTGCTCG AGTCACTGCC CTAGACGCTG GCTCCTC-3′.
−41/+99 ARF: 5′-ATCTTTAAGC TTATGGGTAG GGGGCGGTGC GTGGGT-3′ and 5′-ATCTTGCTCG AGTCACGGGC AGCGTCGTGC ACG-3′.
These constructs were inserted into the pcDNA 4/TO vector (Invitrogen, V1020-20) and used for stable transfection of U2OS-Trex cells. Transfections were performed using FuGENE 6 as per the manufacturer (Promega, E2691). Generated cell lines were cultured in the presence of 0.1 µg/ml doxycycline (Sigma, D3072) to induce ARF, as described.Citation31 Retroviral infections and short hairpin constructs for Trp53, Arf and control vector are described.Citation32,Citation35 Ammonium chloride (Fisher Scientific, AC19997-5000) was used to halt autophagic flux at a concentration of 10 mM, as described.Citation10 CCCP (Carbonyl cyanide m-chlorophenylhydrazone, Sigma-Aldrich, C2759) was used at a concentration of 10 μM.
Mitochondria isolation, western analysis and electron microscopy
Mitochondria were purified using our previously published protocols.Citation9 Western blotting was performed on 100 µg of whole cell lysate and 20 µg of mitochondrial lysate; the following antibodies were used: anti-p19ARF (Abcam, ab80), anti-p14ARF (Sigma, P2610), antisera specific for the N-terminus of human −41/+132 ARF (Abcam, ab14930), anti-ACTB/β−actin (Sigma, AC15), anti-HSPA9/GRP75 (Santa Cruz Biotechnology, C19), anti-PCNA (Santa Cruz Biotechnology, PC10), anti-TP53 (Calbiochem, Ab-6), anti-TOMM20 (Santa Cruz Biotechnology, sc-11415), anti-LC3 (Novus Biologicals, NB100-2331), anti-human SQSTM1 (Santa Cruz Biotechnology, sc-28359) or anti-mouse SQSTM1 (Sigma, P0067), anti-HSPA9 (Santa Cruz Biotechnology sc-1058), and anti-PCNA (Santa Cruz Biotechnology, sc-56). Electron microscopy of autophagosomes was performed in the EM facility at the University of Pennsylvania School of Medicine. The area of autophagosomes was calculated using the NIH ImageJ program. Immunoelectron microscopy was performed as describedCitation36 and visualized by secondary labeling with protein A-colloidal gold.
Immunofluorescence analysis
To visualize GFP-LC3 autophagosomes, cells were transfected for 24 h with 0.1 µg of pEGFP-C1-LC3, followed by treatment with doxycycline for 48 h to induce ARF. The samples were viewed using a Nikon E800 upright microscope with a Bio-Rad Radiance 2000 confocal scanhead (Wistar Institute Imaging Facility) (60 to 100× magnification). The averaged data from three independent experiments in which 200 GFP-positive cells were counted, plus standard deviations, were calculated and analyzed by the Student’s t-test. For the analysis of ARF subcellular localization, 48 h after doxycycline treatment U2OS-ARF cells were fixed in 4% paraformaldehyde, permeabilized in 0.5% Triton X-100, stained with antibodies to p14ARF (Sigma, P2610), p19ARF (Abcam, ab80) and TUBB/β-tubulin (Abcam, ab6046) and then incubated with secondary antibodies. For mitochondria labeling, cells were incubated with 100 nM MitoTracker dye (CMX-Ros, Invitrogen, M7512) or MitoTracker Green FM (Invitrogen, M7514) for 30 min at 37 °C prior to cell fixation. Cells were then stained with DAPI (0.5 µg/ml, Sigma, D9564) prior to confocal microscopy.
Flow cytometric analysis of ΔΨm, RNA isolation, QPCR
Mitochondrial membrane potential was assessed using the MitoPotential kit (Guava Technologies) and analyzed on the Guava EasyCyte System. For QPCR analysis, RNA was isolated from cell lines using RNeasy kit (Qiagen). One microgram of total RNA was reverse-transcribed using oligo(dT) and random primers, using High Capacity cDNA synthesis kit according to the manufacturer's recommendations (Applied Biosystems). Quantitative real-time PCR was performed using SYBR Green QPCR Master Mix (Agilent Technologies). These PCR primers for the ARF genes were used: 5′-CGCGAGTGAG GGTTTTC-3′ and 5′-CTCCTCAGTA GCATCAGCAC-3′. For detection of the N-terminal region of human −41/+132 ARF the following primers were used: 5′-TGCGTGGGTC CCAGTCT-3′ and 5′-TCTCGCCGCC TCCAG-3′. The relative expression of the transcripts was normalized to levels of GAPDH.
| Abbreviations: | ||
| CCCP | = | carbonyl cyanide m-chlorophenylhydrazone |
| doxy | = | doxycycline |
| EM | = | transmission electron microscopy |
| HBSS | = | Hank’s Balanced Salt Solution |
| kDa | = | kilodaltons |
| MEFs | = | mouse embryo fibroblasts |
| OTC | = | ornithine transcarbamylase |
| UTR | = | untranslated region |
Additional material
Download Zip (8.6 MB)Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01 CA139319 to MEM) and by the Wistar Cancer Center Support Grant (P30 CA10815). We acknowledge the expert assistance from James Hayden and Fred Keeney in the Wistar Imaging Facility. RDH was supported by T32 CA009035. MEM was supported by grant R01 CA139319. We thank Joe Testa and Craig Menges (Fox Chase Cancer Center) for ARF knockout mice, from which we made ARF null MEFs. We thank Peter Makhov (Fox Chase Cancer Center) for help cloning full-length ARF.
Disclosure of Potential Conflicts of Interest
The authors declare there are no conflicts of interest.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/25831
Related Research Data
References
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995; 83:993 - 1000; http://dx.doi.org/10.1016/0092-8674(95)90214-7; PMID: 8521522
- Sherr CJ, Bertwistle D, DEN Besten W, Kuo ML, Sugimoto M, Tago K, Williams RT, Zindy F, Roussel MF. p53-Dependent and -independent functions of the Arf tumor suppressor. Cold Spring Harb Symp Quant Biol 2005; 70:129 - 37; http://dx.doi.org/10.1101/sqb.2005.70.004; PMID: 16869746
- Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, Kimchi A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell 2006; 22:463 - 75; http://dx.doi.org/10.1016/j.molcel.2006.04.014; PMID: 16713577
- Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, Palmero I, Ryan K, Hara E, Vousden KH, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J 1998; 17:5001 - 14; http://dx.doi.org/10.1093/emboj/17.17.5001; PMID: 9724636
- Reef S, Kimchi A. Nucleolar p19ARF, unlike mitochondrial smARF, is incapable of inducing p53-independent autophagy. Autophagy 2008; 4:866 - 9; PMID: 18719357
- Abida WM, Gu W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res 2008; 68:352 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-07-2069; PMID: 18199527
- Irvine M, Philipsz S, Frausto M, Mijatov B, Gallagher SJ, Fung C, Becker TM, Kefford RF, Rizos H. Amino terminal hydrophobic import signals target the p14(ARF) tumor suppressor to the mitochondria. Cell Cycle 2010; 9:829 - 39; http://dx.doi.org/10.4161/cc.9.4.10785; PMID: 20107316
- Pimkina J, Humbey O, Zilfou JT, Jarnik M, Murphy ME. ARF induces autophagy by virtue of interaction with Bcl-xl. J Biol Chem 2009; 284:2803 - 10; http://dx.doi.org/10.1074/jbc.M804705200; PMID: 19049976
- Pietsch EC, Leu JI, Frank A, Dumont P, George DL, Murphy ME. The tetramerization domain of p53 is required for efficient BAK oligomerization. Cancer Biol Ther 2007; 6:1576 - 83; http://dx.doi.org/10.4161/cbt.6.10.4719; PMID: 17895645
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010; 6:1090 - 106; http://dx.doi.org/10.4161/auto.6.8.13426; PMID: 20890124
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 2010; 285:27879 - 90; http://dx.doi.org/10.1074/jbc.M110.119537; PMID: 20573959
- Itahana K, Zhang Y. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell 2008; 13:542 - 53; http://dx.doi.org/10.1016/j.ccr.2008.04.002; PMID: 18538737
- Jin S, White E. Tumor suppression by autophagy through the management of metabolic stress. Autophagy 2008; 4:563 - 6; PMID: 18326941
- Koh J, Enders GH, Dynlacht BD, Harlow E. Tumour-derived p16 alleles encoding proteins defective in cell-cycle inhibition. Nature 1995; 375:506 - 10; http://dx.doi.org/10.1038/375506a0; PMID: 7777061
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 2005; 102:8204 - 9; http://dx.doi.org/10.1073/pnas.0502857102; PMID: 15928081
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10:676 - 87; http://dx.doi.org/10.1038/ncb1730; PMID: 18454141
- Tasdemir E, Maiuri MC, Orhon I, Kepp O, Morselli E, Criollo A, Kroemer G. p53 represses autophagy in a cell cycle-dependent fashion. Cell Cycle 2008; 7:3006 - 11; http://dx.doi.org/10.4161/cc.7.19.6702; PMID: 18838865
- Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D’Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis N, Kroemer G. A dual role of p53 in the control of autophagy. Autophagy 2008; 4:810 - 4; PMID: 18604159
- Robertson KD, Jones PA. The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Mol Cell Biol 1998; 18:6457 - 73; PMID: 9774662
- Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci U S A 1998; 95:8292 - 7; http://dx.doi.org/10.1073/pnas.95.14.8292; PMID: 9653180
- Saporita AJ, Maggi LB Jr., Apicelli AJ, Weber JD. Therapeutic targets in the ARF tumor suppressor pathway. Curr Med Chem 2007; 14:1815 - 27; http://dx.doi.org/10.2174/092986707781058869; PMID: 17627519
- Matsuoka M, Kurita M, Sudo H, Mizumoto K, Nishimoto I, Ogata E. Multiple domains of the mouse p19ARF tumor suppressor are involved in p53-independent apoptosis. Biochem Biophys Res Commun 2003; 301:1000 - 10; http://dx.doi.org/10.1016/S0006-291X(03)00080-9; PMID: 12589812
- Paliwal S, Pande S, Kovi RC, Sharpless NE, Bardeesy N, Grossman SR. Targeting of C-terminal binding protein (CtBP) by ARF results in p53-independent apoptosis. Mol Cell Biol 2006; 26:2360 - 72; http://dx.doi.org/10.1128/MCB.26.6.2360-2372.2006; PMID: 16508011
- Kelly-Spratt KS, Gurley KE, Yasui Y, Kemp CJ. p19Arf suppresses growth, progression, and metastasis of Hras-driven carcinomas through p53-dependent and -independent pathways. PLoS Biol 2004; 2:E242; http://dx.doi.org/10.1371/journal.pbio.0020242; PMID: 15314658
- Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, Roussel MF, Sherr CJ, Zambetti GP. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev 2000; 14:2358 - 65; http://dx.doi.org/10.1101/gad.827300; PMID: 10995391
- Bertwistle D, Sugimoto M, Sherr CJ. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol 2004; 24:985 - 96; http://dx.doi.org/10.1128/MCB.24.3.985-996.2004; PMID: 14729947
- Zerrouqi A, Pyrzynska B, Febbraio M, Brat DJ, Van Meir EG. P14ARF inhibits human glioblastoma-induced angiogenesis by upregulating the expression of TIMP3. J Clin Invest 2012; 122:1283 - 95; http://dx.doi.org/10.1172/JCI38596; PMID: 22378045
- di Tommaso A, Hagen J, Tompkins V, Muniz V, Dudakovic A, Kitzis A, Ladeveze V, Quelle DE. Residues in the alternative reading frame tumor suppressor that influence its stability and p53-independent activities. Exp Cell Res 2009; 315:1326 - 35; http://dx.doi.org/10.1016/j.yexcr.2009.01.010; PMID: 19331830
- Tompkins VS, Hagen J, Frazier AA, Lushnikova T, Fitzgerald MP, di Tommaso A, Ladeveze V, Domann FE, Eischen CM, Quelle DE. A novel nuclear interactor of ARF and MDM2 (NIAM) that maintains chromosomal stability. J Biol Chem 2007; 282:1322 - 33; http://dx.doi.org/10.1074/jbc.M609612200; PMID: 17110379
- Datta A, Nag A, Pan W, Hay N, Gartel AL, Colamonici O, Mori Y, Raychaudhuri P. Myc-ARF (alternate reading frame) interaction inhibits the functions of Myc. J Biol Chem 2004; 279:36698 - 707; http://dx.doi.org/10.1074/jbc.M312305200; PMID: 15199070
- Humbey O, Pimkina J, Zilfou JT, Jarnik M, Dominguez-Brauer C, Burgess DJ, Eischen CM, Murphy ME. The ARF tumor suppressor can promote the progression of some tumors. Cancer Res 2008; 68:9608 - 13; http://dx.doi.org/10.1158/0008-5472.CAN-08-2263; PMID: 19047137
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Miceli AP, Saporita AJ, Weber JD. Hypergrowth mTORC1 signals translationally activate the ARF tumor suppressor checkpoint. Mol Cell Biol 2012; 32:348 - 64; http://dx.doi.org/10.1128/MCB.06030-11; PMID: 22064482
- Hemann MT, Fridman JS, Zilfou JT, Hernando E, Paddison PJ, Cordon-Cardo C, Hannon GJ, Lowe SW. An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet 2003; 33:396 - 400; http://dx.doi.org/10.1038/ng1091; PMID: 12567186
- Tokuyasu KT. Immunochemistry on ultrathin frozen sections. Histochem J 1980; 12:381 - 403; http://dx.doi.org/10.1007/BF01011956; PMID: 7440248