Abstract
Cellular stress responses often involve elevation of cytosolic calcium levels, and this has been suggested to stimulate autophagy. Here, however, we demonstrated that agents that alter intracellular calcium ion homeostasis and induce ER stress—the calcium ionophore A23187 and the sarco/endoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin (TG)—potently inhibit autophagy. This anti-autophagic effect occurred under both nutrient-rich and amino acid starvation conditions, and was reflected by a strong reduction in autophagic degradation of long-lived proteins. Furthermore, we found that the calcium-modulating agents inhibited autophagosome biogenesis at a step after the acquisition of WIPI1, but prior to the closure of the autophagosome. The latter was evident from the virtually complete inability of A23187- or TG-treated cells to sequester cytosolic lactate dehydrogenase. Moreover, we observed a decrease in both the number and size of starvation-induced EGFP-LC3 puncta as well as reduced numbers of mRFP-LC3 puncta in a tandem fluorescent mRFP-EGFP-LC3 cell line. The anti-autophagic effect of A23187 and TG was independent of ER stress, as chemical or siRNA-mediated inhibition of the unfolded protein response did not alter the ability of the calcium modulators to block autophagy. Finally, and remarkably, we found that the anti-autophagic activity of the calcium modulators did not require sustained or bulk changes in cytosolic calcium levels. In conclusion, we propose that local perturbations in intracellular calcium levels can exert inhibitory effects on autophagy at the stage of autophagosome expansion and closure.
Introduction
Macroautophagy (referred to as “autophagy” throughout this article) is a fundamental, evolutionarily conserved cellular process characterized by the formation of double-membrane structures termed “autophagosomes” that encircle cellular material destined for degradation via fusion with lysosomes. Whereas basal autophagy performs quality control and turnover of cellular proteins and organelles, autophagy can be induced as a survival mechanism during stressful conditions, such as starvation and hypoxia.Citation1 Alterations in autophagy have been implicated in a number of human pathological conditions, including neurodegenerative diseases and cancer.Citation2
Numerous reports have suggested that elevation of cytosolic calcium levels enhances the formation of autophagosomes and thereby autophagy.Citation3-Citation16 However, these studies are predominantly based on assessments of autophagic markers like LC3, which do not provide information on autophagic activity,Citation17 and observations from four other studies indicate that calcium-modulating agents can have anti-autophagic effects.Citation18-Citation21 Consequently, it still remains to be clarified how intracellular calcium affects the autophagic process. Here, we set out to carefully and systematically examine the effect of calcium modulators on both basal autophagy and autophagy induced by amino acid starvation, by employing functional autophagy assays. We used two naturally occurring agents that elevate cytosolic calcium levels via distinct mechanisms; the bacterially derived antibiotic calcium ionophore A23187, which allows calcium ions to cross biological membranes,Citation22 and the plant-derived sarco/endoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin (TG), which inhibits calcium ions from being transported from the cytosol into the ER.Citation23 Our data demonstrate that these two calcium modulators induce an early block in autophagic flux, manifested by a failure in autophagic sequestration of cytosolic material and a resulting reduction in the autophagic degradation of long-lived proteins. Furthermore, we demonstrate that this can be accomplished independently of ER stress and without sustained or bulk changes in cytosolic calcium levels.
Results
We first aimed to establish whether the calcium modulators A23187 and TG affected autophagy, and, if so, at which concentrations and time points. Potential modulation of autophagy was initially assessed by analyzing protein levels of the autophagosomal marker LC3 (microtubule-associated protein 1 light chain 3), the cytosolic form of which (LC3-I) becomes lipidated (yielding LC3-II) and attached to autophagosomal membranes upon induction of autophagy. As shown in Figure S1, A23187 and TG enhanced LC3-II protein levels in a concentration-dependent manner at a fixed time point of 12 h in LNCaP prostate cancer cells (Fig. S1A), and the effect obtained with 2 µM A23187 or 100 nM TG was reversible with the well-established autophagy inhibitor 3-methyladenine (3-MA) (Fig. S1B). Low concentrations of A23187 (0.75 to 2 µM) and TG (100 nM) were chosen for subsequent experiments to avoid potential unspecific effects that can occur at higher concentrations.Citation24,Citation25 An alteration in the protein levels of LC3-II may mirror an effect on autophagyCitation26 or reflect alterations in the rate of LC3 transcription and translation. To assess this, we performed RT-PCR and western blotting over a time course (1 to 16 h) of A23187 and TG treatment. Strikingly, we observed a very strict correlation between LC3-I and LC3-II protein levels over the whole treatment period (1 to 16 h) as well as between LC3 protein levels and LC3B mRNA levels from 4 h and onwards (Fig S2A–S2D). Similar correlations were observed in the U2OS osteosarcoma cell line (Fig. S2E–S2H). This indicated that the effect of A23187 and TG on LC3-II protein levels was largely mediated by overall alterations in LC3 protein and RNA levels rather than by an effect on LC3 lipidation per se. Combined with the fact that analyses of LC3 lipidation does not assess whether autophagy proceeds to completion or not, this observation led us to undertake a series of functional and imaging assays to further explore the impact of calcium-modulating agents on autophagy.
A quantitative and unbiased way of measuring autophagic activity consists of analyzing the bulk degradation of long-lived proteins, a substantial fraction of which are degraded by autophagy.Citation27-Citation29 We found that A23187 and TG inhibited long-lived protein degradation (LLPD) rates equivalently over short (0 to 6 h) and long (0 to 16 h) time periods in both LNCaP and U2OS cells (). In agreement with previous studies,Citation28,Citation29 however, we observed that long-lived proteins were degraded not only by autophagy, but also by the ubiquitin-proteasomal pathway. This was reflected by a 30% inhibition of LLPD with the lysosomal inhibitors bafilomycin A1 (BafA1) and chloroquine (CQ), and a 50% inhibition of LLPD with the proteasomal inhibitor MG132 (). We therefore asked whether the effects of the calcium modulators A23187 and TG were caused by reduced autophagy or reduced proteasomal degradation. Additive effects of A23187 and TG together with BafA1 or CQ would indicate that the calcium modulators were inhibiting proteasomal activity, whereas additive effects with MG132, and not with BafA1 or CQ, would indicate that A23187 and TG acted to inhibit autophagy. Compellingly, the latter scenario was observed both in LNCaP () and U2OS cells (), strongly indicating that the calcium modulators inhibited autophagic LLPD. In fact, since the inhibitory effects were comparable to those observed with lysosomal inhibitors () the data suggest an exceptionally efficient block in autophagy by these agents. Given this strong inhibitory effect on basal autophagy, we reasoned that calcium mobilization might also inhibit autophagy induced by other conditions. We tested this on cells subjected to the bona fide autophagy induction method of amino acid starvation. Strikingly, A23187 and TG effectively blocked starvation-induced elevation of LLPD in both LNCaP and U2OS cells ().
Figure 1. A23187 (A23) and thapsigargin (TG) block autophagic degradation and sequestration activity under both nutrient-rich and amino acid starvation conditions. LNCaP cells (A) or U2OS cells (B) were radiolabeled with [14C]-valine for 2 to 3 d and chased for 18 h. Subsequently, cells were incubated in complete medium containing 0.2% DMSO (vehicle control; Ctrl), A23 (2 µM) or TG (100 nM) for either 6 h or 16 h as indicated, and the degradation of long-lived proteins was determined as described in Materials and Methods. The columns represent mean degradation rates ± standard error of the mean (SEM) from 3 independent experiments. **P < 0.01 by the Student t-test. (C and D) Degradation of long-lived proteins was measured in LNCaP (C) cells during a 6 h incubation or in U2OS (D) cells during a 16 h incubation with DMSO (Ctrl, 0.1%), A23 (2 µM) or TG (100 nM) alone or in combination with DMSO (Ctrl, 0.1%), BafA1 [Baf, 100 nM in (C); 20 nM in (D)], chloroquine [(CQ, 100 µM in (C); 50 µM in (D)] or MG132 [(MG, 5 µM in (C); 1 µM in (D)] as indicated. Columns represent mean relative degradation rates ± SEM from 3 independent experiments, arbitrarily setting the mean degradation rate in the DMSO/DMSO control condition to 1. **P < 0.01 by the Student t-test. (E and F) LNCaP cells (E) or U2OS cells (F) were radiolabeled with [14C]-valine and chased as in (A). Subsequently, cells were incubated for 6 h in either complete medium (CM) containing DMSO (Ctrl, 0.2%) or in medium devoid of serum and amino acids (EBSS) in the presence of DMSO (Ctrl, 0.2%), A23 (0.75 µM) or TG (100 nM), and the degradation of long-lived proteins during this period was measured. Columns represent mean protein degradation rates ± SEM from 6 (E) or 7 (F) independent experiments. **P < 0.01 by the Student t-test. Note that a lower concentration of A23 (0.75 µM) than that used in (A–D) was sufficient to block the degradation of long-lived proteins induced by amino acid starvation. (G) LNCaP cells were incubated for 3 h or 16 h in complete medium containing DMSO (Ctrl, 0.1%), A23 (2 µM), TG (100 nM) or 3-MA (10 mM) with the additional presence of DMSO (Ctrl, 0.1%) or BafA1 (100 nM for 3 h or 20 nM for 16 h) as indicated. Subsequently, cells were harvested and the amount of sequestered lactate dehydrogenase (LDH) was measured as detailed in Materials and Methods. Mean values from 4 (3 h) or 3 (16 h) independent experiments are shown, with error bars representing SEM *P < 0.05, **P < 0.01 by the Student t-test. (H and I) LNCaP cells (H) or U2OS cells (I) were incubated for 2 h in complete medium containing DMSO (Ctrl, 0.2%) or in medium devoid of serum and amino acids (EBSS) containing DMSO (Ctrl, 0.1%), BafA1 (100 nM), A23 (0.75 µM), TG (100 nM), or 3-MA (10 mM) as indicated. Subsequently, cells were harvested and the amount of sequestered LDH was measured as detailed in Materials and Methods. Mean relative values from five independent experiments are shown, with error bars representing SEM *P < 0.05, **P < 0.01 by the Student t-test.
![Figure 1. A23187 (A23) and thapsigargin (TG) block autophagic degradation and sequestration activity under both nutrient-rich and amino acid starvation conditions. LNCaP cells (A) or U2OS cells (B) were radiolabeled with [14C]-valine for 2 to 3 d and chased for 18 h. Subsequently, cells were incubated in complete medium containing 0.2% DMSO (vehicle control; Ctrl), A23 (2 µM) or TG (100 nM) for either 6 h or 16 h as indicated, and the degradation of long-lived proteins was determined as described in Materials and Methods. The columns represent mean degradation rates ± standard error of the mean (SEM) from 3 independent experiments. **P < 0.01 by the Student t-test. (C and D) Degradation of long-lived proteins was measured in LNCaP (C) cells during a 6 h incubation or in U2OS (D) cells during a 16 h incubation with DMSO (Ctrl, 0.1%), A23 (2 µM) or TG (100 nM) alone or in combination with DMSO (Ctrl, 0.1%), BafA1 [Baf, 100 nM in (C); 20 nM in (D)], chloroquine [(CQ, 100 µM in (C); 50 µM in (D)] or MG132 [(MG, 5 µM in (C); 1 µM in (D)] as indicated. Columns represent mean relative degradation rates ± SEM from 3 independent experiments, arbitrarily setting the mean degradation rate in the DMSO/DMSO control condition to 1. **P < 0.01 by the Student t-test. (E and F) LNCaP cells (E) or U2OS cells (F) were radiolabeled with [14C]-valine and chased as in (A). Subsequently, cells were incubated for 6 h in either complete medium (CM) containing DMSO (Ctrl, 0.2%) or in medium devoid of serum and amino acids (EBSS) in the presence of DMSO (Ctrl, 0.2%), A23 (0.75 µM) or TG (100 nM), and the degradation of long-lived proteins during this period was measured. Columns represent mean protein degradation rates ± SEM from 6 (E) or 7 (F) independent experiments. **P < 0.01 by the Student t-test. Note that a lower concentration of A23 (0.75 µM) than that used in (A–D) was sufficient to block the degradation of long-lived proteins induced by amino acid starvation. (G) LNCaP cells were incubated for 3 h or 16 h in complete medium containing DMSO (Ctrl, 0.1%), A23 (2 µM), TG (100 nM) or 3-MA (10 mM) with the additional presence of DMSO (Ctrl, 0.1%) or BafA1 (100 nM for 3 h or 20 nM for 16 h) as indicated. Subsequently, cells were harvested and the amount of sequestered lactate dehydrogenase (LDH) was measured as detailed in Materials and Methods. Mean values from 4 (3 h) or 3 (16 h) independent experiments are shown, with error bars representing SEM *P < 0.05, **P < 0.01 by the Student t-test. (H and I) LNCaP cells (H) or U2OS cells (I) were incubated for 2 h in complete medium containing DMSO (Ctrl, 0.2%) or in medium devoid of serum and amino acids (EBSS) containing DMSO (Ctrl, 0.1%), BafA1 (100 nM), A23 (0.75 µM), TG (100 nM), or 3-MA (10 mM) as indicated. Subsequently, cells were harvested and the amount of sequestered LDH was measured as detailed in Materials and Methods. Mean relative values from five independent experiments are shown, with error bars representing SEM *P < 0.05, **P < 0.01 by the Student t-test.](/cms/asset/fbf85389-201a-4b8e-bbc9-ae6c6ca9fb52/kaup_a_10925900_f0001.gif)
Another well-established method to measure autophagy relies on quantifying the sequestration of the cytosolic protein lactate dehydrogenase (LDH) into autophagosomes,Citation30,Citation31 and here we adapted this assay to human cancer cell lines. As shown in , an accumulation of sequestered LDH was observed in cells treated with BafA1, and 3-MA completely abolished this effect, thus confirming the assay by showing that the accumulation of LDH was indeed dependent on autophagosome formation. Strikingly, and similarly to 3-MA, A23187 and TG completely inhibited BafA1-induced accumulation of sequestered LDH at both early and late time points (3 h and 16 h). Moreover, and unlike treatment with BafA1, treatment with A23187 or TG alone did not result in any accumulation of sequestered LDH, even at 16 h (). This suggests that A23187 and TG inhibit autophagy prior to closure of the autophagosomes. Next, we repeated this experiment under conditions of amino acid starvation, and we again observed that the calcium modulators inhibited BafA1-induced accumulation of sequestered LDH in both LNCaP and U20S cells (). We also found that A23187 and TG could inhibit LDH sequestration induced by 2 h amino acid starvation in the absence of BafA1 ().
To further corroborate our findings we next employed a quantitative imaging assay to assess autophagosome biogenesis as well as autophagosome maturation using U2OS cells stably expressing mRFP-EGFP-LC3. As shown in Figure S3A–S3C, amino acid starvation for 3 h enhanced the number of EGFP-puncta per cell as well as the number of puncta representing autolysosomes (number of mRFP puncta minus number of EGFP puncta), reflecting enhanced generation of autophagosomes and autophagic flux. Concurrent treatment with A23187 or TG resulted in a decrease in the number of EGFP puncta per cell (Fig. S3B) as well as in the number of puncta representing autolysosomes (Fig. S3C), suggesting that autophagosomes did not fully mature into autolysosomes. Furthermore, we also observed a reduction in the size of the EGFP puncta in cells treated with A23187 or TG (Fig. S3D), indicating that the maturation of autophagosomes was inhibited at an early step. In contrast to A23187 and TG, the late-stage inhibitor BafA1 induced a massive increase in the number of EGFP puncta per cell, the disappearance of puncta representing autolysosomes, and a strong increase in the size of the EGFP puncta—all as expected from an accumulation of mature autophagosomes due to a block imposed at the lysosomal stage. Collectively these results indicate that A23187 and TG elicit an early block in the autophagic process and support the conclusions drawn from the LDH sequestration and LLPD assays.
WIPI1, a phosphatidylinositol-3-phosphate effector at the onset of autophagy, is recruited to forming autophagosomal membranes upstream of LC3-II and is therefore an early marker for autophagosome biogenesis.Citation32 Using EGFP-WIPI1 stably expressed in U2OS cellsCitation33 we found that both the percentage of cells that were positive for EGFP-WIPI1 puncta and the number of EGFP-WIPI1 puncta per cell were increased by treatment with TG under conditions of amino acid starvation (Fig. S4). A similar effect of TG has been reported previously under nutrient-rich and serum-starvation conditions.Citation34 Thus, and taken together, our data indicate that the calcium-modulating agents exert inhibitory effects at a step downstream of WIPI1 recruitment, but upstream of autophagic sequestration of cytoplasmic material and subsequent autolysosome formation and long-lived protein degradation.
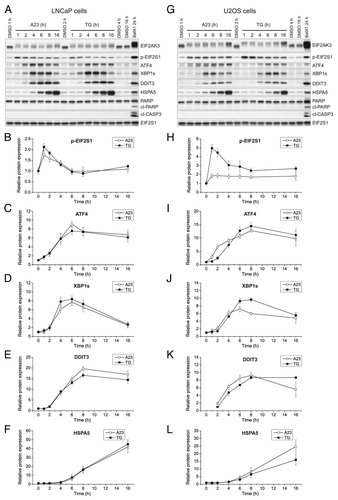
Calcium-modulating agents often impose ER stress,Citation35-Citation37 and elevation of cytosolic calcium levels for extended periods of time can be cytotoxic.Citation38 Furthermore, since there is increasing evidence that both ER stress and cell death mechanisms are co-regulated with autophagy,Citation39-Citation42 we set out to investigate this interplay. When blotting protein extracts from cells treated with A23187 or TG for two markers of apoptosis, cleaved PARP and cleaved CASP3, we observed no induction at any treatment time point (1 to 16 h) in either LNCaP () or U2OS cells (). Moreover, when using a flow cytometry assay employing fluorescent ANXA5/Annexin V to measure the levels of phosphatidylserine on the outer leaflet of the plasma membrane, an early marker of apoptosis, we could not detect any apoptotic response within the time-period that was used in the autophagy assays (up to 16 h) (Fig. S5A and S5B). In line with previous observations,Citation43,Citation44 however, and confirming the assay, we did observe apoptosis induction upon prolonged treatment periods (24 h or more) with A23187 or TG (Fig. S5A and S5B). These observations were also supported by flow cytometry measurements directly assessing the accumulation of propidium iodide, an assay for late apoptotic and/or necrotic cells (Fig. S5C). Thus, the antiautophagic effects that we had observed with A23187 and TG were not caused by induction of cell death. The ER stress-unfolded protein response (UPR) pathway is commonly divided in three arms: the EIF2AK3-EIF2S1-ATF4 arm, the ERN1-XBP1 arm, and the ATF6 arm.Citation45 The former two have been associated with the regulation of autophagy.Citation39 We therefore blotted for a panel of UPR markers to assess their expression and activation in cells treated with A23187 or TG over a 16 h time period (see for representative blots for LNCaP and U2OS cells, respectively). EIF2AK3 and EIF2S1 phosphorylation occurred within 1 h of treatment (), whereas ATF4 was upregulated from between 2 to 4 h and onwards (). Similarly, the expression of the active spliced variant of XBP1 (XBP1s) was increased from 4 h and onwards (). DDIT3 and HSPA5 are target genes for key transcription factors within the unfolded protein response pathways. DDIT3 is regulated by ATF6 and ATF4, while HSPA5 lies downstream of ATF6 and XBP1. The protein levels of DDIT3 and HSPA5 were both increased from 4 () and 6 h (), respectively. Additionally, we found that A23187 and TG enhanced the mRNA levels of PPP1R15A (Fig. S6A and S6D) (which is regulated by ATF4), as well as the mRNA levels of XBP1s (Fig. S6B and S6E) and HSPA5 (Fig. S6C and S6F), and these changes also occurred at early time points. Taken together, our data suggest that all arms of the UPR were activated by A23187 and TG in both LNCaP and U2OS cells.
Figure 2. A23187 (A23) and TG rapidly and persistently induce ER stress in the absence of caspase activation. LNCaP cells (A–F) or U2OS cells (G–L) were incubated with DMSO (0.2%; vehicle control), A23 (2 µM) or TG (100 nM) for various lengths of time as indicated, or BafA1 (100 nM) for 24 h as a positive control for ER stress induction and caspase activation. Subsequently, cell lysates were prepared and subjected to western blot analyses of EIF2AK3, phopsho-EIF2S1 (p-EIF2S1), ATF4, XBP1s, DDIT3, HSPA5, PARP, cleaved CASP3 (cl-CASP3) or EIF2S1 (loading control) as indicated. Representative immunoblots from 4 independent experiments are shown in (A and G), and mean relative protein levels normalized to the loading control are shown in (B–F) and (H–L). The mean values obtained in DMSO control-treated cells were arbitrarily set to 1, except for in (K) where the mean value for the 2 h treatment condition with TG was arbitrarily set to 1, as this was the first time point at which a persistent quantifiable signal appeared. Error bars represent SEM-values. The observed shift toward the more slowly migrating bands that are recognized by the EIF2AK3 antibody is indicative of EIF2AK3 phosphorylation. This was confirmed when using a specific EIF2AK3 inhibitor (see , middle panels). The caspase-specific 89-kDa cleavage product of PARP is indicated by “cl-PARP”.
It was therefore important to determine whether the inhibitory effects of A23187 and TG that we had observed on autophagy were dependent upon activation of the UPR. To that end, we employed siRNAs against ATF4, ERN1, or ATF6 in combination with A23187 or TG, and found that in all cases and in both LNCaP and U2OS cells, the calcium-modulating agents potently inhibited amino acid starvation-induced LLPD () in spite of efficient knockdown of the targets (). Next, we took advantage of two novel selective pharmacological agents that inhibit the EIF2AK3- and ERN1-arms of the UPR, GSK2606414, and MKC8866, respectively. The EIF2AK3 inhibitor, GSK2606414, was developed based on a structure of the target kinase domain and is active and specific in the nanomolar range.Citation46,Citation47 The ERN1 inhibitor, MKC8866, is a more potent variant of a drug that has been recently characterized,Citation48 and another variant of this drug has been characterized in multiple myeloma.Citation49 MKC8866 potently inhibited XBP1 splicing at a 10 µM concentration in both LNCaP and U2OS cells, as assessed by real-time RT-PCR (Fig. S7). Strikingly, as shown in , although the two inhibitors completely blocked A23187- and TG-induced EIF2AK3 phosphorylation and XBP1 splicing, respectively (, middle and lower panels), they had no antagonistic effect on the inhibition of either LDH sequestration (, upper panels) or LLPD () by the calcium-modulating agents in either cell line under amino acid starvation conditions. The same was true for both LDH sequestration and LLPD when the two inhibitors were combined, in spite of efficient simultaneous targeting of both EIF2AK3 phosphorylation and XBP1 splicing (). In conclusion, the inhibition of autophagy by A23187 and TG appeared to be independent of ER stress induction.
Figure 3. A23187 (A23)- and TG-mediated repression of autophagy is independent of ER stress. LNCaP cells (A) or U2OS cells (B) were transfected with a nontargeting control siRNA (siCtrl) or siRNAs targeting ATF4 (siATF4), ERN1 (siERN1), or ATF6 (siATF6). Subsequently, cells were treated for 6 h with DMSO control (Ctrl, 0.2%), A23 (0.75 µM), or TG (100 nM) under amino acid starvation conditions, and long-lived protein degradation rates were determined as described in Materials and Methods. Mean protein degradation rates ± SEM from 3 independent experiments are shown. (C and D) Confirmation of knockdown efficiencies of the siRNAs used in (A and B). LNCaP cells (C) or U2OS cells (D) were transfected with siRNAs as in (A and B) and treated for 6 h with DMSO (Ctrl, 0.2%) or TG (100 nM) under amino acid starvation conditions. Subsequently, RNA was extracted and subjected to real-time RT-PCR analyses of relative mRNA levels of ATF4, ERN1, and ATF6 as indicated. Mean relative mRNA levels ± SEM from 3 independent experiments are shown. The average value of the DMSO control conditions with the control siRNA was arbitrarily set to 1. The mean knockdown efficiency varied between 83% and 92%. (E and F) LNCaP cells (E) or U2OS cells (F) were pretreated for 1 h with DMSO (Ctrl, 0.2%), the EIF2AK3 inhibitor GSK2606414 (EIF2AK3i, 100 nM) + 0.1% DMSO, the ERN1-inhibitor MKC8866 (ERN1i, 10 µM) + 0.1% DMSO, or a combination of the two inhibitors (100 nM EIF2AK3i + 10 µM ERN1i). Thereafter, the cells were washed in EBSS medium and starved for 2 h in EBSS medium containing BafA1 (100 nM) and either DMSO (Ctrl, 0.1%), A23 (0.75 µM) or TG (100 nM). Moreover, to maintain suppression of EIF2AK3 and ERN1 activity, EIF2AK3i, ERN1i, and DMSO were re-added at the same concentrations and combinations as during the pretreatment. Subsequently, cells were harvested and assessed for LDH sequestration (top panels) as detailed in Materials and Methods. Mean relative LDH sequestration values ± SEM from 3 independent experiments are shown. The mean LDH sequestration rate in cells that were starved for amino acids in the absence of BafA1 was arbitrarily set to 1. Second (middle panels), aliquots of the total disruptates that were generated in the LDH sequestration assay were used for western blot analyses of EIF2AK3, XBP1s, and GAPDH (loading control) as detailed in Materials and Methods. The observed shift toward the more slowly migrating bands that are recognized by the EIF2AK3 antibody is indicative of EIF2AK3 phosphorylation. Third (bottom panels), aliquots of the harvested cells were subjected to real-time RT-PCR analysis of XBP1s mRNA levels as described in Materials and Methods. Mean relative values ± SEM from the 3 independent experiments are shown. (G and H) [14C]-valine radiolabelled LNCaP cells (G) or U2OS cells (H) were chased and then treated as in (E and F), but for 6 h and without BafA1 addition. Degradation rates of long-lived proteins were determined as detailed in Materials and Methods. The columns represent mean degradation rates ± SEM from 3 (G) or 4 (H) independent experiments.
![Figure 3. A23187 (A23)- and TG-mediated repression of autophagy is independent of ER stress. LNCaP cells (A) or U2OS cells (B) were transfected with a nontargeting control siRNA (siCtrl) or siRNAs targeting ATF4 (siATF4), ERN1 (siERN1), or ATF6 (siATF6). Subsequently, cells were treated for 6 h with DMSO control (Ctrl, 0.2%), A23 (0.75 µM), or TG (100 nM) under amino acid starvation conditions, and long-lived protein degradation rates were determined as described in Materials and Methods. Mean protein degradation rates ± SEM from 3 independent experiments are shown. (C and D) Confirmation of knockdown efficiencies of the siRNAs used in (A and B). LNCaP cells (C) or U2OS cells (D) were transfected with siRNAs as in (A and B) and treated for 6 h with DMSO (Ctrl, 0.2%) or TG (100 nM) under amino acid starvation conditions. Subsequently, RNA was extracted and subjected to real-time RT-PCR analyses of relative mRNA levels of ATF4, ERN1, and ATF6 as indicated. Mean relative mRNA levels ± SEM from 3 independent experiments are shown. The average value of the DMSO control conditions with the control siRNA was arbitrarily set to 1. The mean knockdown efficiency varied between 83% and 92%. (E and F) LNCaP cells (E) or U2OS cells (F) were pretreated for 1 h with DMSO (Ctrl, 0.2%), the EIF2AK3 inhibitor GSK2606414 (EIF2AK3i, 100 nM) + 0.1% DMSO, the ERN1-inhibitor MKC8866 (ERN1i, 10 µM) + 0.1% DMSO, or a combination of the two inhibitors (100 nM EIF2AK3i + 10 µM ERN1i). Thereafter, the cells were washed in EBSS medium and starved for 2 h in EBSS medium containing BafA1 (100 nM) and either DMSO (Ctrl, 0.1%), A23 (0.75 µM) or TG (100 nM). Moreover, to maintain suppression of EIF2AK3 and ERN1 activity, EIF2AK3i, ERN1i, and DMSO were re-added at the same concentrations and combinations as during the pretreatment. Subsequently, cells were harvested and assessed for LDH sequestration (top panels) as detailed in Materials and Methods. Mean relative LDH sequestration values ± SEM from 3 independent experiments are shown. The mean LDH sequestration rate in cells that were starved for amino acids in the absence of BafA1 was arbitrarily set to 1. Second (middle panels), aliquots of the total disruptates that were generated in the LDH sequestration assay were used for western blot analyses of EIF2AK3, XBP1s, and GAPDH (loading control) as detailed in Materials and Methods. The observed shift toward the more slowly migrating bands that are recognized by the EIF2AK3 antibody is indicative of EIF2AK3 phosphorylation. Third (bottom panels), aliquots of the harvested cells were subjected to real-time RT-PCR analysis of XBP1s mRNA levels as described in Materials and Methods. Mean relative values ± SEM from the 3 independent experiments are shown. (G and H) [14C]-valine radiolabelled LNCaP cells (G) or U2OS cells (H) were chased and then treated as in (E and F), but for 6 h and without BafA1 addition. Degradation rates of long-lived proteins were determined as detailed in Materials and Methods. The columns represent mean degradation rates ± SEM from 3 (G) or 4 (H) independent experiments.](/cms/asset/48bbf023-a00a-4e7d-948e-0eeb7910b888/kaup_a_10925900_f0003.gif)
It remained possible that protein synthesis-driven changes in transcription mediated by other transcription factors than those that are activated by the UPR contributed to the anti-autophagic effect of A23187 and TG. To test this in a global and unbiased manner we used cycloheximide to inhibit nascent protein synthesis. Cycloheximide treatment diminished the extent of LDH sequestration induced by amino acid starvation, but A23187 and TG were still effective inhibitors (Fig. S8). Thus, the inhibitory effect of the calcium-modulating agents did not appear to require translation.
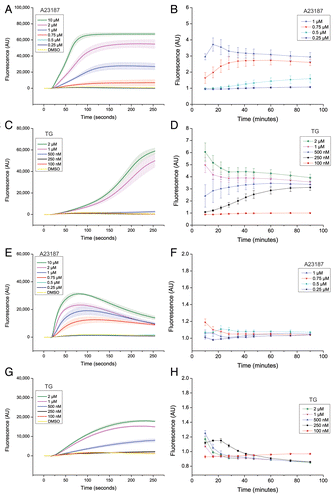
Next, we examined the relationship between elevation of cytosolic calcium levels and autophagy inhibition. First, we aimed to test whether chelation of cytosolic calcium ions with the cell-permeable calcium chelator BAPTA-AM would reverse the anti-autophagic effects of the calcium-modulating agents. However, and in line with previous observations,Citation19,Citation34 treatment with BAPTA-AM by itself completely blocked autophagy, as assessed by LDH sequestration and LLPD, under both nutrient-rich and amino acid starvation conditions (Fig. S9). Thus, we could not use global chelation of cytosolic calcium ions as a straightforward tool to assess the potential relationship between increased cytosolic calcium levels and the anti-autophagic effects observed with A23187 and TG. Therefore, to gain more insight, we next assessed in detail whether A23187- and TG-mediated inhibition of autophagy was proportional to changes in cytosolic calcium levels induced by the treatments. Interestingly, we observed striking differences in both the magnitude and the kinetics of alterations in cytosolic calcium levels induced by treatment with A23187 and TG over a concentration range in the two cell lines (). At the concentration of A23187 that was used in the autophagy assays under amino acid starvation conditions (0.75 µM), A23187 yielded an early (within 1 min) and sustained (over 90 min) elevation of cytosolic calcium levels in LNCaP cells (), and an early (within 30 s) but transient (less than 30 min) increase in U2OS cells (). In both cell lines, the elevation of cytosolic calcium levels observed with 0.75 µM of A23187 was slower and more modest compared with at higher concentrations, with a maximal early increase being observed at 10 µM of A23187 (). In contrast, and to our surprise, at the concentration of TG that was used in the autophagy assays (100 nM) we could not detect any changes in cytosolic calcium levels at any of the time points examined in either LNCaP cells () or U2OS cells (). In fact, bulk alterations in cytosolic calcium levels were only detected at several-fold higher concentrations of TG (250 to 500 nM), and whereas the effect at these higher concentrations was sustained in LNCaP cells (), the effect was more modest and transient in U2OS cells (). Strong and rapid effects of TG were obtained at 1 to 2 µM TG in both cell lines (). In conclusion, the inhibition of autophagy that we observed with A23187 and TG did not correlate with sustained or bulk changes in cytosolic calcium levels.
Figure 4. Suppression of autophagy does not require bulk or sustained elevation of cytosolic calcium levels. LNCaP cells (A–D) or U2OS cells (E–H), loaded with the calcium-sensitive dye “FLIPR calcium V”, were treated simultaneously at the 16 s time point with either DMSO (0.2%; vehicle control) or various concentrations of A23187 or TG as indicated. Changes in cytosolic calcium ion levels were followed over a period of 90 min as detailed in Materials and Methods, first by using the FLIPR384 instrument for imaging at 1 s intervals for the first 4 min after treatment (A, C, E, and G), and second by using a Tecan Infinite F200 Pro plate reader for fluorescence measurements at various time intervals from 10 to 90 min (B, D, F, and H). Each panel shows mean values ± SEM at each time point from the 3 independent experiments. Each experiment was performed in six replicate wells. The graphs in (A, C, E, and G) shows mean values ± SEM of the raw fluorescence values obtained in DMSO- A23187- or TG-treated cells at each time point. In (B, D, F, and H), the mean value obtained in DMSO control-treated cells was arbitrarily set to 1 at each time point. AU = arbitrary units. Of note, unlike in complete culture medium, the two highest concentrations of A23187 (2 and 10 µM) showed signs of toxic effects under amino acid and serum starvation conditions (in EBSS or in the HBSS-HEPES buffer solution used in the calcium measurement assay). Long-term calcium measurements for these two concentrations of A23187 are therefore not shown in (B and F), and were also not used to assess effects of A23187 on autophagy under conditions of amino acid starvation. Also note that several of the lines representing measurements of the DMSO control and concentrations of A23187 and TG which give no discernible increase in fluorescence compared with the DMSO control condition (i.e., no detectable increase in cytosolic calcium levels) often overlap.
Discussion
Autophagy is known to be a multistage process and is tightly regulated at each step from phagophore formation to the fusion of autophagosomes with lysosomes.Citation2,Citation50 By analyzing both early and late stages of autophagy with a variety of established methodsCitation17 we were able to show that calcium-modulating agents were not inducers of ‘committed’ autophagy—defined as the uninterrupted progression from phagophore generation to degradation of sequestered content following fusion with lysosomes—but rather they blocked commitment. The accumulation of lipidated LC3 is often used as a surrogate measure of autophagic activity, but is hazardous to interpret. By blocking commitment it is possible to observe an accumulation of autophagy markers, as well as, depending on the step that is affected, autophagosomes or autophagosome-like structures.Citation17 Calcium-modulating agents have been regarded as autophagy inducers on this basis,Citation3,Citation6,Citation7,Citation10,Citation12,Citation13,Citation51 and on the same basis a number of studies have concluded that enhancement of cytosolic calcium levels induces autophagy.Citation4,Citation5,Citation8,Citation9,Citation11,Citation14-Citation16 Our data strongly argue for a refinement of both possible interpretations.
By using a well-controlled assay for long-lived protein degradation (LLPD), we found that autophagic LLPD was inhibited by calcium-modulating agents in both nutrient-rich and amino acid starvation conditions in two different cell lines. These results are in line with observations from previous studies. First, a recent study by Ganley et al. shows that a high concentration of TG can block amino acid starvation-induced LLPD in mouse embryonic fibroblasts.Citation18 Second, two other recent studies report that longer-term treatment (18 to 24 h) with TG or L-type calcium channel agonists results in the accumulation of ectopically expressed autophagy substrates in neuronal cells.Citation20,Citation21 Third, Gordon et al. reported 20 years ago that A23187 and TG can block LLPD in primary rat hepatocytes, which show a very high rate of autophagy in culture.Citation19 In the study by Ganley et al.,Citation18 immunogold-labeling of LC3 and electron microscopy (EM) have been used to assess whether structures resembling autophagosomes were still generated. EM sections showed discrete circular entities decorated with LC3, but this in itself does not prove the formation of fully intact double-membrane vesicles. A key contribution in the current paper has arisen from adapting an assay for LDH sequestration, developed by Seglen and coworkers and used for decades in primary hepatocytesCitation30,Citation31 to human cancer cell lines. By using this assay we could quantitatively measure the sequestration of LDH, a normally cytosolic enzyme, into autophagic vacuoles, and this was critical in demonstrating that calcium-modulating agents have an inhibitory effect prior to the stage at which nascent autophagosomes seal off their contents (). Furthermore, the observed partial reduction in both the number and size of EGFP-LC3 puncta after treatment with A23187 or TG in starved U2OS-mRFP-EGFP-LC3 cells (Fig. S3), indicate that calcium-modulating agents may in part block early events in autophagosome biogenesis under starved conditions. Reduced numbers of EGFP-LC3 puncta are also noted in the study by Ganley et al. upon TG-treatment of starved mouse embryonic fibroblasts.Citation18 However, in both this and in other studies, EM-visualization indicate that A23187 and TG do allow ring-shaped autophagosomal-like structures to form,Citation3,Citation12,Citation18 and in the current study we found an accumulation, rather than a reduction, in EGFP-WIPI1 puncta in TG-treated cells (Fig. S4). Taken together this indicates that calcium-modulating agents suppress a step in autophagosome biogenesis that lies after the nucleation and initial expansion of the phagophore, but before the final phase of expansion and closure, thus leading to the production of incomplete autophagosomes that are unable to form sealed and stable entities that can sequester cytosolic proteins like LDH.
A23187 and TG are known to not only modulate intracellular calcium ion levels but also to induce ER stress,Citation35,Citation37 and both events reportedly affect autophagy.Citation39,Citation52 Coincidence of calcium perturbation and ER stress is not unusual, as calcium release from the ER can induce ER stress, and ER stress by itself may lead to release of calcium ions from the ER.Citation39 Previous studies have shown inhibitory effects of TG on autophagy,Citation18-Citation21 however, associations between the inhibition of autophagic activity and ER stress induction, or the magnitude of intracellular calcium perturbations and autophagy inhibition by calcium-modulating agents have not been investigated. Here, we addressed this need and carefully distinguished between the roles played by calcium vs. ER stress in the regulation of autophagy. Several downstream effectors of the UPR, in particular of the EIF2AK3- and ERN1-arms, have been linked to enhanced expression and/or altered activation of proteins that are involved in promoting either intracellular signaling for initiation of autophagy or the early steps in the autophagic process related to autophagosome biogenesis.Citation39 This includes induced expression of DDIT4 by the EIF2AK3-ATF4 armCitation53 and negative regulation of the protein kinase AKT,Citation54 both resulting in MTOR inactivation, which again can result in the initiation of autophagy. Moreover, ATF4 has been reported to upregulate the expression of the protein kinase ULK1 (involved in signaling the initiation of autophagy), as well as ATG5 and ATG12 (components of the core autophagic machinery required for autophagosome biogenesis), and LC3B.Citation55-Citation58 Further, XBP1 has recently been shown to control the expression of BECN1/Beclin 1, which is involved in autophagosome biogenesis.Citation59 Other reports have, on the contrary, linked parts of the ERN1-arm to repression of autophagy,Citation20,Citation60 and a recent study reports that XBP1 is required for ATG9-acetylation, which may inhibit ATG9 function and thereby inhibit autophagy.Citation61 Of note, studies on the role of ER stress on autophagy have rarely assessed autophagic activity. Moreover, due to a lack of potent and specific inhibitors of the upstream UPR effectors, most studies have examined the effects of long-term inhibition of UPR signaling using RNAi, dominant-negative constructs, or cells with genetic deletions. Thus, the time-frame and degree of inhibition necessary to affect gene expression patterns and autophagy have not been addressed in detail. Here, we took a more comprehensive approach by combining the use of functional autophagy assays with two novel, potent and specific inhibitors of EIF2AK3 and ERN1, termed GSK2606414 and MKC8866, respectively. These inhibitors represent a new class of drugs with clinical implications, but also for use as research tools to analyze the biology of the UPR. This enabled us to test the effect of rapid and virtually complete inhibition of these two arms of the UPR during the acute ER stress response that is induced by A23187 or TG. In addition, we tested the effect of siRNAs against all three arms of the UPR on the repression of autophagy. By analyzing LDH sequestration and LLPD, we unambiguously show that A23187- and TG-mediated repression of autophagy does not require the UPR (). Additionally, since autophagy was not significantly affected by siRNA-mediated targeting or chemical inhibition of UPR effectors under conditions of amino acid starvation, our results indicate that starvation-induced autophagy is largely independent of UPR effectors. On the basis of these results and the above-mentioned studies on UPR-mediated transcriptional regulation of proautophagic genes, it will be interesting in the future to further delineate the function of this transcriptional control with regard to autophagic capacity vs. autophagic activity in cells.
Having dissociated the activation of the UPR from the inhibitory effect of the calcium-modulating agents on autophagy, we turned to examining the relationship between elevation of cytosolic calcium levels and autophagy inhibition. We found that, first, there was no overall correlation between the magnitude of calcium modulation and inhibition of autophagy, and second, there was no requirement for either sustained or bulk changes in cytosolic calcium levels for autophagy to be inhibited. This is best exemplified by the results with TG, where we observed a complete block in autophagy when measuring LDH sequestration after 2 h of treatment under starved conditions (), but could not detect any increase in cytosolic calcium levels, despite using a sensitive assay and a time-scale up to 90 min (). Our study is the first to show that inhibition of autophagy can be induced by a calcium-modulating agent in the absence of detectable changes in global cytosolic calcium levels, implying that more subtle and local effects are involved, and that this is a regulatory mechanism that may be highly relevant under physiological conditions. The previous studies that have shown inhibition of autophagy with TG used concentrations in the micromolar range,Citation18-Citation21 which results in depletion of intracellular stores and a very strong increase in cytosolic calcium levels.Citation19,Citation21 Also in the current study, we observed rapid and strong elevation in cytosolic calcium levels with 1 to 2 µM TG (). However, our data using a much lower concentration of TG (100 nM) suggest that its antiautophagic activity is not merely caused by a general depletion of intracellular stores, but rather results from local changes in calcium levels. Taken together, our data indicate that local perturbations of calcium levels around intracellular stores that are acted upon by A23187 and TG may be particularly important in autophagy regulation. A23187 and TG both perturb the ER calcium ion gradient by promoting release of calcium ions into the cytosol and hence induce ER stress. Thus, given that the ER appears to be a major site of autophagosome generation,Citation62-Citation66 it is tempting to speculate that localized perturbations of calcium levels around parts of the ER where autophagosomes are synthesized is a major causative effector of the remarkably strong block in autophagosome biogenesis that we observe with A23187 and TG in the current study. Interestingly, however, angiotensin and vasopressin, which stimulate calcium release via the inositol triphosphate receptor (ITPR), have little or no effect on autophagy in primary rat hepatocytes,Citation67 indicating that calcium release from the ER does not block autophagy in all contexts. It has been proposed that in rat hepatocytes autophagy is inhibited due to depletion of calcium ions from intracellular pools other than those related to the ITPR-related ER pools.Citation19,Citation67 A future challenge, which will require spatially and concentration-sensitive assays of calcium release that are presently not available, will be to identify the source(s) of intracellular calcium that are involved in the antiautophagic effects of calcium-modulating agents.
Interestingly, a certain level of free intracellular calcium appears to be required for autophagy, as we found that calcium chelation with BAPTA-AM by itself completely inhibited LDH sequestration and LLPD under both fed and starved conditions (Fig. S9). Similar observations have been made in primary rat hepatocytes.Citation19 Moreover, another study reports that BAPTA-AM completely blocked the generation of EGFP-WIPI1 puncta as well as LC3-II protein abundance under both nutrient-rich and starved conditions.Citation34 This indicates that free intracellular calcium is an absolute requirement for early phagophore formation. Taken together with the results in the current study, it is tempting to speculate that autophagy is only permitted within a certain concentration range of cytosolic calcium levels. Moreover, since our data suggest that very modest and/or local elevations of cytosolic calcium levels may have profound inhibitory effects on autophagic activity, an interesting question is whether also basal cytosolic calcium levels might act to limit autophagy. In favor of this, L-type calcium channel antagonists induce autophagy in neuronal cells,Citation21,Citation68 and lithium-induced autophagy is mediated by a lowering of cellular IP3 levels.Citation69 It should be noted, however, that these studies did not assess whether the treatments actually reduced cytosolic calcium levels. Moreover, and in contrast, suboptimal concentrations of calcium chelators do not elevate autophagy in primary hepatocytes.Citation19 Interestingly, an alternative mechanism has been proposed for how calcium ion channeling via the ITPR can inhibit basal autophagy. Thus, under nutrient-rich and unstressed conditions, it has been shown that a constitutive ITPR-mediated delivery of calcium ions from the ER to the mitochondria acts to maintain optimal bioenergetics, thereby keeping AMPK activity as well as LC3 lipidation low.Citation70 This mechanism does not apply under starved conditions however,Citation70 where autophagy initiation has been reported to be independent ofCitation34 or suppressed byCitation71 AMPK. Interestingly, amino acid starvation has recently been reported to sensitize the ITPRCitation72 and to give a slight increase in cytosolic calcium levels,Citation5 and an involvement of ITPR in starvation-induced autophagy has been indicated, since LC3 lipidation was not further enhanced by amino acid starvation in the presence of the ITPR-inhibitor Xestospongin B.Citation72 Since the calcium-chelator BAPTA-AM inhibited LC3 lipidation, it was suggested that elevation of cytosolic calcium levels is required for starvation-induced autophagy.Citation5,Citation72 However, since BAPTA-AM, as described above, appears to completely block even basal autophagy, this does not give a definitive answer as to whether the slight increase in cytosolic calcium levels observed during amino acid starvationCitation5 promotes autophagy. It would be interesting to test this further with functional autophagy assays and additionally with other means of ITPR inhibition, since Xestospongin B has been reported to also affect aspects of the ITPR that are unrelated to its calcium ion channeling properties.Citation73,Citation74 Thus, it remains to be resolved whether starvation-induced elevation of cytosolic calcium levels acts as fuel or a brake during starvation-induced autophagy. However, based on the observation that amino acid starvation sensitizes the ITPR for calcium releaseCitation72 and our observation that calcium-modulating agents can completely block starvation-induced autophagy, it is tempting to speculate that a sensitization of the ITPR during amino acid starvation may also sensitize the cells to autophagy inhibition upon stress inductions that lead to extra release of calcium ions via the ITPR. This is a fundamental question in resolving cell fate in response to stressors. In a disease context cells are often subjected to an acute array of stresses ranging from hypoxia and nutrient deprivation to chemotherapy.
In conclusion we propose that phagophore expansion and closure are key regulatory steps in the autophagic process, which can be inhibited upon perturbation of the intracellular calcium balance. There are currently few clues as to the identity of regulators of phagophore expansion and closure. Atg8 and LC3 paralogs have been suggested based on EM,Citation75,Citation76 and in vitro membrane fusion assays,Citation77,Citation78 but a recent report has indicated that Atg8 and LC3 are not sufficient for membrane fusion.Citation79 Upstream and downstream of expansion and closure, SNARE proteins are being identified as determinants of critical membrane fusion events in the autophagic process,Citation80 which is in line with the view of SNAREs as regulators of specificity and commitment for membrane fusion events throughout the cell.Citation81 For example, SNAREs are involved in the early membrane fusion processes that are required for autophagosome precursor maturation in both yeast and mammalian cells,Citation79,Citation82 and SNAREs mediate the fusion of autophagosomes with endosomes and with lysosomes in mammalian cells as well as in yeast.Citation80 Interestingly, SNARE-mediated fusion events are in many cases regulated by calcium.Citation81,Citation83 There is now a need for a systematic approach to identify regulators of autophagosome expansion and closure. We propose a primary screen for such candidates, using sequestration assays with “neutral” cargoes (like LDH) that do not have autophagic functions (as do e.g., LC3 and p62). A secondary assay would be imaging for regulators that block sequestration but not the accumulation of markers that build on the nascent autophagosome including WIPI1. Our study also suggests that TG and other drugs may be useful in trapping intermediates, and thus can have vital applications in assay workflow schemes. Such a systematic screening approach will form the basis of future studies.
Materials and Methods
Cells and cell treatments
LNCaP and U2OS cells (ATCC, CRL-1740, and HTB-96) were cultured in RPMI 1640 or DMEM (Gibco, 21875 and 41966) respectively, supplemented with 10% fetal bovine serum (FBS; Gibco, 10500) at 37 °C and 5% CO2. For amino acid starvation experiments, cells were washed with Earle's balanced salt solution (EBSS) (Gibco, 24010) and then incubated in EBSS containing the various test compounds. All cell treatments were performed in calcium-containing media or calcium-containing EBSS, or, for the measurements of cytosolic calcium levels, in calcium-containing Hank’s Balanced Salt Solution (HBSS) (Gibco, 14025050) plus 20 mM HEPES buffer, pH 7.4 (Sigma, H4034).
Chemicals
A23187, thapsigargin (TG) (Sigma C7522 and T9033, respectively), bafilomycin A1 (BafA1; Enzo, BML-CM110), and MG132 (Calbiochem, 474790) were dissolved in DMSO and stored at −20 °C. 3-Methyladenine (3-MA; Sigma, M9281) was dissolved in RPMI or EBSS to 100 mM and stored at −20 °C. Chloroquine (CQ; Sigma C6628) was freshly prepared in water. The EIF2AK3 inhibitor GSK2606414, a kind gift from Dr Jeffrey M Axten (GlaxoSmithKline), and the ERN1 inhibitor MKC8866 (MannKind Corporation) were dissolved in DMSO and kept as aliquots at −80 °C.
Western blot analysis
Cells were washed with PBS (Gibco, 20012) and lysed in RIPA buffer (30 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP40, 0.1% NaDOC, 0.1% SDS) supplemented with protease and phosphatase inhibitor cocktails (Roche, 118361170001, 04906837001), followed by high power sonication for 10 × 30 s using a Bioruptor (Diagenode) water bath sonicator. Protein concentrations of cleared extracts were determined by the BCA assay (Pierce, 23227) and equalized with RIPA buffer. Extracts were mixed with LDS NUPAGE sample buffer (Invitrogen, NP0008) with reducing agent (Invitrogen, NP0009) and incubated for 10 min at 70 °C. Equal amounts of extracts (5 to 15 µg protein per lane) were loaded onto Criterion 4–20% gradient 26-well Tris-HCl gels (Bio-Rad, 345-0034) and wet-blotted (48 mM Tris, 39 mM glycine, 0.02% SDS, 20% methanol) to activated 0.45 µm PVDF membranes (Millipore, IPVH00010) for 100 min at 55V under cooled conditions using a Criterion blotter with plate electrodes (Bio-Rad). Even transfer was confirmed by staining with Ponceau S (Sigma, P7170), which was removed with 0.1 M NaOH. Membranes were blocked in 7.5% nonfat milk (Cell Signaling Technology, 9999S) in TBS-Tween (0.05%) for 30 min followed by incubation with primary antibodies recognizing EIF2AK3/PERK (Cell Signaling Technology, 5683), phosho-EIF2S1/eIF2α (Cell Signaling Technology, 3398), ATF4 (Santa Cruz, sc-200 C-20), DDIT3/CHOP (Cell Signaling Technology, 2895), XBP1s (BioLegend, 647502, clone 143F), HSPA5/BiP (Cell Signaling Technology, 3177), EIF2S1/eIF2α (Cell Signaling Technology, 5324), PARP (Cell Signaling Technology, 9542), cleaved CASP3/caspase 3 (Cell Signaling Technology, 9661) or GAPDH (Cell Signaling Technology, 2118) in 5% BSA/TBS-Tween (0.05%) overnight at 4°C, followed by anti-rabbit- or anti-mouse-HRP (Dako, P0448 and P0447, respectively) in 5% nonfat milk in TBS-Tween (0.05%) for 40 min. Bands were visualized with Novex ECL reagents (Invitrogen, WP20005) and detection with Chemidoc (BioRad), or Hyperfilm (GE Healthcare, 28906844) and developer (AGFA Curix-60). Bands were quantified using the QuantityOne software (Bio-Rad, version 4.6.5). In some experiments (), aliquots of the lysates from total disruptates prepared in the LDH sequestration assays were used for western blot analyses. For this purpose, the 4/5 of the disruptate was mixed with a 5× concentrated RIPA lysis buffer supplemented with 5× concentrated protease and phosphatase inhibitor cocktails (Roche) before high power sonication, determination of protein concentrations and immunoblotting as described above.
Quantitative real-time RT-PCR
Cells were trypsinized and washed in cold PBS/0.5% FBS before total RNA was extracted using the illustra RNAspin Mini Isolation Kit (GE Healthcare, 25-0500-71) according to the manufacturer’s instructions. Total RNA was reverse transcribed using the SuperScript VILO Master Mix (Applied Biosystems, 11754) according to the manufacturer’s manual. Quantitative real-time RT-PCR was performed using TaqMan Gene Expression Assays (Applied Biosystems) (see Table S1 for a list of the assay ID for the probes that were used) with TaqMan Fast Advanced PCR Master Mix (Applied Biosystems, 1208021). PCR amplification was performed in duplicate series using the ABI 7900HT FAST Sequence Detection System (Applied Biosystems). The cycling conditions were 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. Transcript levels relative to those in DMSO control samples were determined using the comparative CT methodCitation84 and normalization to the geometric mean CT value of GAPDH and TBP.Citation85
Degradation of long-lived proteins
Cells were seeded in 6-well plates (BD Falcon, 353046) and radiolabeled with 0.3 µCi [14C]-valine (Perkin Elmer, NEC291EU050UC) in 2 ml complete RPMI 1640 (with 10% FBS) for 2 to 3 d. Subsequently, cells were washed with 2 ml PBS/2.5% FBS to remove unincorporated radioactivity, and chased in 1.5 ml complete RPMI 1640 supplemented with 10 mM cold valine (Sigma, V0513) (to prevent reincorporation of labeled valine) for 18 h. Next, short-lived protein degradation products were washed out with either 2 ml complete RPMI 1640 or 2 ml EBSS, followed by incubation in 1 ml of either complete RPMI 1640 or EBSS (both supplemented with 10 mM cold valine) with the various treatments for another 6 or 16 h, as specified in the figure legends. Cells were scraped in their media and transferred to 2 ml Eppendorf tubes, and the wells were washed with 300 µl PBS/1% BSA (carrier) followed by addition of 300 µl 50% TCA (Sigma, T0699) and overnight incubation at 4 °C to precipitate non-degraded proteins. Precipitates were sedimented by centrifugation at 5,000 × g for 10 min at 4 °C and solubilized in 500 µl 0.2 M KOH (Sigma, 221473). Radioactivity was determined in both the supernatant (the TCA-soluble fraction, containing [14C]-valine from degraded proteins) and in the pellet fraction by liquid scintillation counting. The degradation rate for long-lived proteins was calculated as the percentage of radioactivity in the TCA-soluble fraction relative to the total radioactivity in the TCA-soluble and nonsoluble fractions, divided by the incubation time.
Transfection of cells with siRNAs
Small interfering RNAs (siRNAs) were introduced into LNCaP and U2OS cells by reverse transfection in 6-well plates. The siRNAs were diluted in Opti-Mem Reduced Serum Medium (Gibco, 11058) to 152 nM in a total volume of 336 ul per well. Subsequently, the siRNA-solutions were mixed with Lipofectamine RNAiMax (Invitrogen, 13778) (4 ul per well), and 340 ul of the mixture was transferred to each well of the 6-well culture plate. After 20 to 45 min incubation at room temperature, 1.7 ml cells were added (6 × 105 LNCaP cells or 2.5 × 105 U2OS cells, both in complete medium prewarmed to 37 °C), yielding a final siRNA-concentration of 25 nM. Solutions were mixed by agitation. For LLPD assays, the cells were suspended in complete RPMI 1640 containing [14C]-valine to a final concentration of 0.15 µCi/ml, and after 48 h of transfection the cells were washed and incubated in complete RPMI 1640 supplemented with 10 mM cold valine to start the 18 h chase period. Experimental treatments were initiated after 66 h of transfection. The following siRNAs were used (all Silencer Select siRNAs from Ambion): Silencer® Select Negative Control #1 (4390843), siATF4 (ID 1704), siERN1 (4390824-s200432), and siATF6 (4392420-s223544).
Measurement of autophagic sequestration activity
Autophagic sequestration activity was measured as the transfer of the cytosolic enzyme lactate dehydrogenase (LDH) to sedimentable autophagic vacuoles essentially as described,Citation30,Citation31 except that leupeptin was replaced by BafA1 as a more effective inhibitor of intralysosomal LDH degradation in the cell types used here. U2OS or LNCaP cells were seeded in 10 cm dishes (Corning, 430293) two days before the experiment (2 or 4 million cells, respectively). Subsequently, cells were washed once with 6 ml complete medium or EBSS (for amino acid starvation) followed by incubation at 37°C in 6 ml complete medium or EBSS containing the various treatments in the absence or presence of 100 nM BafA1. Incubation with the sequestration inhibitor 3-MA (10 mM) was routinely used as a negative control. The cells were harvested by trypsination (1.5 ml trypsin-EDTA) for 5 to 10 min at 37 °C and stopped by adding 3.5 ml warm (37 °C) complete medium. Subsequently the cells were resuspended by pipetting until no clumps were visible, and transferred to a 15 ml Falcon tube on ice. The cells were sedimented by centrifugation at 4 °C for 5 min at 150 × g and washed twice by resuspension and recentrifugation (4 min at 600 × g) in 5 ml cold (4 °C) unbuffered, isotonic (10%) sucrose. Finally, the cell pellet was resuspended in 0.3 ml cold 10% sucrose and electrodisrupted by a single high-voltage pulse (2 kV; 1.2 µF) as described elsewhere.Citation86 The resulting disruptate was diluted with 0.3 ml cold phosphate-buffered sucrose (100 mM sodium phosphate, 2 mM dithiothreitol (DTT), 2 mM EDTA, and 1.75% sucrose, pH 7.5), and maintained at 0 °C. Electrodisruption only disrupts the plasma membrane, leaving intracellular organelles (including autophagic vacuoles) intact, trapped in the cytoskeletal meshwork of coherent “cell corpses.” To separate the cell corpses from cytosol, 0.4 ml of disruptate was layered on top of a 4 ml ice-cold density cushion of phosphate-buffered Nycodenz (8% Nycodenz, 50 mM sodium phosphate, 2.2% sucrose, 1 mM EDTA, 1 mM DTT) in 16 ml polycarbonate tubes (Sorvall, 03243). After centrifugation at 4 °C for 15 min at 49,000 × g (Sorvall SM-24, Sorvall®, DuPont Co., at 20,000 rpm) the top half of the supernatant was removed by aspiration, the tube wall was rinsed with 5 ml 10% sucrose (to reduce cytosolic contamination) and finally the supernatant was fully removed. The cell corpse pellets as well as the remaining 0.2 ml of disruptate were exposed to one freeze-thaw cycle (overnight at −80 °C) before measurement of sedimentable (sequestered) and total cellular LDH activity, respectively. Cell corpse pellets were dissolved by vortexing in 0.3 ml detergent-containing phosphate buffer (50 mM sodium phosphate, 1 mM EDTA, 1 mM DTT, 0.3% Triton X-405); the total disruptates were dissolved by 10-fold dilution with the same buffer. Finally, all samples were centrifuged (for cleanup) at 12 °C for 5 min at 21,000 × g (Hettich, 2424A), and LDH activity in the supernatants was measured by the decline in NADH absorbance at 340 nm in a multi-analyzer (Maxmat PL-II, Erba Diagnostics) using an LDH-assay kit (Erba Diagnostics, RM LADH0126V). The amount of LDH sedimenting with the cell corpse pellet (sample value divided by 1.333) relative to the amount in the total disruptate (sample value multiplied by 10) is expressed as “sequestered LDH (% of total)” in the figures. Of note, the 3-MA sensitive net accumulation of LDH in the presence of bafilomycin A1 is taken to represent bona fide autophagic sequestration. Although some of the sequestered LDH observed in the absence of the lysosome inhibitor may represent enzyme en route through the autophagic-lysosomal pathway,Citation30 most of it is regarded as nonspecific “background.”
Measurements of cytosolic calcium levels
Live-cell calcium measurements were made using the FLIPR calcium 5 assay kit (Molecular Devices, R8185) and a FLIPR384 Fluorometric Imaging Plate Reader instrument (Molecular Devices Corp.) for simultaneous addition of compounds and detailed time-resolved imaging at 1 s intervals for the first 4 min after treatment, followed by fluorescence measurements with a Tecan Infinite F200 Pro plate reader (Tecan Systems, Inc.) at various time intervals for up to 90 min after treatment. Cells (1.5 × 104 LNCaP cells or 9 × 103 U2OS cells in 25 µl complete medium) were seeded in black clear-bottomed poly-d-lysine coated 384-well plates (Greiner bio-one, 781946), so that they would reach confluency the next day, at which time the medium was removed and carefully replaced with 25 µl of HBSS plus 20 mM HEPES buffer, pH 7.4. Subsequently, the calcium-sensitive dye was dissolved in an HBSS-HEPES buffer solution containing 5 mM freshly made probenicid (Sigma, P8761) (the final solution adjusted to pH 7.4 with HCl), and 25 µl was carefully added to each well. After incubation for 45 min at 37 °C and 15 min at room temperature, the FLIPR instrument was employed for the simultaneous addition of 12.5 µl of all compounds (i.e., DMSO, A23187, or TG) from a compound plate prepared with 5× concentrated compounds in HBSS-HEPES buffer, at a speed of 5 µl/s and an add height of 20 µl after frame 15, i.e., at the 16 s time point. After the 4 min image-acquisition at the FLIPR instrument with 1 s intervals, the plate was removed for fluorescence measurements with the Tecan Infinite F200 Pro plate reader at 6 min intervals (starting at 10 min) up to 46 min, followed by 14 to 15 min intervals up to 90 min. Each independent experiment was always performed with six replicate wells per condition, and the mean value from the six replicates was used when calculating the mean- and SEM-values depicted in the figures (i.e., for each time point, the 3 mean values from the 3 independent experiments were used in the calculations).
Statistical analyses
For all experiments where tests of significance were performed, approximate normal distribution of the data was assumed and a two-tailed the Student t-test for paired samples (denoted as “Student t-test” in the figure legends) was employed using Excel.
| Abbreviations: | ||
| A23 | = | A23187 |
| ATF4 | = | activating transcription factor 4 |
| ATF6 | = | activating transcription factor 6 |
| AMPK | = | AMP-activated protein kinase |
| ATG | = | autophagy related |
| BafA1 | = | bafilomycin A1 |
| BAPTA-AM | = | 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) |
| HSPA5/BiP | = | heat shock 70 kDa protein 5 (glucose-regulated protein, 78 kDa) |
| DDIT3/CHOP | = | DNA-damage-inducible transcript 3 |
| CQ | = | chloroquine |
| DMSO | = | dimethyl sulfoxide |
| EBSS | = | Earle’s balanced salt solution |
| EGFP | = | enhanced green fluorescent protein |
| EIF2S1/eIF2α | = | eukaryotic translation initiation factor 2, subunit 1 alpha, 35 kDa |
| ER | = | endoplasmic reticulum |
| EM | = | electron microscopy |
| FBS | = | fetal bovine serum |
| PPP1R15A/GADD34 | = | protein phosphatase 1, regulatory subunit 15A |
| GAPDH | = | glyceraldehyde 3-phosphate dehydrogenase |
| ITPR/IP3R | = | inositol 1,4,5-trisphosphate receptor, type 1 |
| ERN1/IRE1 | = | endoplasmic reticulum to nucleus signaling 1 |
| LC3 (MAP1LC3) | = | microtubule-associated protein 1 light chain 3 |
| LDH | = | lactate dehydrogenase |
| LLPD | = | long-lived protein degradation |
| 3-MA | = | 3-methyladenine |
| mRFP | = | monomeric red fluorescent protein |
| MTOR | = | mechanistic target of rapamycin |
| PBS | = | phosphate buffered saline |
| EIF2AK3/PERK | = | eukaryotic translation initiation factor 2-alpha kinase 3 |
| PI | = | propidium iodide |
| DDIT4/REDD1 | = | DNA damage-inducible transcript 4 |
| RNAi | = | RNA interference |
| siRNA | = | small interfering RNA |
| SNARE | = | soluble NSF attachment protein receptor |
| TBP | = | TATA box binding protein |
| TG | = | thapsigargin |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| UPR | = | unfolded protein response |
| WIPI1 | = | WD repeat domain, phosphoinositide interacting 1 |
| XBP1 | = | X-box binding protein 1 |
| XBP1s | = | XBP1 spliced |
Additional material
Download Zip (9.3 MB)Acknowledgments
We thank Dr Jeffrey M Axten (GlaxoSmithKline) for providing the GSK2606414 compound. This research and NE, IJG, SJB, and IGM are supported by funding from the South-Eastern Norway Regional Health Authority, the University of Oslo and the Research Council of Norway (RCN) through the Centre for Molecular Medicine Norway (NCMM). SJB is supported by the Norwegian Cancer Society. MLT and AS are supported by the RCN and the Norwegian Cancer Society. TP-C is supported by funding from the DFG SFB 773 (TP A03).
Disclosure of Potential Conflicts of Interest
The authors declare that they have no conflict of interest.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/25900
Related Research Data
References
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383 - 435; http://dx.doi.org/10.1152/physrev.00030.2009; PMID: 20959619
- Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem 2007; 282:4702 - 10; http://dx.doi.org/10.1074/jbc.M609267200; PMID: 17135238
- Gastaldello A, Callaghan H, Gami P, Campanella M. Ca 2+ -dependent autophagy is enhanced by the pharmacological agent PK11195. Autophagy 2010; 6:607 - 13; http://dx.doi.org/10.4161/auto.6.5.11964; PMID: 20431351
- Ghislat G, Patron M, Rizzuto R, Knecht E. Withdrawal of essential amino acids increases autophagy by a pathway involving Ca2+/calmodulin-dependent kinase kinase-β (CaMKK-β). J Biol Chem 2012; 287:38625 - 36; http://dx.doi.org/10.1074/jbc.M112.365767; PMID: 23027865
- Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, Viollet B, Wesselborg S, Proikas-Cezanne T, Stork B. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal 2010; 22:914 - 25; http://dx.doi.org/10.1016/j.cellsig.2010.01.015; PMID: 20114074
- Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 2007; 25:193 - 205; http://dx.doi.org/10.1016/j.molcel.2006.12.009; PMID: 17244528
- Kandala PK, Srivastava SK. Regulation of macroautophagy in ovarian cancer cells in vitro and in vivo by controlling glucose regulatory protein 78 and AMPK. Oncotarget 2012; 3:435 - 49; PMID: 22564965
- Knöferle J, Koch JC, Ostendorf T, Michel U, Planchamp V, Vutova P, Tönges L, Stadelmann C, Brück W, Bähr M, et al. Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc Natl Acad Sci U S A 2010; 107:6064 - 9; http://dx.doi.org/10.1073/pnas.0909794107; PMID: 20231460
- Law BY, Wang M, Ma DL, Al-Mousa F, Michelangeli F, Cheng SH, Ng MH, To KF, Mok AY, Ko RY, et al. Alisol B, a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca(2+) ATPase pump, induces autophagy, endoplasmic reticulum stress, and apoptosis. Mol Cancer Ther 2010; 9:718 - 30; http://dx.doi.org/10.1158/1535-7163.MCT-09-0700; PMID: 20197400
- Muller C, Salvayre R, Nègre-Salvayre A, Vindis C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ 2011; 18:817 - 28; http://dx.doi.org/10.1038/cdd.2010.149; PMID: 21113143
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26:9220 - 31; http://dx.doi.org/10.1128/MCB.01453-06; PMID: 17030611
- Sakaki K, Wu J, Kaufman RJ. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J Biol Chem 2008; 283:15370 - 80; http://dx.doi.org/10.1074/jbc.M710209200; PMID: 18356160
- Shin DM, Yuk JM, Lee HM, Lee SH, Son JW, Harding CV, Kim JM, Modlin RL, Jo EK. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol 2010; 12:1648 - 65; http://dx.doi.org/10.1111/j.1462-5822.2010.01497.x; PMID: 20560977
- Wang SH, Shih YL, Ko WC, Wei YH, Shih CM. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 2008; 65:3640 - 52; http://dx.doi.org/10.1007/s00018-008-8383-9; PMID: 18850067
- Zhang L, Cui L, Zhou G, Jing H, Guo Y, Sun W. Pterostilbene, a natural small-molecular compound, promotes cytoprotective macroautophagy in vascular endothelial cells. J Nutr Biochem 2013; 24:903 - 11; http://dx.doi.org/10.1016/j.jnutbio.2012.06.008; PMID: 22898568
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell 2011; 42:731 - 43; http://dx.doi.org/10.1016/j.molcel.2011.04.024; PMID: 21700220
- Gordon PB, Holen I, Fosse M, Røtnes JS, Seglen PO. Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J Biol Chem 1993; 268:26107 - 12; PMID: 8253727
- Lee H, Noh JY, Oh Y, Kim Y, Chang JW, Chung CW, Lee ST, Kim M, Ryu H, Jung YK. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum Mol Genet 2012; 21:101 - 14; http://dx.doi.org/10.1093/hmg/ddr445; PMID: 21954231
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4:295 - 305; http://dx.doi.org/10.1038/nchembio.79; PMID: 18391949
- Pressman BC. Biological applications of ionophores. Annu Rev Biochem 1976; 45:501 - 30; http://dx.doi.org/10.1146/annurev.bi.45.070176.002441; PMID: 786156
- Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A 1990; 87:2466 - 70; http://dx.doi.org/10.1073/pnas.87.7.2466; PMID: 2138778
- Qian T, Herman B, Lemasters JJ. The mitochondrial permeability transition mediates both necrotic and apoptotic death of hepatocytes exposed to Br-A23187. Toxicol Appl Pharmacol 1999; 154:117 - 25; http://dx.doi.org/10.1006/taap.1998.8580; PMID: 9925795
- Tóth A, Kedei N, Szabó T, Wang Y, Blumberg PM. Thapsigargin binds to and inhibits the cloned vanilloid receptor-1. Biochem Biophys Res Commun 2002; 293:777 - 82; http://dx.doi.org/10.1016/S0006-291X(02)00293-0; PMID: 12054538
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Bauvy C, Meijer AJ, Codogno P. Assaying of autophagic protein degradation. Methods Enzymol 2009; 452:47 - 61; http://dx.doi.org/10.1016/S0076-6879(08)03604-5; PMID: 19200875
- Fuertes G, Martín De Llano JJ, Villarroya A, Rivett AJ, Knecht E. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem J 2003; 375:75 - 86; http://dx.doi.org/10.1042/BJ20030282; PMID: 12841850
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy 2007; 3:181 - 206; PMID: 17224625
- Kopitz J, Kisen GO, Gordon PB, Bohley P, Seglen PO. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J Cell Biol 1990; 111:941 - 53; http://dx.doi.org/10.1083/jcb.111.3.941; PMID: 2391370
- Seglen PO, Øverbye A, Saetre F. Sequestration assays for mammalian autophagy. Methods Enzymol 2009; 452:63 - 83; http://dx.doi.org/10.1016/S0076-6879(08)03605-7; PMID: 19200876
- Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 2004; 23:9314 - 25; http://dx.doi.org/10.1038/sj.onc.1208331; PMID: 15602573
- Mauthe M, Jacob A, Freiberger S, Hentschel K, Stierhof YD, Codogno P, Proikas-Cezanne T. Resveratrol-mediated autophagy requires WIPI-1-regulated LC3 lipidation in the absence of induced phagophore formation. Autophagy 2011; 7:1448 - 61; http://dx.doi.org/10.4161/auto.7.12.17802; PMID: 22082875
- Pfisterer SG, Mauthe M, Codogno P, Proikas-Cezanne T. Ca2+/calmodulin-dependent kinase (CaMK) signaling via CaMKI and AMP-activated protein kinase contributes to the regulation of WIPI-1 at the onset of autophagy. Mol Pharmacol 2011; 80:1066 - 75; http://dx.doi.org/10.1124/mol.111.071761; PMID: 21896713
- Booth C, Koch GL. Perturbation of cellular calcium induces secretion of luminal ER proteins. Cell 1989; 59:729 - 37; http://dx.doi.org/10.1016/0092-8674(89)90019-6; PMID: 2510935
- Kiviluoto S, Vervliet T, Ivanova H, Decuypere JP, De Smedt H, Missiaen L, Bultynck G, Parys JB. Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim Biophys Acta 2013; 1833:1612 - 24; http://dx.doi.org/10.1016/j.bbamcr.2013.01.026; PMID: 23380704
- Kyriakakis E, Philippova M, Joshi MB, Pfaff D, Bochkov V, Afonyushkin T, Erne P, Resink TJ. T-cadherin attenuates the PERK branch of the unfolded protein response and protects vascular endothelial cells from endoplasmic reticulum stress-induced apoptosis. Cell Signal 2010; 22:1308 - 16; http://dx.doi.org/10.1016/j.cellsig.2010.04.008; PMID: 20457250
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 2003; 4:552 - 65; http://dx.doi.org/10.1038/nrm1150; PMID: 12838338
- Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci 2013; 70:2425 - 41; http://dx.doi.org/10.1007/s00018-012-1173-4; PMID: 23052213
- Gordy C, He YW. The crosstalk between autophagy and apoptosis: where does this lead?. Protein Cell 2012; 3:17 - 27; http://dx.doi.org/10.1007/s13238-011-1127-x; PMID: 22314807
- Heath-Engel HM, Chang NC, Shore GC. The endoplasmic reticulum in apoptosis and autophagy: role of the BCL-2 protein family. Oncogene 2008; 27:6419 - 33; http://dx.doi.org/10.1038/onc.2008.309; PMID: 18955970
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741 - 52; http://dx.doi.org/10.1038/nrm2239; PMID: 17717517
- Engedal N, Korkmaz CG, Saatcioglu F. C-Jun N-terminal kinase is required for phorbol ester- and thapsigargin-induced apoptosis in the androgen responsive prostate cancer cell line LNCaP. Oncogene 2002; 21:1017 - 27; http://dx.doi.org/10.1038/sj.onc.1205167; PMID: 11850819
- Gong Y, Blok LJ, Perry JE, Lindzey JK, Tindall DJ. Calcium regulation of androgen receptor expression in the human prostate cancer cell line LNCaP. Endocrinology 1995; 136:2172 - 8; http://dx.doi.org/10.1210/en.136.5.2172; PMID: 7720667
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012; 13:89 - 102; PMID: 22251901
- Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res 2013; 73:1993 - 2002; http://dx.doi.org/10.1158/0008-5472.CAN-12-3109; PMID: 23333938
- Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, et al. Discovery of 7-methyl-5-(1-[3-(trifluoromethyl)phenyl]acetyl-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 2012; 55:7193 - 207; http://dx.doi.org/10.1021/jm300713s; PMID: 22827572
- Volkmann K, Lucas JL, Vuga D, Wang X, Brumm D, Stiles C, Kriebel D, Der-Sarkissian A, Krishnan K, Schweitzer C, et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J Biol Chem 2011; 286:12743 - 55; http://dx.doi.org/10.1074/jbc.M110.199737; PMID: 21303903
- Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, Hu Y, Fabre C, Minami J, Ohguchi H, et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012; 119:5772 - 81; http://dx.doi.org/10.1182/blood-2011-07-366633; PMID: 22538852
- Wong AS, Cheung ZH, Ip NY. Molecular machinery of macroautophagy and its deregulation in diseases. Biochim Biophys Acta 2011; 1812:1490 - 7; http://dx.doi.org/10.1016/j.bbadis.2011.07.005; PMID: 21787863
- Zahm AM, Bohensky J, Adams CS, Shapiro IM, Srinivas V. Bone cell autophagy is regulated by environmental factors. Cells Tissues Organs 2011; 194:274 - 8; http://dx.doi.org/10.1159/000324647; PMID: 21597271
- Decuypere JP, Bultynck G, Parys JB. A dual role for Ca(2+) in autophagy regulation. Cell Calcium 2011; 50:242 - 50; http://dx.doi.org/10.1016/j.ceca.2011.04.001; PMID: 21571367
- Jin HO, Seo SK, Woo SH, Kim ES, Lee HC, Yoo DH, An S, Choe TB, Lee SJ, Hong SI, et al. Activating transcription factor 4 and CCAAT/enhancer-binding protein-beta negatively regulate the mammalian target of rapamycin via Redd1 expression in response to oxidative and endoplasmic reticulum stress. Free Radic Biol Med 2009; 46:1158 - 67; http://dx.doi.org/10.1016/j.freeradbiomed.2009.01.015; PMID: 19439225
- Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010; 6:239 - 47; http://dx.doi.org/10.4161/auto.6.2.11062; PMID: 20104019
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14:230 - 9; http://dx.doi.org/10.1038/sj.cdd.4401984; PMID: 16794605
- Pike LR, Singleton DC, Buffa F, Abramczyk O, Phadwal K, Li JL, Simon AK, Murray JT, Harris AL. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem J 2013; 449:389 - 400; http://dx.doi.org/10.1042/BJ20120972; PMID: 23078367
- Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 2010; 120:127 - 41; http://dx.doi.org/10.1172/JCI40027; PMID: 20038797
- Rzymski T, Milani M, Singleton DC, Harris AL. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 2009; 8:3838 - 47; http://dx.doi.org/10.4161/cc.8.23.10086; PMID: 19887912
- Margariti A, Li H, Chen T, Martin D, Vizcay-Barrena G, Alam S, Karamariti E, Xiao Q, Zampetaki A, Zhang Z, et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem 2013; 288:859 - 72; http://dx.doi.org/10.1074/jbc.M112.412783; PMID: 23184933
- Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, Caballero B, Kiffin R, Segura-Aguilar J, Cuervo AM, et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum Mol Genet 2012; 21:2245 - 62; http://dx.doi.org/10.1093/hmg/dds040; PMID: 22337954
- Pehar M, Jonas MC, Hare TM, Puglielli L. SLC33A1/AT-1 protein regulates the induction of autophagy downstream of IRE1/XBP1 pathway. J Biol Chem 2012; 287:29921 - 30; http://dx.doi.org/10.1074/jbc.M112.363911; PMID: 22787145
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685 - 701; http://dx.doi.org/10.1083/jcb.200803137; PMID: 18725538
- Dunn WA Jr.. Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol 1994; 4:139 - 43; http://dx.doi.org/10.1016/0962-8924(94)90069-8; PMID: 14731737
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013; 495:389 - 93; http://dx.doi.org/10.1038/nature11910; PMID: 23455425
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 2009; 11:1433 - 7; http://dx.doi.org/10.1038/ncb1991; PMID: 19898463
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. Electron tomography reveals the endoplasmic reticulum as a membrane source for autophagosome formation. Autophagy 2010; 6:301 - 3; http://dx.doi.org/10.4161/auto.6.2.11134; PMID: 20104025
- Seglen PO, Gordon PB, Holen I, Høyvik H, Kovács AL, Strømhaug PE, et al. Autophagic-endocytic interactions in hepatocytes. In: Courtoy PJ, ed. Endocytosis. Berlin: Springer-Verlag, 1992:247-54.
- Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A 2007; 104:19023 - 8; http://dx.doi.org/10.1073/pnas.0709695104; PMID: 18024584
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170:1101 - 11; http://dx.doi.org/10.1083/jcb.200504035; PMID: 16186256
- Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung KH, Yang J, Parker I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142:270 - 83; http://dx.doi.org/10.1016/j.cell.2010.06.007; PMID: 20655468
- Samari HR, Møller MT, Holden L, Asmyhr T, Seglen PO. Stimulation of hepatocytic AMP-activated protein kinase by okadaic acid and other autophagy-suppressive toxins. Biochem J 2005; 386:237 - 44; http://dx.doi.org/10.1042/BJ20040609; PMID: 15461583
- Decuypere JP, Welkenhuyzen K, Luyten T, Ponsaerts R, Dewaele M, Molgó J, Agostinis P, Missiaen L, De Smedt H, Parys JB, et al. Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy 2011; 7:1472 - 89; http://dx.doi.org/10.4161/auto.7.12.17909; PMID: 22082873
- Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, Molgó J, Díaz J, Lavandero S, Harper F, et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 2007; 14:1029 - 39; PMID: 17256008
- Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, Joza N, Vitale I, Morselli E, Tailler M, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 2009; 16:1006 - 17; http://dx.doi.org/10.1038/cdd.2009.34; PMID: 19325567
- Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell 2008; 19:4651 - 9; http://dx.doi.org/10.1091/mbc.E08-03-0312; PMID: 18768752
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010; 29:1792 - 802; http://dx.doi.org/10.1038/emboj.2010.74; PMID: 20418806
- Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007; 130:165 - 78; http://dx.doi.org/10.1016/j.cell.2007.05.021; PMID: 17632063
- Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell 2011; 20:444 - 54; http://dx.doi.org/10.1016/j.devcel.2011.02.006; PMID: 21497758
- Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, et al. SNARE proteins are required for macroautophagy. Cell 2011; 146:290 - 302; http://dx.doi.org/10.1016/j.cell.2011.06.022; PMID: 21784249
- Moreau K, Renna M, Rubinsztein DC. Connections between SNAREs and autophagy. Trends Biochem Sci 2013; 38:57 - 63; http://dx.doi.org/10.1016/j.tibs.2012.11.004; PMID: 23306003
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science 2009; 323:474 - 7; http://dx.doi.org/10.1126/science.1161748; PMID: 19164740
- Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC. Autophagosome precursor maturation requires homotypic fusion. Cell 2011; 146:303 - 17; http://dx.doi.org/10.1016/j.cell.2011.06.023; PMID: 21784250
- Jena BP. Role of SNAREs in membrane fusion. Adv Exp Med Biol 2011; 713:13 - 32; http://dx.doi.org/10.1007/978-94-007-0763-4_3; PMID: 21432012
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) Method. Methods 2001; 25:402 - 8; http://dx.doi.org/10.1006/meth.2001.1262; PMID: 11846609
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3:H0034; http://dx.doi.org/10.1186/gb-2002-3-7-research0034; PMID: 12184808
- Gordon PB, Seglen PO. Autophagic sequestration of [14C]sucrose, introduced into rat hepatocytes by reversible electro-permeabilization. Exp Cell Res 1982; 142:1 - 14; http://dx.doi.org/10.1016/0014-4827(82)90402-5; PMID: 7140848