Abstract
DCN (decorin), an extracellular matrix constituent and archetypical small leucine-rich proteoglycan (SLRP), acts as a soluble tumor repressor. DCN exerts high-affinity binding interactions with receptor tyrosine kinases and evokes receptor internalization consequent with lysosomal degradation for tumorigenic and angiogenic suppression. In our recent study, we discovered that DCN evokes synthesis of PEG3 (paternally expressed 3), an imprinted gene often silenced in various forms of cancer. Upon DCN stimulation, PEG3 relocalizes to BECN1- and LC3-positive phagophores. Importantly, PEG3 physically associates with BECN1- and LC3-containing supramolecular complexes, in a DCN-inducible manner, and PEG3 is necessary to maintain homeostatic levels of BECN1. Furthermore, DCN evokes a protracted autophagic program via transactivation of the BECN1 and MAPLC3A loci that is critically dependent on PEG3 expression. Mechanistically, DCN directly binds to the Ig domains 3–5 of the KDR/VEGFR2 ectodomain, in a region that partially overlaps with the canonical binding site for VEGFA. Therefore, we have unveiled a novel mechanism for a secreted proteoglycan to induce endothelial cell autophagy in a PEG3-dependent manner. These findings are consistent with earlier preclinical studies focusing on DCN-mediated tumorigenic and angiogenic suppression and may represent the mechanism of action to achieve these effects. Therefore, DCN and perhaps other members of this class of matrix constituents may represent a novel control of autophagy from the outside of the cells.
Microenvironmental interactions between tumor and surrounding stroma are emerging as crucial factors underlying tumor development and progression. DCN, a class I SLRP, regulates collagen fibrillogenesis, hence its eponym as “decorator” of fibrils, during development. However, in the adult there is a pool of soluble, circulating DCN induced during inflammation and cancer. Biologically, DCN, as the “guardian from the matrix,” acts as an endogenous pan-receptor tyrosine kinase (RTK) inhibitor to attenuate cancer growth and angiogenesis and could act directly on the tumor proper via RTK antagonism. However, a paradigmatic shift occurred following high-resolution transcriptome profiling of triple-negative breast carcinoma xenografts treated with systemically-administered DCN. We found a small subset of DCN-inducible genes exclusively modulated within the tumor stroma. Among the most upregulated genes is PEG3, a Krupple-like zinc finger-containing transcription factor commonly silenced in cancer. Utilizing endothelial cells as a genetically stable proxy of the tumor stroma, we found a prolonged and dynamic induction of PEG3 evoked by DCN in both human umbilical vein endothelial cells and mouse microvascular dermal endothelial cells. Additionally, DCN-evoked endothelial cell autophagy is sensitive to therapeutic inhibition of PIK3C3/VPS34 with 3-methyladenine (3-MA) (). Further delving into the spatial-temporal induction of BECN1 and LC3 mRNA and protein that DCN elicits, we performed co-immunoprecipitation of PEG3 and found a novel protein-protein interaction with BECN1 and LC3 in a DCN-inducible fashion. Recruitment of PEG3 into a BECN1-positive scaffolding complex has the potential to regulate the initial formation of the phagophore and its eventual expansion into an autophagosome. The biological significance of a DCN-inducible PEG3-BECN1 complex is currently being investigated and may function as a mutually exclusive switch for autophagy activation by disrupting the repressive association of BCL2-BECN1 at the outer mitochondrial membrane.
Figure 1. Soluble DCN evokes endothelial cell autophagy. (A) Schematic representation showing how high-affinity interactions between DCN and KDR ectodomain (Ig3–5) trigger a signal cascade simultaneously activating and suppressing PIK3C3 and MTOR, respectively. This DCN-evoked process is blocked by siRNA targeting KDR (siKDR) or by KDR tyrosine kinase-inhibitor SU5416. The rapidly-induced PEG3 physically associates with BECN1 and LC3 for autophagic induction. Moreover, unbound PEG3 presumably undergoes nuclear translocation to transactivate key genetic loci including BECN1 and LC3, and this process is inhibited by siRNA targeting PEG3 (siPEG3). This reinforced network, with PEG3 serving as the primary node, permits DCN to evoke protracted endothelial cell autophagy as the underlying basis for angiostasis. (B) Differential interference contrast microscopy of a mouse microvascular endothelial cell following an 18 h treatment with DCN (200 nM) in nutrient-rich conditions. The nucleus is stained in blue by DAPI. Notice the formation of multiple cytoplasmic autophagic vacuoles (AV).
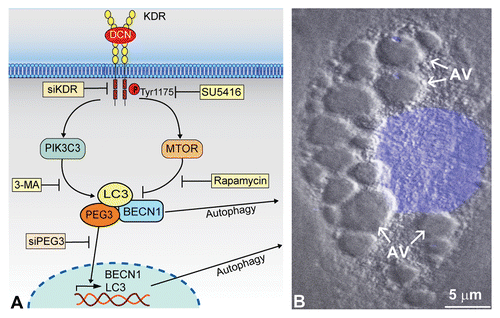
As DCN-evoked autophagy occurs exclusively within endothelial cells and not tumor cells, we postulated that DCN is directly interacting with KDR/VEGFR2 [kinase insert domain receptor (a type III receptor tyrosine kinase)], the primary receptor orchestrating endothelial cell homeostasis and angiogenic signaling. As such, we found a high-affinity binding of DCN to the KDR ectodomain (Kd ~2 nM) in a region partially overlapping with VEGFA between Ig domains 3–5 (). Mechanistically, DCN requires direct KDR interactions and downstream signaling for autophagic induction, presumably to induce PIK3C3 activation and MTOR inactivation (). Indeed, genetic and pharmacological inhibition with either siRNA specific for KDR (siKDR) or the small molecule inhibitor (SU5416) of KDR tyrosine kinase results in a complete blockade of BECN1 and MAPLC3A expression.
Unexpectedly, differential interference contrast microscopy done on endothelial cells exposed to DCN revealed the formation of large cytosolic structures reminiscent of autophagosomes (). Subsequent immunofluorescence studies revealed a surprising localization of PEG3 to these intracellular structures. Further corroborating the evidence of DCN-evoked vesicular macroautophagy and a potential role for PEG3, we found that PEG3 strongly colocalizes with two canonical autophagy markers, BECN1 and LC3. Moreover, these data were reinforced as DCN disrupts mitochondrial membrane potential coincident with mitochondrial fragmentation, strong harbingers of autophagic processes.
As DCN is able to dynamically recruit PEG3 to BECN1-containing complexes and colocalizes to the surface of mature autophagosomes, we hypothesized that PEG3 would be involved in BECN1 and MAPLC3A expression. Importantly, depletion of PEG3 with a targeting siRNA (siPEG3) revealed that not only is PEG3 required for DCN-mediated induction of BECN1 and LC3 at the mRNA and protein levels, but PEG3 is also critically necessary to maintain homeostatic levels of BECN1 (). Therefore, we have identified PEG3 as a novel master regulatory component necessary for endothelial cell autophagy.
The novel ability of DCN to evoke a PEG3-dependent macroautophagy in endothelial cells, the cells intimately responsible for neovascularization of the growing tumors, led us to hypothesize the biological function and relevance of evoking autophagy. We surmised that induction of autophagy underlies the mechanistic basis for DCN to inhibit angiogenesis in an endothelial cell-autonomous manner. Capillary morphogenesis and endothelial cell migration are processes critical for successful vascularization. Thus, we found that DCN inhibits tubulogenesis and migration concomitant with autophagy. The induction of BECN1 and formation of PEG3-BECN1 complexes might be critical for blunting endothelial cell migration and capillary morphogenesis. Identification of the downstream effectors or alternate binding partners for PEG3 will be required to understand this connection in greater detail.
All in all, we have identified a novel role for a soluble matrix constituent, and perhaps a novel paradigmatic function for the SLRP family, to evoke a fundamental cell process, autophagy, in the context of suppressing endothelial cell-mediated angiogenesis. Moreover, the discovery of a PEG3-dependent autophagy and the transcriptional role PEG3 exerts over key genetic loci will have a profound effect on the macroautophagic program, signaling and gene expression networks as a whole.
| Abbreviations: | ||
| PEG3 | = | paternally expressed 3 |
| RTK | = | receptor tyrosine kinase |
| SLRP | = | small leucine-rich proteoglycan |
| KDR | = | kinase insert domain receptor (a type III receptor tyrosine kinase) |
Acknowledgments
This work was supported in part by National Institutes of Health Grants RO1 CA39481, RO1 CA47282 and RO1 CA164462 (to RVI). TN was supported by NIH training grant T32 AA07463. AT was supported by a Diversity Research Supplement NIH Grant CA39481-26S1.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.