Abstract
Metastasis is one of the main causes of poor prognosis for hepatocellular carcinoma (HCC), which has been linked to cell-death resistance. Autophagy is an important survival mechanism under conditions of cell stress. We hypothesized that autophagy may play a role in HCC metastasis due to its prosurvival effect. Highly metastatic HCC cell lines with stable autophagy inhibition were established via lentivirus-mediated silencing of BECN1 and ATG5 genes. Mouse models of pulmonary metastasis were then developed using the cells with or without autophagy inhibition. The analysis of lung metastasis by histopathological examination and small animal imaging showed that autophagy inhibition significantly decreased the incidence of pulmonary metastases in vivo. Further invasion, migration, detachment, lung colonization, and epithelial-mesenchymal transition (EMT) assays indicated that autophagy inhibition did not affect cell invasiveness, migration or EMT but attenuated the anoikis-resistance and lung colonization of HCC cells. Investigation of the molecular mechanisms underlying showed that the autophagy-inhibition-mediated anoikis-resistance attenuation was associated with the regulation of apoptotic signaling. As autophagy inhibition was shown to be able to suppress HCC metastasis, an autophagy-based HCC tissue-specific target therapy system (AFP-Cre/LoxP-shRNA) was constructed. In vitro and in vivo analyses showed that the system was able to efficiently inhibit autophagy of HCC cells and tissue in a tissue-specific manner. Further in vivo metastasis assay showed that intratumoral administration of the system could significantly suppress lung metastasis. Together, our findings suggest that autophagy may be involved in HCC metastasis through facilitating anoikis resistance and lung colonization of HCC cells. Autophagy-based HCC tissue-specific target therapy may be a new strategy for the management of HCC metastasis.
Introduction
Hepatocellular carcinoma (HCC) is one of the most fatal cancers. It is the third most common cause of death from cancer worldwide and the second most common in China.Citation1,Citation2 Metastasis is the major cause of death from HCC. Although advances have been made in HCC therapies, it remains the major obstacle to improving the prognosis of HCC.Citation3
Metastasis is a complex, multistep process in which tumor cells invade neighboring tissues, intravasate into the systemic circulation, translocate through the vasculature, extravasate from the circulation to colonize at distant sites, and finally form a secondary tumor.Citation4 It has been demonstrated that metastasis is an inefficient process, as many of the tumor cells die during the process.Citation5,Citation6 To successfully metastasize, tumor cells must be capable of both maintaining their viability throughout the process and adapting to the constantly changing foreign microenvironments before they establish the metastatic foci.Citation7 Understanding of the cell survival mechanisms responsible for metastasis is of great importance for the management of HCC.
Autophagy appears to be one of the cell survival mechanisms. Autophagy is a self-degradative process by which cells break down cytoplasmic materials. It is essential for maintaining homeostasis through the management of bioenergetics and the housekeeping role of removing damaged cell components. Under conditions of stress, autophagy serves as an important cytoprotective mechanism that promotes cell survival. It has been demonstrated that autophagy can protect cells against hypoxia, metabolic stress, detachment-induced anoikis, and diverse cellular damage, as well as apoptosis or necrosis induced by antitumor therapy or other cell death stimuli. Inhibition of autophagy can promote tumor cell death.Citation7-Citation15 Therefore, we postulated that autophagy might serve as an important adaptive prosurvival mechanism that mediates cancer cell survival during metastasis. Autophagy may mitigate metabolic stress under the nutrient-deprivation conditions that are often faced by metastatic tumor cells, especially in the formation of metastases in remote organs.Citation16,Citation17 Tumor cells may utilize autophagy-mediated cell death resistance to resist death induction during metastasis. For example, metastatic tumor cells detached from the extracellular matrix may utilize autophagy to resist anoikis and facilitate tumor cell dissemination and dormancy.Citation11 Tumor cells may also acquire TNFSF10/TRAIL resistance via autophagy to resist the induction of apoptosis caused by TNSF10 exposure during metastasis.Citation7 With the aid of the autophagy-mediated cell survival mechanisms, tumor cells can overcome diverse unfavorable stresses and adapt to microenvironment transformations to successfully metastasize.
The purpose of this study is to evaluate whether autophagy plays a role in HCC metastasis and how it influences metastasis. After the role of autophagy was confirmed, an autophagy-based target therapy for HCC metastasis was designed and examined.
Results
Autophagy inhibition decreases pulmonary metastasis of HCC in mice
To explore whether autophagy plays a role in HCC metastasis, we examined the effect of autophagy inhibition on HCC metastasis in a nude mouse model of pulmonary metastasis. First, two highly metastatic HCC cell lines with stable autophagy inhibition (HCCLM3-ATGi and HCCLM3-R-ATGi) were established via lentivirus-mediated knockdown of BECN1 and ATG5 (). Then a nude mouse model of pulmonary metastasis was established with HCCLM3-ATGi/HCCLM3-R-ATGi cells and cells without autophagy inhibition (HCCLM3, HCCLM3-Mock, HCCLM3-R and HCCLM3-R-Mock cells). All mice successfully formed liver tumors, and there were no significant differences in tumor size among the groups (). The number of lung metastases was 192.0 ± 21.2 in HCCLM3-ATGi group (grade I, 95.2 ± 7.4; grade II, 62.0 ± 16.0; grade III, 26.2 ± 5.7; grade IV, 8.7 ± 3.1), 223.0 ± 26.5 in HCCLM3-Mock group (grade I, 91.5 ± 11.3; grade II, 74.0 ± 12.2; grade III, 38.7 ± 5.9; grade IV, 18.8 ± 5.3), and 247.5 ± 25.1 in HCCLM3 group (grade I, 95.7 ± 13.9; grade II, 85.8 ± 9.4; grade III, 39.8 ± 8.5; grade IV, 26.2 ± 3.6). The number of lung metastasis in HCCLM3-ATGi group was significantly less than those in the HCCLM3 and HCCLM3-Mock groups (HCCLM3-ATGi vs. HCCLM3, P = 0.001; HCCLM3-ATGi vs. HCCLM3-Mock, P = 0.044), while there was no significant difference between HCCLM3 and HCCLM3-Mock groups (P = 0.102). There was a prevalence of smaller-sized metastatic foci in the HCCLM3-ATGi group; the percentage of grade III/IV metastatic foci in the HCCLM3-ATGi group (18.2%) was lower than those in the HCCLM3 group (26.7%) and in the HCCLM3-Mock group (25.8%). Small-animal imaging analysis in the nude mouse model was established using RFP-expressing HCCLM3-R, and HCCLM3-ATGi cells confirmed the results from histopathological analysis ().
Figure 1. Generation of HCC cell lines with stable autophagy inhibition via lentivirus-mediated knockdown of BECN1 and ATG5. (A and B) Quantitative real-time PCR and western blot analysis of BECN1 and ATG5 knockdown. Effective shRNAs targeting BECN1 and ATG5 were selected and constructed into 2 lentiviral vectors. The knockdown efficiency of the cotransfection was analyzed by western blot. (C and D) HCC cell lines with stable autophagy inhibition (HCCLM3-ATGi and RFP-expressing HCCLM3-R-ATGi). GFP-LC3 transfection analysis and western blot analysis showed that rapamycin (Rapa) treatment did not induce autophagy in HCCLM3-ATGi or HCCLM3-R-ATGi cells while it induced intense autophagy in HCCLM3 and HCCLM3-R cells without autophagy inhibition.
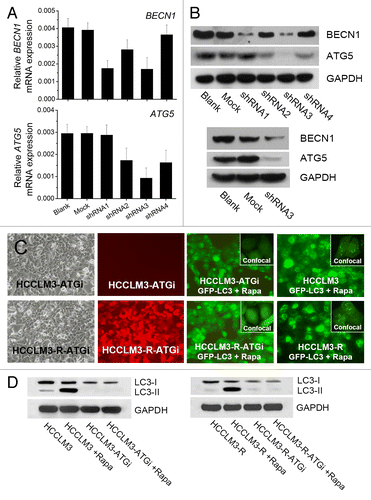
Figure 2. In vivo analysis of the effect of autophagy inhibition on HCC metastasis. (A) Nude mouse model of pulmonary metastasis was established using HCC cells with or without autophagy inhibition (HCCLM3-ATGi or HCCLM3/HCCLM3-Mock). Histopathological analysis showed that there were significantly fewer lung metastases of mice receiving HCCLM3-ATGi cells than in mice subjected to HCCLM3 and HCCLM3-Mock cells. *P = 0.001, **P = 0.044. (From left to right) Mice receiving orthotopic implantation of HCCLM3 / HCCLM3-ATGi / HCCLM3-Mock cells, lung and liver of the mice, representative panoramic image of lung section, lung metastasis (100×) and lung metastasis (200×); (B) Small-animal imaging analysis using RFP-expressing HCCLM3-R-ATGi and HCCLM3-R cells confirmed the histopathological analysis. The images of fluorescence signals were pseudocolored (pink, least intense; red, most intense), and the images of lung metastases and primary HCC tumors were merged onto the corresponding X-ray images.
Metastasis is a complex process that features a series of steps. To clarify in which step the suppressive effect of autophagy inhibition occurs, we next examined the effect of autophagy inhibition on several steps in the metastatic cascade.
Autophagy inhibition has no effect on cell migration and invasion
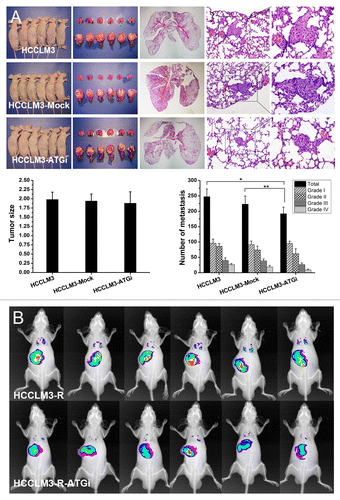
We first examined the effect of autophagy inhibition on cell migration and invasion. In vitro migration assays, including transwell and scratch assays, showed that autophagy inhibition had no effect on cell migration (). The in vitro matrigel invasion assay indicated that autophagy inhibition had no significant effect on cell invasiveness (HCCLM3-ATGi vs. HCCLM3, P = 0.553; HCCLM3-ATGi vs. HCCLM3-Mock, P = 0.425) (). These data suggest that the suppressive effect of autophagy inhibition on pulmonary metastasis might not be associated with cell migration and invasion.
Figure 3. Effect of autophagy inhibition on migration and invasion of HCC cells. (A) Transwell migration and invasion assays showed no effect of autophagy inhibition on the migration and invasiveness of HCCLM3 cells. (B) The scratch assay showed results similar to the transwell migration assay.
Autophagy inhibition attenuates the anoikis resistance of HCC cells via regulation of apoptotic signaling
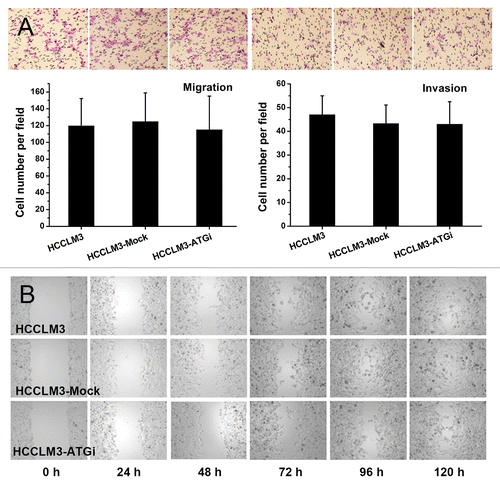
We next tested the effect of autophagy inhibition on the anoikis resistance of HCCLM3 cells. Anoikis resistance is responsible for the survival of tumor cells when they detach from the extracellular matrix (ECM) and disseminate into the circulatory system. The in vitro anoikis assay indicated that HCCLM3 cells were anoikis-resistant (). The anoikis rates of HCCLM3 cells were 4.06 ± 1.00%, 15.06 ± 1.93% and 20.37 ± 2.51% at 12 h, 24 h and 48 h after cell detachment, respectively. When autophagy was inhibited, the anoikis rate significantly increased to 8.4 ± 0.93% (12 h), 19.96 ± 1.9% (24 h) and 41.55 ± 9.3% (48 h) (HCCLM3-ATGi vs. HCCLM3, P = 0.001, 0.023, and 0.005; HCCLM3-ATGi vs. HCCLM3-Mock, P = 0.000, 0.007, and 0.014, respectively) (). We then repeated the anoikis assay in another HCC cell line MHCC97H. After inhibition of autophagy, the anoikis rate of MHCC97H cells also significantly increased (). The effect of autophagy inhibition by RNA silencing of BECN1 and ATG5 on anoikis was also validated by knockdown of another essential autophagy gene ATG7 in combination with BECN1 or ATG5 in the two HCC cell lines (Fig. S1). Furthermore, an in vivo anoikis assay was performed to verify the in vitro finding using a nude mouse model of peritoneal dissemination.Citation18 After intraperitoneal inoculation of HCCLM3 and HCCLM3-ATGi cells, no mice (0/6) in the HCCLM3-ATGi group formed peritoneal metastases and ascites, while 2 mice (2/6) in the HCCLM3 group did (). These data suggest that the suppressive effect of autophagy inhibition on pulmonary metastasis might be attributed to the attenuation of anoikis resistance. Further investigation of the underlying molecular mechanisms showed that cell detachment did not activate the autophagy-associated integrated stress response, growth factor and nutrient-sensing, and energy sensing pathways in HCCLM3 cells (Fig. S2). However, the levels of BAX, BAK1, and FADD [Fas (TNFRSF6)-associated via death domain] proteins increased while the level of BCL2L1 protein decreased after stable inhibition of autophagy, which indicated an increase of the ratio of proapoptotic to antiapoptotic proteins (Fig. S3). The results suggested that the autophagy-inhibition mediated anoikis-resistance attenuation was associated with the regulation of apoptotic signaling while the autophagy-associated integrated stress response, growth factor and nutrient-sensing, and energy sensing pathways was not involved.
Figure 4. Autophagy inhibition attenuates anoikis resistance of HCC cells via regulation of apoptotic signaling. (A) The in vitro anoikis assay indicated that autophagy inhibition significantly increased the anoikis rate of HCCLM3 cells after cell detachment. (B) In vivo anoikis assays showed that no mice receiving HCCLM3 cells with autophagy inhibition (HCCLM3-ATGi) formed peritoneal metastases and ascites while mice subjected to HCCLM3 cells without autophagy inhibition (HCCLM3) could establish. The HCCLM3 ascites cells were determined by smear test (ST) and flow cytometric DNA analysis (FCM).
Autophagy inhibition does not induce epithelial–mesenchymal transition
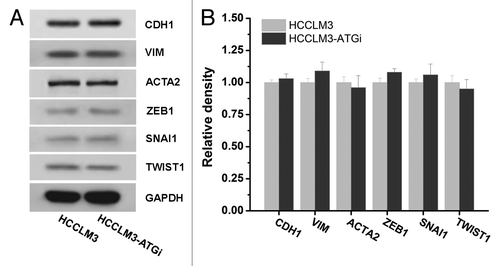
We subsequently examined whether autophagy inhibition induced EMT. Western blot analysis of key EMT markers expression [CDH1 (cadherin 1, type 1, E-cadherin (epithelial)), VIM (vimentin), ACTA2 (actin, α 2, smooth muscle, aorta), ZEB1 (zinc-finger E-box binding homeobox 1), SNAI1 (snail family zinc finger 1) and TWIST1 (twist basic helix-loop-helix transcription factor 1)] showed no significant changes after autophagy inhibition in HCCLM3 cells, which suggested that autophagy inhibition did not induce EMT ().
Figure 5. Effect of autophagy inhibition on epithelial mesenchymal transition (EMT) of HCC cells. Western blot analysis of EMT markers expressions (CDH1, VIM, ACTA2, ZEB1, SNAI1, and TWIST1) in HCCLM3 cells with or without autophagy inhibition (HCCLM3 or HCCLM3-ATGi) revealed no significant differences between HCCLM3 and HCCLM3-ATGi cells. Cell lysates from HCCLM3-ATGi and HCCLM3 cells were immunoblotted with indicated antibodies (left). Protein levels were determined by densitometry measurements and normalized (right).
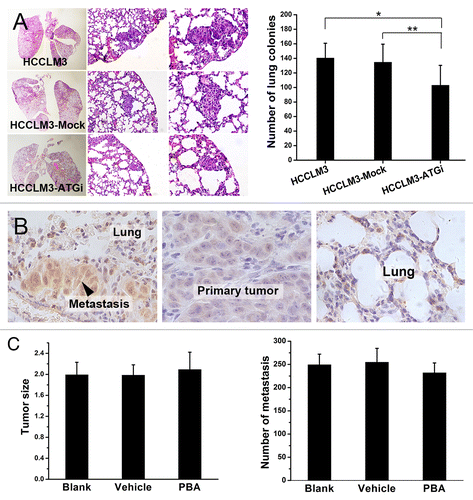
Autophagy inhibition decreases lung colonization in HCC metastasis
We finally examined the role of autophagy in metastatic colonization. Lung colonization assay indicated that lung colonies in the mice receiving HCCLM3-ATGi cells were significantly fewer (103.0 ± 27.4) than in mice subjected to HCCLM3 and HCCLM3-Mock cells (140.5 ± 20.3 and 134.7 ± 24.9, respectively) (HCCLM3-ATGi vs. HCCLM3: P = 0.018; HCCLM3-ATGi vs. HCCLM3-Mock: P = 0.040; HCCLM3 vs. HCCLM3-Mock: P = 0.684) (). This result suggests that the negative effect of autophagy inhibition on metastasis may also be associated with colonization.As endoplasmic reticulum stress (ER stress) was speculated to be induced in metastatic colonization and subsequently triggered autophagy to promote metastasis, we further examined whether the negative effect of autophagy inhibition on metastasis was associated with ER stress. Immunohistochemical analysis of ER stress marker HSPA5/GRP78 [heat shock, 70 kDa protein 5 (glucose-regulated protein, 78 kDa)] expression in metastases and primary HCC tumors showed a higher level of ER stress in lung metastases than in primary tumors (P = 0.001), indicating that ER stress was induced in metastasis of HCC and might play a role (). However, further ER stress blocking test using ER stress inhibitor PBA (4-phenyl butyric acid) in mouse model of pulmonary metastasis showed that inhibition of ER stress did not suppress metastasis. The in vivo metastasis assay indicated that the number of lung metastases of mice receiving PBA (PBA group, 232 ± 21.22) was not significantly different from that of mice subjected to either PBA vehicle (Vehicle group, 251.67 ± 29.90) or no treatment (Blank group, 249.33 ± 22.71) (PBA vs. Vehicle, P = 0.136; PBA vs. Blank, P = 0.247). These suggested that ER stress was not involved in metastatic colonization though it was induced ().
Figure 6. Autophagy inhibition suppresses lung colonization of HCC cells and ER stress is not involved in. (A) Lung colonization assays indicated that autophagy inhibition significantly suppressed lung colonization of HCCLM3 cells (*P = 0.018, **P = 0.040). (From left to right) representative panoramic image of lung section, lung metastasis (100×), and lung metastasis (200×). (B and C) ER stress was induced in metastatic colonies but the suppressive effect of autophagy inhibition on metastatic colonization was not associated with ER stress. Immunohistochemical analysis of ER stress marker HSPA5 expression in lung metastasis and paired primary tumor showed significantly higher HSPA5 expression in lung metastases, indicating that ER stress was induced in metastatic colonization (B). Further in vivo metastasis assays showed that inhibition of ER stress by PBA did not affect lung metastasis (C). The number of lung metastases of mice receiving PBA (PBA group) was not significantly different from those of mice subjected to PBA vehicle (Vehicle group) or no treatment (Blank group). (PBA vs. Blank, P = 0.247; PBA vs. Vehicle, P = 0.136).
Autophagy-based target therapy is effective for HCC metastasis
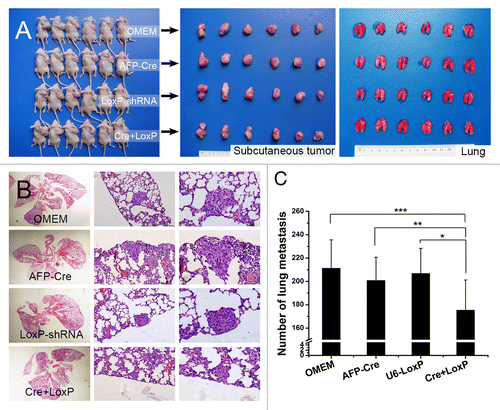
The fact that autophagy played a role in HCC metastasis indicated its potential value for HCC metastasis management. To reveal the efficacy of autophagy-based target therapy in HCC, we constructed an HCC-specific autophagy-based target therapy system (AFP-Cre/LoxP-shRNA) (). In vitro and in vivo analyses showed that the AFP-Cre/LoxP-shRNA system was able to efficiently knock down BECN1 or ATG5 and inhibit autophagy in a HCC tissue-specific manner (). We then evaluated the effectiveness of the system for HCC metastasis treatment. Intravenous administration in the nude mouse model of pulmonary metastasis (established via orthotopic implantation) showed that the system could not reach the orthotopic liver tumor, even if a lethal dose was given, which was thought to be attributed to the limited intake of the system. Thus, the effectiveness of the system was then evaluated by intratumoral injection in nude mouse model of pulmonary metastasis established via subcutaneous inoculation. All mice successfully formed subcutaneous tumors, and there was no significant difference in tumor weight among all groups (Cre+LoxP group, 2.47 ± 0.73 g; AFP-Cre group, 2.67 ± 0.67 g; LoxP-shRNA group, 2.61 ± 0.72 g; OMEM group, 2.83 ± 0.81 g, P = 0.671) (). The number of lung metastases in the mice receiving the AFP-Cre/LoxP-shRNA system (175.4 ± 15.8) was significantly lower compared with other groups (AFP-Cre group, 199.8 ± 19.9; LoxP-shRNA group, 206.9 ± 21.4; OMEM group, 211.3 ± 24.3; Cre+LoxP vs. AFP-Cre, P = 0.022; Cre+LoxP vs. LoxP-shRNA, P = 0.015, Cre+LoxP vs. OMEM, P = 0.007), while there was no significant difference between the AFP-Cre, LoxP-shRNA and OMEM groups (P = 0.874) ().
Figure 7. Construction of HCC tissue-specific autophagy-based target therapy system (AFP-Cre/LoxP-shRNA). (A) The AFP-Cre/LoxP-shRNA system consisted of AFP-Cre vector and LoxP-shRNA vector. A CMV-eGFP reporter in the system could indicate whether the system worked. (B) In vitro analysis showed that the system worked efficiently in HCC cells (HCCLM3/HepG2) but did not work in non-HCC cells (L-02/Hela). The disappearance of GFP fluorescence indicated that the Cre recombinase driven by the AFP promoter had cut the LoxP-CMV-eGFP-LoxP in U6 promoter and activated expression of shRNAs targeting BECN1 and ATG5. Western blot analysis showed that the system efficiently silenced target genes BECN1 and ATG5. Cre+LoxP or Cre+LoxP NC: cotransfection of AFP-Cre and LoxP-shRNA (AFP-Cre/LoxP-shRNA system); NC, negative control; AFP-Cre or LoxP-shRNA: transfection of AFP-Cre or LoxP-shRNA alone (control). (C) In vivo analysis showed that intratumoral injection of AFP-Cre/LoxP-shRNA system in HCCLM3 subcutaneous tumors worked efficiently in vivo.
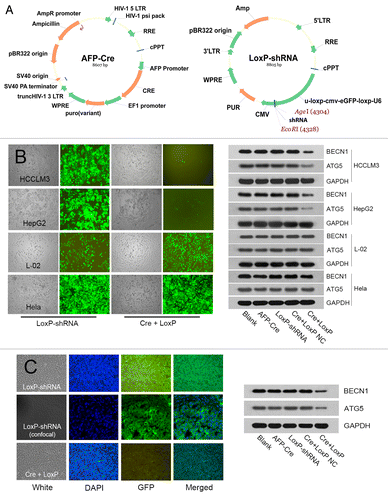
Figure 8. In vivo evaluation of HCC-tissue-specific autophagy-based target therapy for HCC metastasis. (A) A nude mouse model of pulmonary metastasis via subcutaneous injection was established. The target therapy system was intratumorally administrated. Cre+LoxP: AFP-Cre/LoxP-shRNA system; OMEM: Opti-MEM (control); AFP-Cre: AFP-Cre alone (control), LoxP-shRNA: LoxP-shRNA alone (control). From left to right: mice bearing subcutaneous HCCLM3 tumors, subcutaneous tumors of the mice, lungs of the mice; (B and C) The number of lung metastases in the Cre+LoxP group was significantly less than those in the other 3 groups (*P = 0.015, **P = 0.022, ***P = 0.007). (From left to right) representative panoramic image of lung section, lung metastasis (100×), and lung metastasis (200×).
Discussion
A myriad of studies have established that autophagy is an important cell survival mechanism.Citation7-Citation11 It has been postulated that the prosurvival effect and stress-adaptive capability of autophagy are involved in metastasis.Citation7 However, few studies have confirmed this hypothesis, especially regarding the exact role of autophagy in HCC metastasis. This was largely due to a lack of in vivo study with a reliable assay.Citation19 In our study, we used a well-established nude mouse model of pulmonary metastasis and lentivirus-mediated stable autophagy inhibition to obtain an in vivo analysis. For the first time, the role of autophagy in HCC metastasis was examined. Our data showed that autophagy inhibition suppressed the pulmonary metastasis of HCCLM3 cells, suggesting that autophagy might play a role in HCC metastasis. The nude mouse model used in this study is well established and has been widely used.Citation3,Citation20,Citation21 It provided a good model for in vivo study of HCC metastasis. To obtain stable autophagy inhibition in vivo, we chose lentivirus-mediated knockdown of BECN1 and ATG5 because lentivirus-mediated RNA interference allowed persistent and heritable gene silencing. The commonly used transient BECN1 and ATG5 siRNAs were not applied because their inhibition of autophagy only lasts for 8 to 12 d, while our lung metastasis model took at least 3 to 4 wk to develop detectable metastasis. Autophagy inhibitors, such as chloroquine, were not used because they are not specific and might produce off-target effects.Citation22 Besides, we knocked down both BECN1 and ATG5 instead of single BECN1 or ATG5 because our pilot study demonstrated that silencing a single gene (BECN1 or ATG5) could not achieve a satisfactory level of autophagy inhibition (Fig. S4) and subsequent suppression of pulmonary metastasis (Figs. S5 and S6). In this study, lentivirus-mediated double knockdown of BECN1 and ATG5 was utilized to achieve stable and long-lasting autophagy inhibition in vivo, which provided a reliable in vivo analysis of the role of autophagy in metastasis.
Metastasis is a complex, multistep process.Citation4 The role of autophagy in the individual steps of the metastatic cascade has been postulated to be complex.Citation7 Our study indicated that autophagy inhibition did not affect cell migration, invasion and EMT but attenuated anoikis resistance and colonization. Invasion and migration are the key steps of metastasis. Autophagy appeared to not be involved in these steps, which was similar to findings in other malignant tumors.Citation23,Citation24 Anoikis resistance is responsible for the survival of tumor cells when they detach from the ECM and disseminate in the circulatory system. Recent studies indicated that autophagy may be one mechanism of anoikis resistance.Citation25,Citation26 ECM detachment can robustly induce autophagy in epithelial cells, and the inhibition of autophagy results in increased anoikis.Citation25,Citation26 In this study, we found that autophagy inhibition could attenuate the anoikis resistance of HCCLM3 cells in vitro and in vivo, suggesting that autophagy might indeed play an important role in anoikis resistance. This effect might protect HCC cells against ECM-detachment stress and favor HCC dissemination.Citation7 Thus far, little is known about the crosstalk between anoikis and autophagy. Recent studies showed that some signaling pathways may be involved in the autophagy-associated anoikis-resistance.Citation11 The candidate intracellular signaling pathways include integrated stress response; growth factor and nutrient-sensing pathway; and energy-ensing pathways.Citation11 Avivar-Valderas et al. report that EIF2AK3/PERK (eukaryotic translation initiation factor 2-α kinase 3) integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment.Citation13 In this study, we examined all the 3 candidate pathways but found no correlation with the autophagy-associated anoikis-resistance in HCC cells. Our results were different from Avivar-Valderas et al.’s findings. The difference may be attributed to the different cell lines and study models. The cell line used in Avivar-Valderas et al.’s study was MCF 10A cell, which is an anoikis-sensitive, nontumorigenic epithelial cell line, while the HCC cell lines used in our study were anoikis-resistant tumor cells. Besides, our study was performed in a simple in vitro anoikis model which was different from their study.Citation13 As the candidate autophagy-associated pathways were shown not to be involved, we then asked whether apoptosis-related proteins were responsible for the autophagy-associated anoikis-resistance. It has been demonstrated that autophagy inhibition could significantly enhance apoptosis induction in HCC, breast adenocarcinoma, lymphoma, and metastatic skin carcinoma cells.Citation14,Citation15,Citation27-Citation29 As anoikis is one form of apoptosis, the crosstalk between anoikis and autophagy might be similar. Further study showed that stable autophagy inhibition resulted in an increase of the ratio of proapoptotic to antiapoptotic proteins, which indicated a shift of the balance of apoptosis. The shift of apoptotic balance could subsequently sensitize HCC cells to apoptotic inducer (ECM detachment), which might in part explain why autophagy inhibition attenuated the anoikis-resistance of HCC cells. However, the exact mechanisms underlying still remain elusive and need further in-depth study.
In this study, we attempted an HCC tissue-specific, autophagy-based target therapy for HCC metastasis. Intratumoral administration of the AFP-Cre/LoxP-shRNA system was effective, suggesting its value in the management of HCC metastasis. However, intravenous injection of the system was not effective, which might be due to several reasons. First, intake of the system was limited. Due to the disadvantages of the lentiviral vector and nude mouse model, the maximum concentration and volume of the system was 2 × 109 TU/ml and 180 µl. Thus, the maximum intake was only 3.6 × 108 TU, which might be unable to achieve a sufficient transfection. Second, there was a considerable loss of the system during intravenous administration. The lentiviral vector used for the system can quickly transfect any type of cells, although the system only worked in AFP-positive HCC cells. After being intravenously injected, it may transfect blood and endothelial cells immediately and therefore considerably diminished before it reaches the tumor in liver. However, our data showed that autophagy-based target therapy is promising for HCC metastasis. The problems of the current system are expected to be overcome by further improvements, including using an AAV vector to achieve sufficient intake (> 1012 TU/ml), and modifying the vector so that it can only transfect HCC cells. Meanwhile, it should be noted that autophagy inhibition alone may not be sufficient for the treatment of HCC metastasis. Therefore, combining autophagy inhibition with other agents, such as sorafenib, may be required to achieve a satisfactory efficacy.Citation30
One limitation of this study is that we did not use different cell lines, which does not completely exclude the possibility of clonal differences. This is largely due to a lack of different cell lines available. To offset this disadvantage, we applied the following methodology as rigorously as possible to guarantee unbiased evaluations. All of the procedures were performed with cells derived from the same origin. All of the procedures were performed in the same way by one independent operator, and all of the observers were blinded to the experimental design. Therefore the only difference between groups is expected to be the treatment under investigation.
In conclusion, our data suggest that autophagy may play a role in HCC metastasis through facilitating the anoikis resistance and colonization of HCC cells. The inhibition of autophagy can suppress HCC metastasis, and autophagy-based HCC tissue-specific target therapy may be a new strategy for the management of HCC metastasis.
Materials and Methods
Cell lines and animals
Human HCC cell lines with high metastatic potential established by our institute, HCCLM3, RFP-expressing HCCLM3-R and MHCC97H,Citation31,Citation32 human HCC cell line HepG2 (ATCC, HB-8065), and cervical cancer cell line HeLa (ATCC, CCL-2) were routinely maintained.Citation20,Citation21,Citation32,Citation33 Male BALB/c nu/nu mice (6 wk old, Chinese Academy of Science) were bred in specific pathogen-free conditions. All mice were cared for according to the Guide for the Care and Use of Laboratory Animals. Experimental protocol was approved by Shanghai Medical Experimental Animal Care Committee.
Generation of cell lines with stable autophagy inhibition
HCC cell lines with stable autophagy inhibition were generated using lentivirus-mediated knockdown of ATG5 and BECN1. Lentiviral vectors encoding shRNA targeting ATG5 or BECN1 (pMAGic-Puro-shRNA-ATG5 and pLKO.1-Puro/GFP-shRNA-BECN1) were separately constructed. The HCCLM3 or HCCLM3-R cells were cotransfected by BECN1 and ATG5 lentivirus particle mixtures (all multiplicity of infections = 20) and then selected in media containing 2 μg/ml puromycin (Invivogen, ant-pr-1). The stable transfectants were amplified, and Q-PCR and western blot analysis were performed to determine the efficiency of gene silencing. Autophagy inhibition was determined by GFP-LC3 transfection analysis with autophagy induction (rapamycin, 300 nM) (Invivogen, tlrl-rap). The stability of the autophagy inhibition was determined by repeating an autophagic responsiveness assay 2 mo after transfection. The HCCLM3 and HCCLM3-R cells with stable autophagy inhibition were termed as HCCLM3-ATGi and HCCLM3-R-ATGi, respectively. Transfectants receiving empty lentiviral vectors served as controls (HCCLM3-Mock and HCCLM3-R-Mock).
The shRNA sequences for BECN1 and ATG5 were as follow: BECN1 shRNA: CCGGCCCGTG GAATGGAATG AGATTCTCGA GAATCTCATT CCATTCCACG GGTTTTTG; AATTCAAAAA CCCGTGGAAT GGAATGAGAT TCTCGAGAAT CTCATTCCAT TCCACGGG; ATG5 shRNA: CCGGGCTAGC TGGCTGTCCA TATTTCAAGA GAATATGGAC AGCCAGCTAG CTTTTTTG; AATTCAAAAA AGCTAGCTGG CTGTCCATAT TCTCTTGAAA TATGGACAGC CAGCTAGC.
Quantitative real-time PCR analysis
The quantitative real-time PCR (Q-PCR) analyses of ATG5 and BECN1 expression were performed as previously described.Citation33
Western blot analysis
Western blot analysis was performed as previously described.Citation33 The following antibodies were used: anti-human antibodies against BECN1 (1:1000; Cell Signaling, 3495), ATG5 (1:500; Cell Signaling, 8540), LC3B/MAP1LC3B (1:200; Abcam, ab48394), EIF2AK3/PERK, p-PERK/p-EIF2AK3 (1:500; Cell Signaling, 5683), EIF2S1/eIF2α, p-EIF2S1/p-eIF2α (p-EIF2S1) (1:500; Cell Signaling, 9079, 9721), AKT1, p-AKT1 (1:500; Abcam, ab8805, ab81283), MTOR, p-MTOR (1:500; Abcam, ab137340, ab109268), PRKAA2/AMPK, p-PRKAA2/p-AMPK (1:500; Cell Signaling, 2532, 2531), BCL2L1/Bcl-xl, BCL2/Bcl-2, MCL1/Mcl-1, BAX, BAK1/Bak, BID (1:500; Abcam, ab2568, ab59348, ab32087, ab53154, ab32371, ab32060), FADD (1:300; Cell Signaling, 2782), CDH1/E-cadherin, VIM/vimentin (1:500; Cell Signaling, 3195, 5741), ACTA2/α-SMA (1:500; Abcam, ab32575), ZEB1 (1:300; Santa Cruz, sc-25388), SNAI1/Snail1, TWIST1 (1:300; Abcam, ab135708, ab49254) or GAPDH (1:5000; Millipore, AB2302). Densitometry was measured using ImageJ. Mean ± SD was calculated from 3 individual experiments.
Metastasis assays in vivo
Metastasis assay via orthotopic implantation
The nude mouse model of pulmonary metastasis via orthotopic implantation was established as previously described.Citation20,Citation31 Briefly, 3.0 × 106 HCCLM3-ATGi, HCCLM3-Mock, HCCLM3, HCCLM3-R-ATGi, HCCLM3-R-Mock, or HCCLM3-R cells were suspended in 100 µl DMEM (Gibco, 11965-092) and Matrigel (BD, 354234)(1:1) and then inoculated into the liver parenchyma of nude mice (n = 6 per group). The animals were monitored every day and sacrificed 6 wk later. The lungs and livers of the mice were examined histopathologically. The number of lung metastases was counted under microscope as described previously.Citation20,Citation31 In addition, the pulmonary metastases of mice subjected to RFP-expressing HCCLM3-R, HCCLM3-R-ATGi, or HCCLM3-R-Mock cells were visualized using Kodak Multi-modal Imaging System IS4000MM (Carestream, USA). In vivo whole-body imaging and ex vivo imaging of the lung and liver were performed. Images of signals were analyzed with Kodak Molecular Imaging Software.
Metastasis assay via subcutaneous injection
The nude mouse model of pulmonary metastasis via subcutaneous injection was established as previously described.Citation31 Briefly, 1 × 107 HCCLM3 cells were suspended in PBS and subcutaneously inoculated into nude mice. The mice were monitored every day and sacrificed 6 wk later. The lung metastases were analyzed histopathologically as described previously.Citation31
In vitro cell migration and invasion assays
Transwell migration assay
Two × 105 cells were suspended in 200 μl of DMEM with 1% BSA and seeded on top chamber of the transwell (Millicell, PI8P01250). Full medium (900 μl) was added to the bottom chamber. The cells were allowed to migrate for 12 h and then stained with Giemsa stain and counted under microscope.
Scratch assay
Cells were cultured to form a tight cell monolayer and then incubated with 25 µg/ml mitomycin C (Sigma, M4287-2MG) for 3 h. A linear wound was made by scraping a 10-μl pipette tip across the cell monolayer. Cells were routinely maintained for 120 h. The cell motility was quantified as described previously.Citation34
Invasion assay
In vitro matrigel invasion assays were performed as described previously.Citation34
In vitro and in vivo anoikis assays
In vitro anoikis assays
Cells were cultured on either ultra-low attachment plates (Corning, 3471) or plastic plates (Corning, 3516) for 12 to 72 h. After incubation, the suspended and adherent cells were harvested, and apoptosis was measured using ANXA5/annexin V and propidium iodide flow cytometry
In vivo anoikis assays
The nude mouse model of peritoneal dissemination was employed to evaluate anoikis in vivo.Citation18,Citation35 The effect of autophagy inhibition on formation of peritoneal metastasis and ascites as a measure of anoikis were performed. Twelve mice were randomly divided into 2 groups (n = 6 per group); 1 × 107 HCCLM3-ATGi or HCCLM3 cells in 0.5 ml PBS were intraperitoneally injected into peritoneal cavity of each nude mouse. The mice were sacrificed 6 wk later, and the peritoneal metastases were examined by laparotomy.
Lung colonization assays
Eighteen nude mice were randomly divided into 3 groups (n = 6 per group). Five × 105 HCCLM3-ATGi, HCCLM-Mock or HCCLM3 cells were suspended in 0.1 ml PBS and intravenously injected via lateral tail veins of the mice. The mice were sacrificed 4 wk later, and the lung metastases were analyzed histopathologically as described above.
GFP-LC3 analysis of autophagy
A lentiviral vector containing a GFP-LC3 reporter (pLV-Puro-GFP-LC3) was constructed (SunBio). Cells were transfected with lentivirus particles at a multiplicity of infection of 40. The number of GFP-LC3 dots per cell was determined using Top-Hat algorithm of Image-Pro plus 6 (MediaCybernetics) and manual counting.
ER stress test
ER stress was evaluated by examining the protein expression of the ER stress marker HSPA5. The nude mouse model of lung metastasis was established using HCCLM3 cells as described above. Immunohistochemical analysis of HSPA5 expression in lung metastases and paired primary HCC samples was performed using anti-HSPA5 antibody (1:100, Abcam, ab32618) as described.Citation36 The HSPA5 expression was quantitatively analyzed by integrated optical density calculation using computer-assisted digital image analysis (Image-pro plus 6 software). Further, the ER stress blocking test was performed using the ER stress inhibitor PBA (Calbiochem, 567616-100MG) as described.Citation37 Eighteen mice with orthotopic implantation of HCCLM3 cells were divided into 3 groups (n = 6 per group): Blank group (mice received no treatment), PBA group (mice were orally treated with PBA for 6 wk, 1g/kg/d) and Vehicle group (mice were subjected to the same volume of PBA vehicle at the same treatment points). The animals were monitored every day and sacrificed 6 wk later. The lung metastases were analyzed histopathologically as described.Citation20,Citation31
Autophagy-based HCC-tissue-specific target therapy
Construction of AFP-Cre/LoxP-shRNA system
The AFP-Cre/LoxP-shRNA system consisted of AFP-Cre lentiviral vector and LoxP-shRNA lentiviral vector. The AFP-Cre vector contained an AFP promoter followed by Cre gene. The AFP promoter could drive Cre recombinase expression. The LoxP-shRNA vector had a modified U6 promoter followed by shRNA targeting BECN1 or ATG5. The modified U6 promoter harbored a LoxP-CMV-eGFP-LoxP inside. When the AFP-Cre and LoxP-shRNA vectors cotransfected cells, the Cre recombinase cut the LoxP-CMV-eGFP-LoxP inside the U6 promoter and activated it. The activated U6 promoter then drove the expression of the shRNAs and knocked down BECN1 and ATG5. The CMV-eGFP inside the U6 promoter served as a reporter; when CMV-eGFP was cut by the Cre enzyme, the GFP fluorescence would disappear. The effectiveness and tissue specificity of the system were evaluated in AFP-positive HCC cells (HCCLM3/HepG2) and AFP-negative cells (L-02/Hela). Gene silencing and autophagy inhibition were analyzed as mentioned above, and the CMV-eGFP reporter was simultaneously analyzed for consistency with the other two assays.
In vivo evaluation of the AFP-Cre/LoxP-shRNA system: Intravenous injection
Nude mouse model of pulmonary metastasis via orthotopic implantation of HCCLM3 cells was established as described above. One week later, the AFP-Cre/LoxP-shRNA system was intravenously injected via tail vein (LoxP-shRNA group: 1.8 × 108 TU LoxP-shRNA-BECN1 + 1.8 × 108 TU LoxP-shRNA-ATG5 vectors in 180 µl Opti-MEM; Cre+LoxP group: 1.8 × 108 TU AFP-Cre + 0.9 × 108 TU LoxP-shRNA-BECN1 + 0.9 × 108 TU LoxP-shRNA-ATG5 vectors in 180 µl Opti-MEM). The mice were sacrificed 5 wk later.
Intratumoral injection
36 mice were randomly divided into four groups (n = 6 per group), and subcutaneous injection of HCCLM3 cells was performed as described above. When the tumor reached 4 mm in diameter, the AFP-Cre/LoxP-shRNA system was intratumorally injected (OMEM group: 50 µl Opti-MEM (Invitrogen, 31985-070); AFP-Cre group: 1 × 108 TU AFP-Cre vector in 50 µl Opti-MEM; LoxP-shRNA group: 0.5 × 108 TU LoxP-shRNA-BECN1 + 0.5 × 108 TU LoxP-shRNA-ATG5 in 50 µl Opti-MEM; Cre+LoxP group: 0.5 × 108 TU AFP-Cre + 0.25 × 108 TU LoxP-shRNA-BECN1 + 0.25 × 108 TU LoxP-shRNA-ATG5 vectors in 50 µl Opti-MEM). The mice were sacrificed 6 wk later. All of the tumors were resected, and the frozen sections were stained with DAPI (Invitrogen, D3571) and observed under fluorescence microscope. The lung metastases were examined histopathologically as mentioned above.
Statistical analysis
Statistical analysis was performed using SPSS 13.0 software (SPSS). Comparisons of the quantitative data were made using the Student t test or Wilcoxon signed-rank test between 2 groups or by one-way ANOVA for multiple groups. Statistical significance was set at P < 0.05.
| Abbreviations: | ||
| ACTA2 | = | actin, alpha 2, smooth muscle, aorta |
| CDH1 | = | cadherin 1, type 1, E-cadherin (epithelial) |
| ECM | = | extracellular matrix |
| EIF2AK3 | = | eukaryotic translation initiation factor 2-alpha kinase 3 |
| EIF2S1 | = | eukaryotic translation initiation factor 2, subunit 1 alpha, 35 kDa |
| EMT | = | epithelial–mesenchymal transition |
| ER stress | = | endoplasmic reticulum stress |
| FADD | = | Fas (TNFRSF6)-associated via death domain |
| HCC | = | hepatocellular carcinoma |
| HSPA5/GRP78 | = | heat shock, 70 kDa protein 5 (glucose-regulated protein, 78 kDa) |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| MTOR | = | mechanistic target of rapamycin |
| p-MTOR | = | phosphorylated MTOR |
| PBA | = | 4-phenyl butyric acid |
| p-EIF2AK3 | = | phosphorylated EIF2AK3 |
| PRKAA2 | = | protein kinase, AMP-activated, alpha 2 catalytic subunit |
| Q-PCR | = | quantitative real-time polymerase chain reaction |
| SNAI1 | = | snail family zinc finger 1 |
| TWIST1 | = | twist basic helix-loop-helix transcription factor 1 |
| VIM | = | vimentin |
| ZEB1 | = | zinc-finger E-box binding homeobox 1 |
Additional material
Download Zip (371 KB)Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (No. 81030038, 81001060, 81272389, 81001056), National Key Sci-Tech Project (2012ZX10002011-002), China Postdoctoral Science Foundation (No. 20100470639, 201104240), Shanghai Postdoctoral Science Foundation (No. 11R21410300), the Shanghai Morning Light Program (No. 10CG02) and the PhD Programs Foundation of the Ministry of Education of China (No. 20100071120063).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/26398
References
- Ferenci P, Fried M, Labrecque D, Bruix J, Sherman M, Omata M, Heathcote J, Piratsivuth T, Kew M, Otegbayo JA, et al, World Gastroenterology Organization. Hepatocellular carcinoma (HCC): a global perspective. J Clin Gastroenterol 2010; 44:239 - 45; http://dx.doi.org/10.1097/MCG.0b013e3181d46ef2; PMID: 20216082
- Ministry of Health of the People's Republic of China. Report of the Third National Mortality Retrospective Sampling Survey. Peking Union Medical College Press, 2008.
- Tang ZY, Ye SL, Liu YK, Qin LX, Sun HC, Ye QH, Wang L, Zhou J, Qiu SJ, Li Y, et al. A decade’s studies on metastasis of hepatocellular carcinoma. J Cancer Res Clin Oncol 2004; 130:187 - 96; http://dx.doi.org/10.1007/s00432-003-0511-1; PMID: 14685850
- Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 2003; 3:453 - 8; http://dx.doi.org/10.1038/nrc1098; PMID: 12778135
- Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 2006; 12:895 - 904; http://dx.doi.org/10.1038/nm1469; PMID: 16892035
- Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2:563 - 72; http://dx.doi.org/10.1038/nrc865; PMID: 12154349
- Kenific CM, Thorburn A, Debnath J. Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol 2010; 22:241 - 5; http://dx.doi.org/10.1016/j.ceb.2009.10.008; PMID: 19945838
- Moreau K, Luo S, Rubinsztein DC. Cytoprotective roles for autophagy. Curr Opin Cell Biol 2010; 22:206 - 11; http://dx.doi.org/10.1016/j.ceb.2009.12.002; PMID: 20045304
- Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy 2007; 3:28 - 31; PMID: 16969128
- White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 2009; 15:5308 - 16; http://dx.doi.org/10.1158/1078-0432.CCR-07-5023; PMID: 19706824
- Lock R, Debnath J. Extracellular matrix regulation of autophagy. Curr Opin Cell Biol 2008; 20:583 - 8; http://dx.doi.org/10.1016/j.ceb.2008.05.002; PMID: 18573652
- Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann Surg Oncol 2009; 16:761 - 71; http://dx.doi.org/10.1245/s10434-008-0260-0; PMID: 19116755
- Liu D, Yang Y, Liu Q, Wang J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med Oncol 2011; 28:105 - 11; http://dx.doi.org/10.1007/s12032-009-9397-3; PMID: 20041317
- Han J, Hou W, Goldstein LA, Lu C, Stolz DB, Yin XM, Rabinowich H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J Biol Chem 2008; 283:19665 - 77; http://dx.doi.org/10.1074/jbc.M710169200; PMID: 18375389
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326 - 36; http://dx.doi.org/10.1172/JCI28833; PMID: 17235397
- Witz IP. Tumor-microenvironment interactions: dangerous liaisons. Adv Cancer Res 2008; 100:203 - 29; http://dx.doi.org/10.1016/S0065-230X(08)00007-9; PMID: 18620097
- Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol 2009; 3:366 - 75; http://dx.doi.org/10.1016/j.molonc.2009.05.007; PMID: 19559660
- Yawata A, Adachi M, Okuda H, Naishiro Y, Takamura T, Hareyama M, Takayama S, Reed JC, Imai K. Prolonged cell survival enhances peritoneal dissemination of gastric cancer cells. Oncogene 1998; 16:2681 - 6; http://dx.doi.org/10.1038/sj.onc.1201792; PMID: 9632144
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Ke AW, Shi GM, Zhou J, Wu FZ, Ding ZB, Hu MY, Xu Y, Song ZJ, Wang ZJ, Wu JC, et al. Role of overexpression of CD151 and/or c-Met in predicting prognosis of hepatocellular carcinoma. Hepatology 2009; 49:491 - 503; http://dx.doi.org/10.1002/hep.22639; PMID: 19065669
- Shi YH, Ding WX, Zhou J, He JY, Xu Y, Gambotto AA, Rabinowich H, Fan J, Yin XM. Expression of X-linked inhibitor-of-apoptosis protein in hepatocellular carcinoma promotes metastasis and tumor recurrence. Hepatology 2008; 48:497 - 507; http://dx.doi.org/10.1002/hep.22393; PMID: 18666224
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy 2007; 3:181 - 206; PMID: 17224625
- Ito S, Koshikawa N, Mochizuki S, Takenaga K. 3-Methyladenine suppresses cell migration and invasion of HT1080 fibrosarcoma cells through inhibiting phosphoinositide 3-kinases independently of autophagy inhibition. Int J Oncol 2007; 31:261 - 8; PMID: 17611681
- Yang Z, Lei Z, Li B, Zhou Y, Zhang GM, Feng ZH, Zhang B, Shen GX, Huang B. Rapamycin inhibits lung metastasis of B16 melanoma cells through down-regulating alphav integrin expression and up-regulating apoptosis signaling. Cancer Sci 2010; 101:494 - 500; http://dx.doi.org/10.1111/j.1349-7006.2009.01412.x; PMID: 19922502
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19:797 - 806; http://dx.doi.org/10.1091/mbc.E07-10-1092; PMID: 18094039
- Debnath J. Detachment-induced autophagy during anoikis and lumen formation in epithelial acini. Autophagy 2008; 4:351 - 3; PMID: 18196957
- Ko H, Kim YJ, Park JS, Park JH, Yang HO. Autophagy inhibition enhances apoptosis induced by ginsenoside Rk1 in hepatocellular carcinoma cells. Biosci Biotechnol Biochem 2009; 73:2183 - 9; http://dx.doi.org/10.1271/bbb.90250; PMID: 19809182
- Longo L, Platini F, Scardino A, Alabiso O, Vasapollo G, Tessitore L. Autophagy inhibition enhances anthocyanin-induced apoptosis in hepatocellular carcinoma. Mol Cancer Ther 2008; 7:2476 - 85; http://dx.doi.org/10.1158/1535-7163.MCT-08-0361; PMID: 18723493
- Claerhout S, Verschooten L, Van Kelst S, De Vos R, Proby C, Agostinis P, Garmyn M. Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma. Int J Cancer 2010; 127:2790 - 803; http://dx.doi.org/10.1002/ijc.25300; PMID: 21351258
- Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW, Wang XY, Dai Z, Peng YF, Gu CY, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011; 7:1159 - 72; http://dx.doi.org/10.4161/auto.7.10.16818; PMID: 21691147
- Li Y, Tang Y, Ye L, Liu B, Liu K, Chen J, Xue Q. Establishment of a hepatocellular carcinoma cell line with unique metastatic characteristics through in vivo selection and screening for metastasis-related genes through cDNA microarray. J Cancer Res Clin Oncol 2003; 129:43 - 51; PMID: 12618900
- Yang BW, Liang Y, Xia JL, Sun HC, Wang L, Zhang JB, Tang ZY, Liu KD, Chen J, Xue Q, et al. Biological characteristics of fluorescent protein-expressing human hepatocellular carcinoma xenograft model in nude mice. Eur J Gastroenterol Hepatol 2008; 20:1077 - 84; http://dx.doi.org/10.1097/MEG.0b013e3283050a67; PMID: 19047839
- Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, Shi GM, Wang XY, Ke AW, Wu B, et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res 2008; 68:9167 - 75; http://dx.doi.org/10.1158/0008-5472.CAN-08-1573; PMID: 19010888
- Ding ZB, Shi YH, Zhou J, Shi GM, Ke AW, Qiu SJ, Wang XY, Dai Z, Xu Y, Fan J. Liver-intestine cadherin predicts microvascular invasion and poor prognosis of hepatitis B virus-positive hepatocellular carcinoma. Cancer 2009; 115:4753 - 65; http://dx.doi.org/10.1002/cncr.24513; PMID: 19626651
- Sood AK, Armaiz-Pena GN, Halder J, Nick AM, Stone RL, Hu W, Carroll AR, Spannuth WA, Deavers MT, Allen JK, et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J Clin Invest 2010; 120:1515 - 23; http://dx.doi.org/10.1172/JCI40802; PMID: 20389021
- Giatromanolaki A, Koukourakis MI, Harris AL, Polychronidis A, Gatter KC, Sivridis E. Prognostic relevance of light chain 3 (LC3A) autophagy patterns in colorectal adenocarcinomas. J Clin Pathol 2010; 63:867 - 72; http://dx.doi.org/10.1136/jcp.2010.079525; PMID: 20876316
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG Jr., Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 2009; 9:35 - 51; http://dx.doi.org/10.1016/j.cmet.2008.12.004; PMID: 19117545