Abstract
Prion protein (PRNP) has been implicated in various types of neurodegenerative diseases. Although much is known about prion diseases, the function of cellular PRNP remains cryptic. Here, we show that PRNP mediates amyloid β1–42 (Aβ42)-induced autophagy activation through its interaction with BECN1. Treatment with Aβ42 enhanced autophagy flux in neuronal cells. Aβ42-induced autophagy activation, however, was impaired in prnp-knockout primary cortical neurons and Prnp-knockdown or prnp-knockout neuronal cells. Immunoprecipitation assays revealed that PRNP interacted with BECN1 via the BCL2-binding domain of BECN1. This interaction promoted the subcellular localization of BECN1 into lipid rafts of the plasma membrane and enhanced activity of PtdIns3K (whose catalytic subunit is termed PIK3C3, mammalian ortholog of yeast VPS34) in lipid rafts by generating PtdIns3P in response to Aβ42. Further, the levels of lipid rafts that colocalized with BECN1, decreased in the brains of aged C57BL/6 mice, as did PRNP. These results suggested that PRNP interacts with BECN1 to recruit the PIK3C3 complex into lipid rafts and thus activates autophagy in response to Aβ42, defining a novel role of PRNP in the regulation of autophagy.
Introduction
Macroautophagy, or simply autophagy, is an evolutionarily conserved intracellular degradation system. Autophagy is initiated by the formation of a double-membrane vesicle, called an autophagosome, which finally fuses with lysosomes to degrade cytoplasmic components. Macromolecules, generated by lysosomal degradation, act as nutrients or useful materials in response to cellular metabolism or diverse stress conditions.Citation1 In various stressful situations, including nutrient starvation, oxidative stress, inflammation, and endoplasmic reticulum (ER) stress, autophagy is activated to overcome the harmful conditions.Citation2 The molecular signaling of these processes relies largely on the ULK1 complex (ULK1-ATG13-RB1CC1-C12orf44) for the induction of autophagy, class III phosphatidylinositol 3-kinase (PtdIns3K; whose catalytic subunit is known as PIK3C3) complex for vesicle nucleation, and 2 ubiquitin-like conjugation systems (ATG7, ATG10, ATG12-ATG5-ATG16L1 and ATG4, ATG7, ATG3, LC3/GABARAP) for vesicle elongation.Citation3
BECN1, which was initially identified as an essential autophagy regulatory protein in mammals, associates with the PIK3C3 core complex.Citation4 In mammalian systems, the PIK3C3 complex is composed of PIK3C3, BECN1, and PIK3R4 (the mammalian ortholog of VPS15 [phosphoinositide-3-kinase, regulatory subunit 4]) and generates phosphatidylinositol-3-phosphate (PtdIns3P), which is required for the initial steps of vesicle nucleation.Citation5 Recently, additional BECN1-interacting partners were identified that also regulate various autophagic events through the PIK3C3 core complex. ATG14 (also known as BARKOR), UVRAG (UV radiation resistance associated), AMBRA1 (autophagy/Beclin 1 regulator 1), and SH3GLB1/BIF-1 (SH3-domain GRB2-like endophilin B1) are autophagy-inducing partners of BECN1.Citation6 In contrast, other proteins, including KIAA0226/RUBICON, BCL2, and BCL2L1, negatively regulate autophagy.Citation7 These BECN1-interacting proteins act as fine-tuning regulators of complicated cellular responses. Thus, identifying new BECN1-interacting proteins and unveiling their physiological functions under various cellular responses is needed.
The cellular prion protein (PrPc; hereafter referred to as PRNP) is anchored to the cell surface by glycosylphosphatidylinositol (GPI). Although PRNP is expressed in various cell types and tissues, it is highly expressed within the nervous system.Citation8 Since the 1980s, PRNP-related studies have mainly focused on the mechanisms by which PRNP is converted into the abnormal protease-resistant isoform (PrPsc) that is responsible for transmissible spongiform encephalopathies and causes fatal neurodegeneration and protein aggregation. Concomitantly, the physiology of PRNP has also been studied, including its role in the cellular trafficking of copper ions, hippocampal morphology, cognition, oxidative stress and apoptosis.Citation9 Depending on cell context and/or physiological conditions, PRNP is beneficial or harmful to the cell. Many studies have indicated that PRNP can rescue neurons from various stressful situations.Citation10 Evidence from prnp-knockout (KO) studies has provided information regarding subtle phenotypes, for example, mild neuropathological, cognitive and behavioral deficitsCitation11 and enhanced brain injury after ischemia.Citation12 In addition, the N-terminal region of PRNP displays a neuroprotective function.Citation13 Conversely, PRNP also tends to function as a neurotoxic protein.Citation14,Citation15
Similar to other GPI-anchored proteins, PRNP is attached to low-density, detergent-resistant membrane (DRM) domains, which are rich in cholesterol and sphingolipids and are termed lipid rafts.Citation16 These specialized membrane microdomains compartmentalize cellular processes, for instance, the assembly of signaling molecules, and influence membrane fluidity, membrane protein trafficking and receptor trafficking.Citation17 Likewise, PRNP located in lipid rafts participates in cell signaling pathways, such as trafficking and stress responses.Citation9 Many reports have identified putative ligands of PRNP, such as ribosomal protein SA/laminin receptor precursor (RPSA/LRP), GRB2, and CAV1/caveolin 1.Citation18-Citation20 Recently, Aβ42, which is associated with neurodegeneration in Alzheimer disease (AD), was also reported to act as a ligand of PRNP.Citation21 However, the physiological function of PRNP as an Aβ42-binding protein is not clear.
In the present study, we showed that PRNP is critical in Aβ42-mediated autophagy in neurons. The interaction of PRNP with BECN1 facilitates the localization of BECN1 into lipid rafts and thus allows the activation of PIK3C3 complex in response to Aβ42, showing a beneficial role of PRNP as a positive regulator of BECN1-PIK3C3 complex in lipid rafts.
Results
Identification of PRNP as an autophagy regulator: PRNP deficiency impairs autophagy activation by Aβ42
To identify new regulators of autophagy, we employed a functional screening approach using a cell-based assay in which HeLa cells grown on a multiwell culture plate were transiently cotransfected with a GFP-LC3 fusion protein, an autophagosomal marker, and cDNAs in mammalian expression vectors. In this approach, we screened 5,678 cDNAs and the cDNAs that enhanced the formation of GFP-LC3 dots were then isolated. Among them, the cDNA encoding PRNP strongly increased the number of GFP-LC3 dots. To unveil the link between PRNP and LC3 dot formation, we examined whether the ablation of PRNP expression in neuronal cells affected autophagy under various conditions. The mouse hippocampal neuronal HpL3-4(E) cell line, which is derived from prnp-KO murine hippocampal cells, and HpL3-4(PrP) cells, in which HpL3-4 cells were reconstituted with PRNP,Citation22 were employed in an autophagy assay. Using mRFP-LC3, we found that treatment with autophagy inducers, such as Aβ42, serum starvation (starved), EBSS (amino acid starvation), hydrogen peroxide, tunicamycin, CuSO4, A23187, tumor necrosis factor-α (ΤΝF), propyl gallate and carbonyl cyanide m-chlorophenylhydrazone, greatly induced the formation of LC3 dots in HpL3-4(PrP) neuronal cells (Fig. S1A). Unlike the other stress signals, however, Aβ42 did not significantly increase LC3 dot formation in prnp-deficient HpL3-4(E) cells.
We then determined whether Aβ42 affected the formation of autophagosomes. When SH-SY5Y human neuroblastoma cells expressing GFP-LC3 were treated with Aβ42 (Fig. S1B, right), the number of GFP-LC3 dots was increased but this increase was blocked by 3-methyladenine (3-MA), an autophagy inhibitor (Fig. S1B, left). Western blot analysis also showed that Aβ42 increased LC3-II level, while it decreased SQSTM1 level (Fig. S1C), an indicator of ubiquitinated protein. The effect of Aβ42 on autophagy flux was examined using mCherry-GFP-LC3. In the mCherry-GFP-LC3 assay, mCherry+/GFP+ double-positive puncta imply the localization of LC3 on phagophores or autophagosomes, as the autophagosome fused with a lysosome forms autolysosomes, and acid-sensitive GFP signals subsequently disappear.Citation23 Treatment with Aβ42 increased the number of mCherry-LC3 puncta only in HpL3-4(PrP) cells, but there was no drastic accumulation of GFP-LC3 by Aβ42, resulting from the increase of autophagy flux (). Compared with that observed during starvation which increased mCherry-LC3 puncta in both HpL3-4(PrP) and HpL3-4(E) cells, these results clearly show that Aβ42 increases autophagy flux in a PRNP-dependent manner.
Figure 1. Autophagy induction by Aβ42 is impaired by PRNP deficiency. (A) HpL3-4(PrP) and HpL3-4(E) cells were transfected with mCherry-GFP-LC3 for 24 h and then incubated in serum-free medium (Starved) or treated with 0.5 μM Aβ42 for 12 h. The cells were stained with Hoechst dye and observed under a confocal microscope. Scale bars: 10 μm. (B) The number of cells with mCherry-LC3 and/or GFP-LC3 puncta in confocal images, including (A), was quantified (mean values ± S.D., n = 12; *P < 0.01, **P < 0.005 vs. control cells; ns, not-significant). (C) HpL3-4(PrP) and HpL3-4(E) cells were incubated with 0.5 μM Aβ42 for 12 h and then subjected to electron microscopic analysis. Asterisks indicate autophagic vacuoles (AVs). AV and mitochondria (M) are also shown in enlarged view. (D) The numbers of cytoplasmic AVs were calculated (mean values ± S.D., n = 20; **P < 0.005 vs. control cells). (E) HpL3-4(PrP) and HpL3-4(E) cells were incubated in serum-free medium (Starved) or treated with 0.5 μM Aβ42 for 12 h in the absence or presence of 20 nM bafilomycin A1 (BafA1) for 3 h before harvesting, and the cell lysates were analyzed by western blotting using the indicated antibodies. (F) Three-month-old WT and prnp-KO mice were injected i.c.v. with PBS or 2 μg Aβ42. After 12 and 48 h, hippocampal lysates were prepared and subjected to western blot. The signals on the blot were quantified using densitometric analysis and the relative ratios of LC3-II to ACTB are shown at the bottom of the blot.
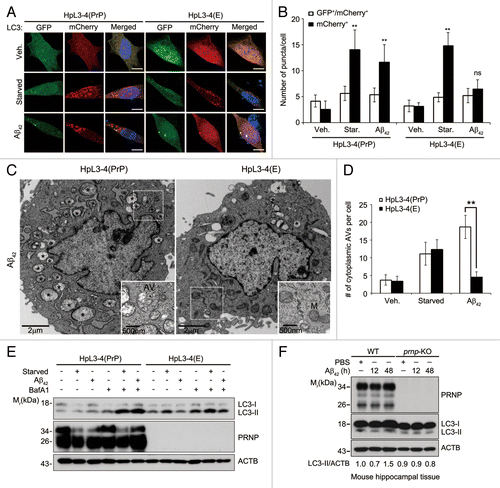
We further analyzed the intracellular structures of HpL3-4(PrP) and HpL3-4(E) cells using transmission electron microscope. Aβ42 treatment drastically increased the numbers of typical autophagic vacuoles (AVs) with double membranes and cellular contents in HpL3-4(PrP) cells but not in HpL3-4(E) cells, whereas starvation increased the numbers of AVs in both cells (; Fig. S2A). Consistently, western blot analysis showed that Aβ42 further increased LC3-II levels in the presence of bafilomycin A1, an inhibitor of lysosome proteolysis,Citation24 in HpL3-4(PrP) cells (). This Aβ42-induced PRNP-dependent autophagy activation was also observed in human SH-SY5Y neuroblastoma cells (Fig. S2B–S2D). Synthetic Aβ42 at low concentrations, i.e., less than 5 μM, was not usually toxic to neuronal cells in vitro but apparently increased autophagy flux (Fig. S3A–S3C). Furthermore, evaluation of the in vivo effect of exogenously added Aβ42 on autophagy revealed that the intracerebroventricular (i.c.v.) administration of Aβ42 increased LC3-II levels at 48 h by 1.5-fold in the hippocampal tissue of WT mice (). On the contrary, there was no significant change of LC3-II levels in prnp-KO mice. Together, these results suggest that PRNP is required for Aβ42-induced autophagy in neuronal cells.
PRNP colocalizes and interacts with the BECN1-PIK3C3 complex
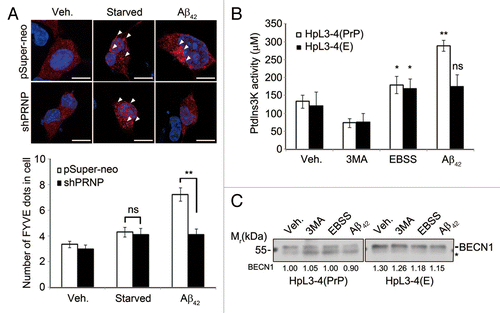
To gain insight into the molecular mechanism by which PRNP mediates Aβ42-induced autophagy, we employed an FYVE-RFP probe, which binds to PtdIns3P and thus monitors the activation of BECN1-PIK3C3 complex.Citation25 When SH-SY5Y cells expressing FYVE-RFP were incubated with Aβ42, the punctate structure of FYVE-RFP was increased and readily detected in SH-SY5Y cells (). By contrast, these puncta were not observed in SH-SY5Y/PRNP-knockdown cells. Under this condition, starvation slightly increased the formation of FYVE-RFP puncta in both SH-SY5Y and SH-SY5Y/PRNP-knockdown cells. In addition, an ELISA of the BECN1-PIK3C3 complex showed that PtdIns3P generation was increased by 2-fold in HpL3-4(PrP) cells in response to Aβ42, but not in prnp-null HpL3-4(E) cells (). In this assay, the PtdIns3P activity was normalized by the immunoprecipitated BECN1 (). These results imply that Aβ42 activates the BECN1-PIK3C3 complex to generate PtdIns3P in a PRNP-dependent manner.
Figure 2. Aβ42 enhances PIK3C3 complex activity via PRNP. (A) SH-SY5Y cells were transfected with FYVE-RFP and either pSuper-neo or PRNP shRNA for 48 h and then left untreated (Veh.) or incubated with either serum-free medium for 12 h (Starved) or 0.5 μM Aβ42 for 12 h. The cells were observed under a confocal microscope (upper) and the number of FYVE-RFP puncta per cell was quantified. The bars represent mean values ± S.D. (n = 30 cells) (lower). The arrowheads indicate FYVE-RFP puncta. Scale bars: 10 μm. (B and C) HpL3-4(PrP) and HpL3-4(E) cells were left untreated (Veh.) or incubated with 5 mM 3MA for 3 h, EBSS for 3 h, or 0.5 μM Aβ42 for 12 h. Cell lysates were subjected to an immunoprecipitation assay using anti-BECN1 antibody. The immunoprecipitates bound to protein G-beads were used for the PtdIns3K assay (B) and the immunoprecipitated BECN1 was detected with western blotting using anti-BECN1 antibody (C). The signal of BECN1 on the blot was quantified by densitometric analysis and used to normalize the PtdIns3K activity. The asterisk indicates nonspecific signal and bars represent mean values ± S.E. (n = 3).
We then assessed the functional interaction between PRNP and the BECN1-PIK3C3 complex. Because the availability of antibodies for immunocytochemical analysis was limited, we utilized GFP- or RFP-fused protein for subcellular localization analysis. Interestingly, when we coexpressed mRFP-BECN1 and GFP-PRNP, a small but significant amount of mRFP-BECN1 was also detected in the plasma membrane (Fig. S4A). In addition, in discontinuous iodixanol density gradient centrifugation analysis to separate cytosol from ER and membrane fraction, we found that BECN1 and PRNP were located in the same membrane fractions (Fig. S4B). These observations, that PRNP was required for the activation of the PIK3C3 complex and colocalized with BECN1, led us to examine the physical association of PRNP with BECN1. Using immunoprecipitation analysis, we found that GFP- or HA-tagged PRNP interacted with BECN1 in the transfected HEK293T cells (). However, PRNP-HA did not interact with ATG5-GFP or GFP-LC3 (Fig. S5A and S5B). Because PRNP is highly expressed in brain tissue but expressed at a low level in neuronal cell lines, we performed an in vivo immunoprecipitation assay using cortical extracts from mouse brain. In western blot analysis, we detected endogenous PRNP in the BECN1-containing immunoprecipitates (). In addition, we found that PRNP, PIK3C3, and BCL2, but not ATG5, formed protein complexes with BECN1 in an overlay assay using purified His-tagged BECN1 protein (). Furthermore, fractionation assay revealed that the interaction of BECN1 with PRNP occurred in the membrane fraction (Fig. S5C).
Figure 3. PRNP interacts with BECN1. (A and B) HEK293T cells were cotransfected with PRNP-GFP and BECN1-HA (A) or PRNP-HA and BECN1 (B) for 24 h, after which the cell lysates were subjected to an immunoprecipitation (IP) assay using an anti-HA antibody (bottom). The immunoprecipitates were then analyzed with western blotting using anti-GFP (A) and anti-BECN1 (B) antibodies (top). Whole cell lysates (WCLs) were examined with western blotting as a reference. (C) Tissue lysates prepared from the cortex of 3-mo-old C57BL/6 mice were analyzed with an immunoprecipitation (IP) assay using preimmune serum of rat (control) or anti-BECN1 antibody (#1: From Novus; #2: This study). The immunoprecipitates were examined with immunoblotting using anti-PRNP and anti-BECN1 antibodies. The arrows and asterisks indicate PRNP and non-specific signals, respectively. (D) BECN1 (10 μg) purified from E. coli was separated by SDS-PAGE and transferred to a PVDF membrane. After an overnight incubation with 3% BSA or mouse whole brain lysates, the membrane was subjected to western blot analysis using the indicated antibodies. Bovine serum albumin (BSA; 10 μg) was used as a control. (E) SH-SY5Y cell lysates were prepared and analyzed with an immunoprecipitation (IP) assay using anti-rabbit serum (control) and anti-CAV1 antibody. (F) SH-SY5Y cells were cotransfected with CAV1-HA, PRNP and BECN1for 24 h and the cell lysates were then subjected to a co-IP assay using anti-HA antibodies. The immunoprecipitates were analyzed with western blotting using the indicated antibodies. (G) SH-SY5Y cells were cotransfected with PRNP, BECN1-HA and CAV1 shRNA (shCAV1), as indicated, for 48 h and the cell lysates were then subjected to an immunoprecipitation assay (IP) using anti-HA antibody. The immunoprecipitates were analyzed with western blotting using the indicated antibodies. WCL, whole cell lysate.
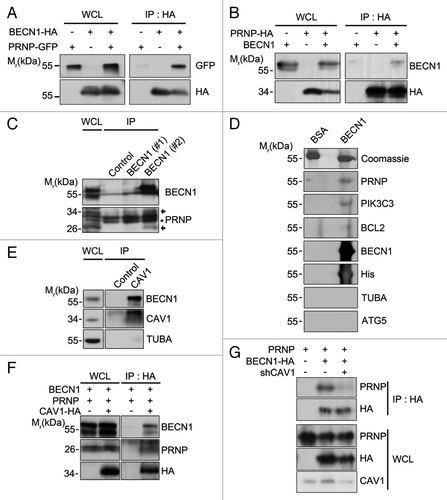
As it was predicted that GPI-anchored PRNP could not directly bind to cytosolic BECN1, we found that the purified PRNP protein did not bind to the purified BECN1 protein in vitro. It seems that PRNP interacts with an as-yet unidentified protein to form a protein complex with BECN1-PIK3C3. Until now, several proteins such as RPSA/LRP, GRB2, and CAV1 have been reported to bind to PRNP. Among them, a physical interaction between PRNP and CAV1 may explain the localization of PRNP in membrane lipid rafts.Citation18-Citation20 In the immunoprecipitation assays, we found that CAV1 and, to a lesser extent, RPSA/LRP, but not GRB2, bound to BECN1 in the transfected cells (Fig. S6A and S6B). We also found that endogenous BECN1 bound to CAV1 () and CAV1 interacted with both PRNP and BECN1 (). Further, from an immunoprecipitation assay, we found that reduced expression of CAV1 apparently decreased the interaction between BECN1 and PRNP (). Consistently, ectopic expression of CAV1 enhanced LC3 dot formation in Aβ42-treated SH-SY5Y cells (Fig. S6C and S6D). Together, these results demonstrated that PRNP may form a protein complex with BECN1 through CAV1 in the membrane.
PRNP recruits BECN1 into lipid rafts to generate PtdIns3P in response to Aβ42
Since GPI-anchored PRNP mainly localizes in lipid rafts,Citation8 we examined whether PRNP recruited BECN1 into lipid rafts. Using a sucrose gradient fractionation assay of HpL3-4(PrP) and HpL3-4(E) cell extracts, we separated the DRM fractions (usually containing lipid rafts) from the non-DRM fractions. Western blotting of the fractions prepared from HpL3-4(PrP) cells revealed that both PRNP and small but significant amount of BECN1 were present in the DRM fractions (fractions #4 to 6), which also contained FLOT1/Flotillin 1, a lipid raft marker (, top and left). Conversely, little BECN1 was detected in the DRM fractions from prnp-deficient HpL3-4(E) cells (, top and right). Interestingly, treatment of HpL3-4(PrP) cells with Aβ42 markedly increased the amount of BECN1 detected in the DRM fractions, while this change was not observed in HpL3-4(E) cells (, bottom; ). The amounts of FLOT1 detected in the DRM fractions were not significantly different between HpL3-4(PrP) and HpL3-4(E) cells regardless of Aβ42 treatment. Similarly, BECN1 and Aβ42 were detected in the DRM fractions (fractions #4 to 6) prepared from WT primary cortical neurons, but much less was observed in prnp-KO primary cortical neurons. Also, treatment with Aβ42 increased the levels of BECN1 in the lipid rafts of WT primary cortical neurons (Fig. S7A and S7B).
Figure 4. Enhanced localization of BECN1 in lipid rafts by Aβ42 in the presence of PRNP. (A) HpL3-4(PrP) and HpL3-4(E) cells were left untreated (Veh.) or incubated with 0.5 μM Aβ42 for 12 h. Cell homogenates were subjected to sucrose gradient ultracentrifugation. Equal volumes of the fractions were subjected to western blot analysis using the indicated antibodies. (B) The intensity of BECN1 on the western blot images in (A) was quantified (mean values ± S.E., n = 3). (C) HpL3-4(PrP) and HpL3-4(E) cells were transfected with BECN1-GFP (green) for 24 h. After incubation with Alexa Fluor-594-conjugated cholera toxin subunit B (red) for 15 min at 4 °C, the cells were further incubated for 30 min at 37 °C for uptake. Cell nuclei were stained with Hoechst dye (blue) and observed under a confocal microscope. (D and E) HpL3-4(PrP) and HpL3-4(E) cells were left untreated or incubated with 0.5 μM Aβ42 for 12 h, and cell extracts were subjected to sucrose gradient ultracentrifugation as in (A). DRM fractions (fractions #4 to 6) and non-DRM fractions (fractions #11 to 12) were then pooled and subjected to an immunoprecipitation assay using an anti-BECN1 antibody. The immunoprecipitates were assayed for PtdIns3K activity and bars represent mean values ± S.E. (n = 3) (D) or proved with western blotting using anti-BECN1 antibody (E).
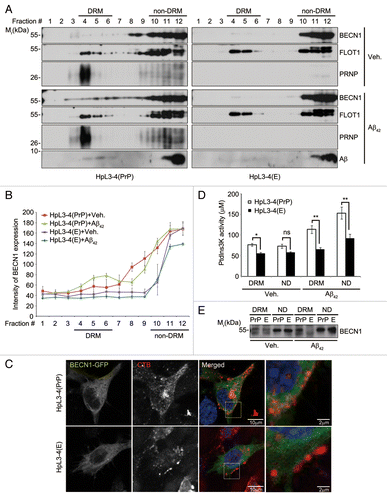
Likewise, we found that TRITC-conjugated cholera toxin B (CTxB), which labels cellular lipid rafts,Citation26 colocalized much more with BECN1 in HpL3-4(PrP) cells than in prnp-deficient HpL3-4(E) cells (). An ELISA showed that PtdIns3P levels were significantly higher in the DRM fractions prepared from HpL3-4(PrP) cells than HpL3-4(E) cells (). Strikingly, the levels of PtdIns3P were further increased by Aβ42 in the DRM fractions, as well in the non-DRM fractions prepared from HpL3-4(PrP) cells, but were not significantly increased in the fractions prepared from prnp-deficient HpL3-4(E) cells. Similar to the levels of PtdIns3P, the amount of immunoprecipitated BECN1 was higher in the DRM fractions from Aβ42-treated HpL3-4(PrP) cells than untreated cells or Aβ42-treated HpL3-4(E) cells (). These results suggest that BECN1 localizes in lipid rafts of neuronal cells in the presence of PRNP and this localization is increased by Aβ42.
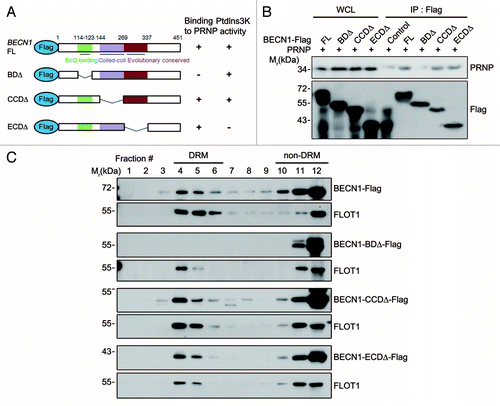
We then determined which domain of BECN1 was responsible for its interaction with PRNP. We constructed BECN1 deletion (Δ) mutants based on the proposed domains, including the BCL2-binding domain (BD), coiled-coil domain (CCD), and evolutionarily conserved domain (ECD) (). Immunoprecipitation assays revealed that full-length BECN1 and the CCDΔ and ECDΔ mutants could bind to PRNP in the transfected cells, while the BDΔ mutant was unable to (), demonstrating that the BD motif of BECN1 is required for its interaction with PRNP. With the help of the domain map, we addressed whether the protein-protein interaction between PRNP and BECN1 was related to the lipid raft localization of BECN1. Sucrose gradient fractionation analysis revealed the presence of full-length BECN1 in the DRM fractions of SH-SY5Y cells. Accordingly, the CCDΔ and ECDΔ mutants, which could bind to PRNP, were also detected in the DRM fractions. In contrast, the BDΔ mutant, which failed to interact with PRNP, was not found in the DRM fractions, but was detected in the non-DRM fractions (). Furthermore, CTxB assays revealed that the BDΔ mutant failed to localize on the lipid rafts (Fig. S7C). We conclude that the interaction between BECN1 and PRNP is critical for the localization of BECN1 in lipid rafts.
Figure 5. BCL2-binding domain of BECN1 is required for its interaction with PRNP and lipid rafts localization. (A) Schematic diagram of BECN1 domains and its deletion mutants. The binding ability to PRNP and the PtdIns3K activity of BECN1 and its deletions are summarized. (B) HEK293T cells were cotransfected with PRNP and either BECN1-flag (FL), BECN1−BDΔ-flag (BDΔ), BECN1−CCDΔ-flag (CCDΔ), or BECN1−ECDΔ-flag (ECDΔ) for 24 h. Cell lysates were analyzed with immunoprecipitation (IP) using anti-flag beads and immunoblotting using anti-PRNP and anti-flag antibodies. (C) SH-SY5Y cells were transfected with BECN1 or its mutant for 24 h and cell homogenates were subjected to sucrose gradient ultracentrifugation and western blotting as described in . WCL, whole cell lysate.
BECN1 in lipid rafts decreases in aged brain and is necessary to activate autophagy in response to Aβ42
During aging, autophagy activity is known to be impaired via different ways,Citation27 while the pathogenesis of age-dependent neuronal diseases, such as AD, increases.Citation28 We thus investigated whether the lipid rafts-localized BECN1 is changed during aging. A comparison of LC3 and SQSTM1 levels in tissue lysates using western blot analysis showed that the levels of LC3-II were slightly lower in the hippocampus of 24-mo-old mice than that in 3-mo-old mice, whereas SQSTM1 was relatively higher in the old mouse hippocampus (). On the other hand, the levels of BECN1 and PRNP were not significantly different between these two groups. Interestingly, sucrose fractionation analysis revealed that the levels of BECN1 in the DRM fractions (fractions #4 to 6) were reduced in the hippocampal tissues of old mice compared with young mice. PRNP seemed also to be reduced in the DRM fractions and scattered in the non-DRM fractions of the aged hippocampal tissues. Quantification of BECN1 and PRNP in the pooled DRM and non-DRM fractions using western blot analysis showed that the levels of BECN1 and PRNP in the DRM fractions were decreased by 2.0 and 1.5 fold, respectively, in the hippocampus of the aged mice (). Moreover, the PtdIns3P generation was also reduced in the aged hippocampus (Fig. S8). Observations that lipid raft-localized BECN1 decreases in the hippocampal neurons during aging may, thus, underlie age-dependent decline of autophagy activity.
Figure 6. Lipid raft-localized BECN1 and PRNP decrease in aged hippocampus, and disruption of lipid rafts by MβCD impairs Aβ42-induced autophagy. (A) Hippocampal neurons were purified by discontinuous iodixanol gradient assay from 3- and 24-mo-old WT C57BL/6 mice and their lysates and whole brain lysates (WBL) were subjected to western blot analysis using the indicated antibodies. (B) The signals on the blots were quantified by densitometric analysis using the ScienceLab software. The bars represent mean values ± S.E. (n = 3). (C) DRM (fractions #4 to 5) and non-DRM (fractions #11 to 12) fractions of each mouse were pooled and then subjected to western blot analysis. (D) The signals on the blot shown in (C) were quantified by densitometric analysis. The bars represent the relative ratios of BECN1 in the DRM to the non-DRM fractions with mean values ± S.D. (n = 3). (E) SH-SY5Y cells were transiently transfected with mCherry-GFP-LC3 for 24 h and left untreated (Veh.) or treated with 1 mg/mL MβCD for 12 h. The cells were then incubated with serum-free DMEM (Starved) or 1 μM Aβ42 for 12 h, and then observed under a fluorescence microscope. Scale bars: 10 μm. (F) The number of mCherry-LC3- and GFP-LC3-positive puncta per cell was counted and represented as mean values ± S.D. (n = 12). (G) SH-SY5Y cells were pretreated with PBS (Veh.) or 1 mg/mL MβCD for 12 h and then incubated with serum-free DMEM (Starved) or 1 μM Aβ42 for 3 h. Cell extracts were analyzed with western blotting using anti-SQSTM1, anti-LC3, and anti-ACTB antibodies. (H) The signals on the blots were quantified by densitometric analysis. The bars represent the relative ratios of the LC3-II signal to ACTB with mean values ± S.D. (n = 3).
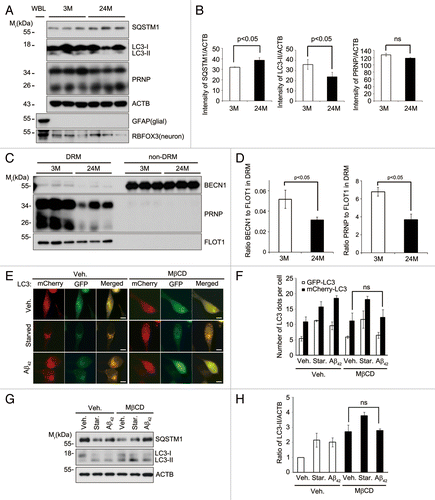
We then assessed the effect of the reduction of BECN1 in lipid rafts of cultured cells on autophagy activity. We first examined the effect of cholesterol depletion on the distribution of lipid raft-resident proteins, such as BECN1 and FLOT1. In sucrose gradient fraction assays, we found that treatment of SH-SY5Y cells with methyl-β-cyclodextrin (MβCD), a compound to remove cholesterol from cultured cells,Citation29 caused a shift of BECN1 from the DRM fractions (fractions #4 to 6) to the non-DRM fractions (fractions #11 to 12), as FLOT1 had also shifted (Fig. S9). Under the same condition using MβCD, treatment with Aβ42 no longer induced autophagy in SH-SY5Y cells, as analyzed using mCherry-GFP-LC3, while starvation still enhanced autophagy flux (). Likewise, western blot assays revealed that treatment with MβCD inhibited the conversion of LC3 triggered by Aβ42, but not that by starvation, and concomitantly rescued SQSTM1 to the control level in SH-SY5Y cells exposed to Aβ42 (). These results demonstrate that lipid raft-localized BECN1 is important to activate autophagy in response to Aβ42.
In summary, BECN1 is found in lipid rafts and lipid raft-localization of BECN1 is regulated by Aβ42 in a PRNP-dependent manner. Lipid raft-localized BECN1, which declines with aging, plays a crucial role in Aβ42-induced autophagy.
Discussion
For the precise regulation of autophagy under various conditions, BECN1 plays a crucial role by forming distinct protein complexes. In addition to the core complex (BECN1, PIK3C3, and PIK3R4/VPS15), approximately 20 proteins regulate the function of the PIK3C3 complex.Citation30 Among the BECN1-interacting proteins, PRNP is the only membrane-anchored protein located in lipid rafts. Because the localized production of PtdIns3P and a PtdIns3P-enriched membrane compartment are essential for autophagy induction and the nucleation of autophagosomes,Citation31 we believe that lipid raft-localized PIK3C3 complex activates autophagy. In this study, we provide evidence that lipid rafts are where the BECN1-PIK3C3 complex is anchored to PRNP and thus activates autophagy in the plasma membrane in response to external stresses, such as Aβ42. When the Aβ42 burden is below the cytotoxic level, neuronal cells may respond to the stress of Aβ42 by activating autophagy as an adaptation process to this burden, as also reported previously.Citation32,Citation33 In the same context, we also observed that the inhibition of autophagy by 3-MA enhanced the stress of Aβ42 in neuronal cells, leading to appearance of apoptotic cells. The discovery that PRNP activates autophagy via BECN1 in response to Aβ42 may, thus, explain one aspect of the proposed protective function of PRNP in neuronal cells.Citation9,Citation11
Then, important questions remain as to why and how PRNP responds to Aβ42 to induce autophagy. Bearing in mind that Aβ42 may act as a ligand for GPI-anchored PRNP,Citation21 one plausible model is that Aβ42 binds to PRNP, causing a conformational change of PRNP and thereby allowing more PRNP to bind to BECN1 and recruiting BECN1 into lipid rafts, as seen in our analysis in which Aβ42 increases the amount of DRM-resident BECN1 in a PRNP-dependent manner. Alternatively, another unidentified receptor of Aβ42 may transduce the autophagy signal through PRNP. In either case, PRNP plays an essential role in mediating the Aβ42-induced autophagy. In addition to the findings for Aβ42, the monomeric PRNP or PrPsc itself may affect autophagy with distinct mechanisms, while the induction of autophagy by them remains to be further analyzed.Citation34,Citation35 Then, it will be interesting to examine whether a sublethal dose of PrPsc is also able to bind to BECN1 for neuronal adaption.
The PRNP-dependent activation of autophagy in response to Aβ42, proposed here, is apparently distinct from autophagy triggered by starvation in prnp-deficient neuronsCitation36 or doppel overexpression in Ngsk Prnp-deficient (NP0/0) mice.Citation37 Prnd, which is part of the prion protein subfamily and is neurotoxic, can activate autophagy during cell death. Interestingly, identified function of PRNP and PRND in autophagy is consistent with their neuroprotective and neurotoxic roles. It will be interesting to know how autophagy activity and cellular toxicity are regulated by PRNP overexpression in PRND-expressing NP0/0 purkinje cells. In addition, our observation that PRNP is also involved in autophagy activation triggered by hydrogen peroxide is consistent with a recent report.Citation38 Unlike Aβ42, however, it is not clear yet how hydrogen peroxide regulates PRNP-mediated autophagy.
In addition to the transcriptional regulation of Atg genes and the decrease of chaperone-mediated autophagy,Citation27 a reduction in the amount of DRM-resident BECN1 may explain a decline in autophagy activity during aging. Because autophagy is generally considered as a survival signal, the age-related decline of a selective form of autophagy often leads to fatal defects in cellular defense mechanisms.Citation27,Citation39,Citation40 Therefore, the age-dependent decline of autophagy may result in neurotoxicity due to a cellular defect in the adaptation to Aβ42 burden. In particular, as Aβ42 burden is frequently observed in the brains of AD patients and gradually increases for the pathogenesis during aging, neuronal activation of autophagy via PRNP-BECN1 in early stages may be beneficial to neurons and provide a new therapeutic window to prevent the pathogenesis of neurotoxic Aβ42 in AD. Thus, we consider lipid raft-localized BECN1 as a “stepping stone” between autophagy and age-dependent neuronal disorders.
In conclusion, our study suggested that lipid rafts may contribute to autophagosome formation through the interaction of PRNP with BECN1 and lipid rafts-localized BECN1 serves as a scout that detects extracellular stress, such as Aβ42, to overcome Aβ42 burden by activating autophagy machinery.
Materials and Methods
Reagents and plasmid constructions
The following chemicals were used, including Aβ42 (Sigma-Aldrich, A9810), A23187 (Sigma-Aldrich, C7522), MβCD (Sigma-Aldrich, 332615), PG (Sigma-Aldrich, P3130), Tunicamycin (Sigma-Aldrich, T7765), bafilomycin A1 (Calbiochem, 196000), CCCP (Calbiochem, 555-60-2), hydrogen peroxide (H2O2; DUKSAN, 0817-2260) and TNF (CHEMICON, GF023). Human BECN1 and CAV1 cDNAs were subcloned into pcDNA3-HA, pEGFP, monomeric RFP (Invitrogen), and p3XFLAG-CMV-10 vector (Sigma-Aldrich). BECN1 deletion mutants were generated by subcloning the PCR products [BDΔ (BH3 domain (114 to 123) deletion)/CCDΔ [coiled coil domain (144 to 269) deletion)/ECDΔ [Evolutionary conserved domain (244 to 337) deletion)]. The ss-GFP-PRNP, pEGFP-LC3, ATG5-pEGFP, and mCherry-GFP-LC3 have been described previously.Citation41,Citation42 For PRNP shRNA, target sequences were cloned into pSuper-Neo vector (pshRNA, OligoEngine). The target sequences for PRNP were (shPRNP#1: 5′-GAACAAGCCG AGTAAGCCA-3′, 5′-TGGCTTACTC GGCTTGTTC-3′, shPRNP#2: 5′-GTGAAAACAT GCACCGTTA-3′, 5′-TAACGGTGCA TGTTTTCAC-3′) and CAV1 was (shCAV1: 5′-GCATTTGGAA GGCCAGCTT-3′, 5′-AAGCTGGCCT TCCAAATG C-3′) .
Antibodies, immunoprecipitation assay, and western blot analysis
The primary antibodies used include anti-LC3 (Novus Biologicals, NB600-1384), anti-PRNP (Abcam, ab52604), anti-ACTB/β-actin (Sigma-Aldrich, A1978), anti-TUBA/α-tubulin (Sigma-Aldrich, T6074), anti-BECN1 #1 (Novus Biologicals, NB110-87318), anti-GFP (Santa Cruz Biotechnology, sc-8334), anti-HA, anti-PIK3C3/VPS34 (Cell Signaling, #3358), anti-BCL2, anti-His, anti-ATG5 (Novus Biologicals, NB110-53818), anti-FLOT1 (BD Biosciences, 610821), anti-Flag (Sigma-Aldrich), anti-SQSTM1 (Abnova, H00008878-M01), anti-NFKBIA (Santa Cruz Biotechnology, sc-203), anti-HSPA5 (Santa Cruz Biotechnology) and anti-ATG9A (Novus Biologicals, NB110-56893) antibodies. Anti-BECN1 antibody #2 was generated by following standard immunization procedures using the purified BECN1 protein from E. coli. Western blot analysis was performed using standard techniques. Cells were lysed with sample buffer (10% glycerol, 2% SDS, 62.5 mM Tris-HCl, 2% β-mercaptoethanol, pH 6.8). Immunoprecipitation assays were performed as previously described;Citation42 briefly, cells were lysed with modified RIPA buffer (10 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 1 mM Na3VO4, 1% CHAPS). After pulldown with the appropriate antibodies, the same amounts of protein were separated by SDS-PAGE and transferred onto the polyvinylidene fluoride membrane (ATTO, AE-6667-P). Immunoblot analysis was then performed and visualized by the enhanced chemiluminescence method.
Cell culture and generation of stable cell lines
SH-SY5Y cells (human neuroblastoma cells) and HEK293T cells (human embryonic kidney cells) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone, SH30243.01) with 10% (v/v) fetal bovine serum (Hyclone, SH30919.03). HpL3-4(E) and HpL3-4(PrP) cells were cultured as described previously.Citation22 To generate SH-SY5Y cell lines stably expressing pSuper or shPRNP, cells were transfected with pSuper-Neo and pSuper-shPRNP vector (Polyfect; QIAGEN, 1015586) for 24 h and incubated with 1,100 μg/ml G418 (Invitrogen) for 2 more weeks to generate stable cells. Single clones of the stable cells were then selected by western blotting following standard protocol. Hep3B/GFP-LC3 stable cells were generated as described above.
Maintenance of prnp KO mice
All of the experiments were performed on C57BL/6J mice, prnp KO miceCitation43 were maintained under a 12:12 h light:dark cycle and were approved by the Seoul National University Standing Committees on Animals.
Primary culture of cortical neurons
Cortical tissues of mouse E14.5 embryonic brain were dissociated by incubating with 0.01% trypsin-ethylenediaminetetraacetic acid (EDTA) (Gibco, 25300-054) and were plated on culture dishes coated with poly-l-lysine (0.01% in 100 mM borate buffer, pH 8.5) (Sigma-Aldrich, P4707) in NeurobasalTM medium containing 2% B27 supplement (Invitrogen, 17504-044). Neurons grown in vitro for 7 d were transfected with appropriate DNA using LipofectAMINETM 2000 Reagent (Invitrogen, 11668-027).
Sucrose density fractionation
Lipid rafts were isolated from neuronal cells (Primary cortical neurons, SH-SY5Y, and HpL3-4 cells) or mouse hippocampal tissue as described previously.Citation44 Samples were washed in PBS and lysed in 1 ml of TNEV lysis buffer containing 1% CHAPS (GenDEPOT, C0660) in 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM Na3VO4 and 5 mM EDTA. The TNEV lysis buffer was added with a 1 mM PMSF (USB, 20203). Cells were homogenized by 20 passages through a 22-gauge needle and then 26-gauge needle. The homogenized samples were spun down using centrifugation, adjusted to 45% final concentration of sucrose (final volume, 1 ml), and transferred to a 10 ml ultracentrifuge tube. A discontinuous sucrose gradient was then formed by sequentially layering 35% sucrose (4.5 ml) and 5% sucrose (2.5 ml), and the tubes were subject to ultracentrifugation at 200,000 g for 20 h in a Beckman SW32.1Ti rotor (Beckman Coulter) at 4 °C. Twelve fractions were collected from the top of the gradient and equal volume of each fraction was analyzed by western blotting. In some case, samples were concentrated by methanol/chloroform precipitation or subjected to immunoprecipitation assays before analysis.
Analysis of PtdIns3K activity
HpL3-4 cells were washed with ice-cold PBS and extracted for 30 min at 4 °C with extract buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM PMSF, 1× inhibitor cocktail, and 1% Triton X-100). Cells were subjected to centrifugation at 10,000 g and the supernatants were then immunoprecipitated for 6 h at 4 °C with 5 μg/ml anti-BECN1 antibody preabsorbed on Protein G-Sepharose (GE Healthcare, 17-0618-01). The pellets were washed three times with extraction buffer and once with reaction buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl and 0.2 mM EDTA). The immobilized proteins were subjected to PtdIns3K ELISA as described in the user’s manual (Echelon, K-3000).
Cholera toxin B staining
To observe glycosphingolipid-enriched membranes, cells grown into coverslip were incubated with cholera toxin B subunit, Alexa Fluor 594 conjugate (Molecular Probes, C-34777) for 15 min at 4 °C and after 30 min at 37 °C, to allow its internalization, and cells were then washed twice with PBS. Cells were fixed with 4% paraformaldehyde, blocked with 3% BSA, and processed to immunofluorescence analysis. Images were acquired with a confocal microscope (Carl Zeiss Ltd, LSM510), subjected to deconvolution with the manufacturer’s software, and prepared using Adobe Photoshop CS2 software.
Overlay assay
BECN1 protein (10 μg), which was expressed in E. coli and purified, was resolved by SDS-PAGE and transferred to nitrocellulose membranes. After blocking, the membranes were incubated at 4 °C for 12 h with whole brain lysates (lysed by modified RIPA buffer as described above) of 3-mo-old C57BL/6 mice. Membranes were then processed for western blotting as mentioned above.
Discontinuous iodixanol gradients for isolation of neuron
Tissues were cut in 0.5-mm slices, digested with trypsin-EDTA, and dissociated into a single cell suspension. The cell suspension was enriched for neurons by centrifugation on a density gradient of OptiPrep (Sigma Aldrich, D1556). OptiPrep was first diluted with DMEM to produce a density of 1.15. The diluted OptiPrep was further diluted with DMEM (v/v) to make four steps of 1 ml in a 15 ml centrifuge tube: bottom (0.35:0.65), 0.25:0.75, 0.2:0.8, top (0.15:0.85). Up to 6 ml of cell suspension was layered over the OptiPrep step gradient. Although neurons and glia are present throughout the gradient, the fraction between the pellet and the dense band of debris were collected for the highest enrichment of neurons.Citation45
Transmission electron microscope analysis
Cells were fixed with 2% paraformaldehyde/2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4), followed by 1% OsO4. After dehydration, cells were stained with uranyl acetate and lead citrate for observation under a JEM1010 transmission electron microscope (JEOL, Japan).
Statistics
All experiments were performed in triplicate parallel instances and repeated at least three times. Statistical analyses were performed using the Microsoft Office 2007 Excel software package (Microsoft Corporation). Mean values were compared using the unpaired Student t tests.
| Abbreviations: | ||
| Aβ42 | = | amyloid beta1-42 |
| ACTB | = | β-actin |
| AD | = | Alzheimer disease |
| ATG5 | = | autophagy-related 5 |
| AV | = | autophagic vacuoles |
| BafA1 | = | bafilomycin A1 |
| BCL2 | = | B-cell CLL/lymphoma 2 |
| BD | = | BCL2-binding domain |
| BECN1 | = | Beclin 1, autophagy related |
| CAV1 | = | Caveolin-1, caveolae protein |
| CCD | = | coiled-coil domain |
| CTxB | = | cholera toxin B |
| DRM | = | detergent-resistant membrane |
| ECD | = | evolutionarily conserved domain |
| ER | = | endoplasmic reticulum |
| FLOT1 | = | Flotillin 1 |
| GFP | = | green fluorescent protein |
| GPI | = | glycosylphosphatidylinositol |
| GRB2 | = | growth factor receptor-bound protein 2 |
| KO | = | knockout |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| MβCD | = | methyl-beta-cyclodextrin |
| mRFP | = | monomeric red fluorescent protein |
| PIK3C3 | = | VPS34, phosphatidylinositol 3-kinase, catalytic subunit type 3 |
| PRNP | = | prion protein |
| PRND | = | prion protein dublet |
| PtdIns | = | phosphatidylinositide |
| RPSA | = | LRP, ribosomal protein SA |
| SQSTM1/p62 | = | sequestosome 1 |
| TUBA | = | α-tubulin |
| WT | = | wild type |
Additional material
Download Zip (1.3 MB)Acknowledgments
We thank Dr C Weissmann (Scripps Florida, USA) for providing prnp KO mice. Dr J Nah and Dr JO Pyo were supported by the Brain Korea 21 program. This work was supported by the grants from Global Research Laboratory Program (K21004000002-12A0500-00210) and the grants funded by the the Korea government (MSIP) (NRF-2013R1A2A1A01016896) and Alzheimer disease grant (A092058-1113-0000400) funded by the Ministry of Human Health and Welfare of the Korean government.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/26118
References
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 77; http://dx.doi.org/10.1016/S1534-5807(04)00099-1; PMID: 15068787
- Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol 2011; 13:453 - 60; http://dx.doi.org/10.1038/ncb2204; PMID: 21394080
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814 - 22; http://dx.doi.org/10.1038/ncb0910-814; PMID: 20811353
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 1998; 72:8586 - 96; PMID: 9765397
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol 2010; 20:355 - 62; http://dx.doi.org/10.1016/j.tcb.2010.03.002; PMID: 20356743
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447:1121 - 5; PMID: 17589504
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385 - 96; http://dx.doi.org/10.1038/ncb1846; PMID: 19270696
- Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987; 51:229 - 40; http://dx.doi.org/10.1016/0092-8674(87)90150-4; PMID: 2444340
- Guillot-Sestier MV, Checler F. Cellular prion and its catabolites in the brain: production and function. Curr Mol Med 2012; 12:304 - 15; http://dx.doi.org/10.2174/156652412799218912; PMID: 22272726
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Matsumoto Y, Yokoyama T, Itohara S, et al. Prions prevent neuronal cell-line death. Nature 1999; 400:225 - 6; http://dx.doi.org/10.1038/22241; PMID: 10421360
- Criado JR, Sánchez-Alavez M, Conti B, Giacchino JL, Wills DN, Henriksen SJ, Race R, Manson JC, Chesebro B, Oldstone MB. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol Dis 2005; 19:255 - 65; http://dx.doi.org/10.1016/j.nbd.2005.01.001; PMID: 15837581
- Weise J, Sandau R, Schwarting S, Crome O, Wrede A, Schulz-Schaeffer W, Zerr I, Bähr M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 2006; 37:1296 - 300; http://dx.doi.org/10.1161/01.STR.0000217262.03192.d4; PMID: 16574930
- Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 2009; 284:35973 - 86; http://dx.doi.org/10.1074/jbc.M109.051086; PMID: 19850936
- Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Neurotoxicity of a prion protein fragment. Nature 1993; 362:543 - 6; http://dx.doi.org/10.1038/362543a0; PMID: 8464494
- Westaway D, DeArmond SJ, Cayetano-Canlas J, Groth D, Foster D, Yang SL, Torchia M, Carlson GA, Prusiner SB. Degeneration of skeletal muscle, peripheral nerves, and the central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell 1994; 76:117 - 29; http://dx.doi.org/10.1016/0092-8674(94)90177-5; PMID: 8287472
- Taylor DR, Hooper NM. The prion protein and lipid rafts. Mol Membr Biol 2006; 23:89 - 99; http://dx.doi.org/10.1080/09687860500449994; PMID: 16611584
- Korade Z, Kenworthy AK. Lipid rafts, cholesterol, and the brain. Neuropharmacology 2008; 55:1265 - 73; http://dx.doi.org/10.1016/j.neuropharm.2008.02.019; PMID: 18402986
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science 2000; 289:1925 - 8; http://dx.doi.org/10.1126/science.289.5486.1925; PMID: 10988071
- Rieger R, Edenhofer F, Lasmézas CI, Weiss S. The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat Med 1997; 3:1383 - 8; http://dx.doi.org/10.1038/nm1297-1383; PMID: 9396609
- Spielhaupter C, Schätzl HM. PrPC directly interacts with proteins involved in signaling pathways. J Biol Chem 2001; 276:44604 - 12; http://dx.doi.org/10.1074/jbc.M103289200; PMID: 11571277
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 32; http://dx.doi.org/10.1038/nature07761; PMID: 19242475
- Sakudo A, Lee DC, Saeki K, Nakamura Y, Inoue K, Matsumoto Y, Itohara S, Onodera T. Impairment of superoxide dismutase activation by N-terminally truncated prion protein (PrP) in PrP-deficient neuronal cell line. Biochem Biophys Res Commun 2003; 308:660 - 7; http://dx.doi.org/10.1016/S0006-291X(03)01459-1; PMID: 12914801
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A 2007; 104:19023 - 8; http://dx.doi.org/10.1073/pnas.0709695104; PMID: 18024584
- Bastiaens PI, Majoul IV, Verveer PJ, Söling HD, Jovin TM. Imaging the intracellular trafficking and state of the AB5 quaternary structure of cholera toxin. EMBO J 1996; 15:4246 - 53; PMID: 8861953
- Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem 2000; 275:31505 - 13; http://dx.doi.org/10.1074/jbc.M002102200; PMID: 10806201
- Coria F, Rubio I, Bayón C. Alzheimer’s disease, beta-amyloidosis, and aging. Rev Neurosci 1994; 5:275 - 92; http://dx.doi.org/10.1515/REVNEURO.1994.5.4.275; PMID: 7697197
- Ohtani Y, Irie T, Uekama K, Fukunaga K, Pitha J. Differential effects of alpha-, beta- and gamma-cyclodextrins on human erythrocytes. Eur J Biochem 1989; 186:17 - 22; http://dx.doi.org/10.1111/j.1432-1033.1989.tb15171.x; PMID: 2598927
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571 - 80; http://dx.doi.org/10.1038/cdd.2010.191; PMID: 21311563
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685 - 701; http://dx.doi.org/10.1083/jcb.200803137; PMID: 18725538
- Son SM, Jung ES, Shin HJ, Byun J, Mook-Jung I. Aβ-induced formation of autophagosomes is mediated by RAGE-CaMKKβ-AMPK signaling. Neurobiol Aging 2012; 33:1006.e11 - 23; http://dx.doi.org/10.1016/j.neurobiolaging.2011.09.039
- Pajak B, Songin M, Strosznajder JB, Orzechowski A, Gajkowska B. Ultrastructural evidence of amyloid beta-induced autophagy in PC12 cells. Folia Neuropathol 2009; 47:252 - 8; PMID: 19813145
- Liberski PP, Brown DR, Sikorska B, Caughey B, Brown P. Cell death and autophagy in prion diseases (transmissible spongiform encephalopathies). Folia Neuropathol 2008; 46:1 - 25; PMID: 18368623
- Zhou M, Ottenberg G, Sferrazza GF, Lasmézas CI. Highly neurotoxic monomeric α-helical prion protein. Proc Natl Acad Sci U S A 2012; 109:3113 - 8; http://dx.doi.org/10.1073/pnas.1118090109; PMID: 22323583
- Oh JM, Shin HY, Park SJ, Kim BH, Choi JK, Choi EK, Carp RI, Kim YS. The involvement of cellular prion protein in the autophagy pathway in neuronal cells. Mol Cell Neurosci 2008; 39:238 - 47; http://dx.doi.org/10.1016/j.mcn.2008.07.003; PMID: 18674620
- Heitz S, Grant NJ, Leschiera R, Haeberlé AM, Demais V, Bombarde G, Bailly Y. Autophagy and cell death of Purkinje cells overexpressing Doppel in Ngsk Prnp-deficient mice. Brain Pathol 2010; 20:119 - 32; http://dx.doi.org/10.1111/j.1750-3639.2008.00245.x; PMID: 19055638
- Oh JM, Choi EK, Carp RI, Kim YS. Oxidative stress impairs autophagic flux in prion protein-deficient hippocampal cells. Autophagy 2012; 8:1448 - 61; http://dx.doi.org/10.4161/auto.21164; PMID: 22889724
- Kiffin R, Kaushik S, Zeng M, Bandyopadhyay U, Zhang C, Massey AC, Martinez-Vicente M, Cuervo AM. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J Cell Sci 2007; 120:782 - 91; http://dx.doi.org/10.1242/jcs.001073; PMID: 17284523
- Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008; 4:176 - 84; PMID: 18059160
- Lee KS, Magalhães AC, Zanata SM, Brentani RR, Martins VR, Prado MA. Internalization of mammalian fluorescent cellular prion protein and N-terminal deletion mutants in living cells. J Neurochem 2001; 79:79 - 87; http://dx.doi.org/10.1046/j.1471-4159.2001.00529.x; PMID: 11595760
- Pyo JO, Nah J, Kim HJ, Lee HJ, Heo J, Lee H, Jung YK. Compensatory activation of ERK1/2 in Atg5-deficient mouse embryo fibroblasts suppresses oxidative stress-induced cell death. Autophagy 2008; 4:315 - 21; PMID: 18196969
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992; 356:577 - 82; http://dx.doi.org/10.1038/356577a0; PMID: 1373228
- Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem 2004; 279:44945 - 54; http://dx.doi.org/10.1074/jbc.M407986200; PMID: 15322084
- Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods 1997; 71:143 - 55; http://dx.doi.org/10.1016/S0165-0270(96)00136-7; PMID: 9128149