
Abstract
Degradation of signaling proteins is one of the most powerful tumor-suppressive mechanisms by which a cell can control its own growth, its survival, and its motility. Emerging evidence suggests that autophagy limits several signaling pathways by degrading kinases, downstream components, and transcription factors; however, this often occurs under stressful conditions. Our recent studies revealed that constitutive autophagy temporally and spatially controls the RHOA pathway. Specifically, inhibition of autophagosome degradation induces the accumulation of the GTP-bound form of RHOA. The active RHOA is sequestered via SQSTM1/p62 within autolysosomes, and accordingly fails to localize to the spindle midbody or to the cell surface, as we demonstrate herein. As a result, all RHOA-downstream responses are deregulated, thus driving cytokinesis failure, aneuploidy and motility, three processes that directly have an impact upon cancer progression. We therefore propose that autophagy acts as a degradative brake for RHOA signaling and thereby controls cell proliferation, migration, and genome stability.
Some components of the autophagic machinery are commonly mutated or downregulated in human cancers.Citation1,Citation2 Despite a huge amount of work, the roles of autophagy in cancer still remain controversial; it may act either as a tumor-suppressive or a tumor-promoting pathway. The current consensus is that the autophagic process initially acts to prevent tumor initiation. As a safeguard mechanism against cancer, it has been proposed that constitutive autophagy (i.e., under physiological nutrient conditions) can dispose of old and damaged organelles, such as mitochondria, which would otherwise cause genotoxic stress resulting in DNA mutations. Autophagy may also promote cellular senescence in response to oncogenic signaling, thus inhibiting cellular transformation. Moreover, autophagy can prevent inflammation, a trigger of tumorigenesis.Citation1 Conversely, once the cancer is established, autophagy is dramatically induced to meet the needs of cancer cells. As a tumor promoter, autophagy may sustain cell survival and tumor dormancy in response to hypoxia and nutrient limitation in the inner area of the tumor.Citation1 Later, autophagy enables cancer cells to survive anoikis during metastasisCitation3,Citation4 and to evade cell death during chemotherapy. As a result, modulation of autophagy in cancer therapy can be a “double-edged sword,” promoting or preventing the cytotoxic effect of anticancer drugs.Citation5
Even though the tumor-suppression function of autophagy was first described in 2003,Citation6,Citation7 its precise role in tumorigenesis is not understood. Key observations of Eileen White et al. described 5 years ago the role of autophagy in genomic stability,Citation8,Citation9 but the underlying mechanisms still remain unknown. This issue is critical given the importance of genomic integrity in cell homeostasis, and the correlation between defects in genomic integrity with tumorigenesis.Citation10 Foremost among the signaling players that influence genome stability is the small GTPase RHOA. Decades of studying small GTPases have led to the concept that RHOA must be tightly controlled by activating (a guanine nucleotide exchange factor, GEF) and inhibiting (a GTPase activating protein) factors. Of particular interest, recent accumulating evidence suggests that the levels of inactive RHOA are also controlled by proteasomal degradation,Citation11-Citation16 but whether RHOA-GTP is similarly degraded remains, so far, elusive.
Instead of the proteasome, however, we recently demonstrated that active RHOA is constitutively maintained at low levels by autophagy.Citation17 Indeed, through targeted manipulation of the autophagy pathway (such as Atg5, Atg7, Sqstm1, and Tcirg1 [v-ATPase a3] knockdowns, gene deletion, and use of chemical inhibitors), we established that autophagy is the most important mechanism for restricted RHOA activation at the midbody during cytokinesis. Inhibition of autophagosome degradation (by the loss of the TCIRG1 subunit) allowed us to demonstrate the accumulation of RHOA-GTP within autolysosomes, close to the midbody of cells undergoing cytokinesis. At the molecular level, we identified SQSTM1 as the molecular adaptor that targets the active and ubiquitinated RHOA to autophagosomes. As a result, a failure in autophagy (irrespective of the studied defects: formation, sequestration, or degradation) deregulates all RHOA downstream responses, driving cytokinesis failure, and aneuploidy, one hallmark of aggressive cancer. Importantly, the control of RHOA by autophagy is remarkably specific as the closely related GTPase RAC as well as the upstream regulators required for proper activation of RHOA such as the kinesin KIF23/MKLP1, the RHOA GEF ECT2, and ARHGDIA/RHOGDI are not affected.Citation17
Considering the apparent connection between autophagy and RHOA, a key issue is how defects in autophagy might affect cell migration, another RHOA-controlled cell response relevant for cancer progression. Remarkably, the v-ATPase TCIRG1-dependent autophagy defect was characterized by an increase in cell size, a dramatic remodeling of the ACTIN cytoskeleton with the loss of stress fibers, and the formation of ACTIN-rich lamellipodia (). Correlated with the mesenchymal spreading, another striking hallmark of Tcirg1-null cells was a punctate staining of RHOA within autophagic vesicles, just under the lamella of crawling Tcirg1-null cells (, left panel), one region where RHOA inactivation is essential in cell migration.Citation18,Citation19 We thus followed the wild-type (WT) and Tcirg1-null cells by time-lapse microscopy and observed that the Tcirg1-null cells migrated 7 times as fast as the WT cells (; Vid. S1). A role for autophagy in controlling the localization of RHOA-GTP was then demonstrated by the shRNA-mediated inhibition of autophagosome formation: expression of ATG5 shRNA released active RHOA at the plasma membrane of Tcirg1-null cells, which allowed ACTIN polymerization into filaments, and impaired formation of cell protrusions ().Citation17 In light of these findings, we propose that autophagy might be absolutely necessary for maintaining the appropriate amount of RHOA at the lamellipodia to allow cell motility ().
Figure 1. Cell motility is induced by v-ATPase Tcirg1 loss. (A) Confocal images of v-ATPase Tcirg1-null cells showing colocalization of RHOA with autophagosomes (LC3-positive, arrowhead) and autolysosomes (LC3- and LAMP1-positive, arrows), close to highly dynamic lamellipodia (right). As a result of the apparent RHOA sequestration, Tcirg1-null cells lacked ACTIN stress fibers and developed aberrant lamellipodia (arrowhead, left). (B) Time-lapse video microscopy (right) and XY migration tracks (left) showing that the Tcirg1-null cells established short-lived cellular contacts, and migrated rapidly at 15.2 ± 3.9 µm/h over long distances (still-images from Vid. S1; intervals in h:min). Arrows indicate the direction of cell movement. Inset: representative ACTIN (green) and microtubule (red) staining showing that Tcirg1-null cells displayed a classical front-rear polarized morphology, with membrane protrusion at the leading edge (arrowhead). (C) Left. Representative confocal images showing in WT cells the presence of RHOA at the plasma membrane (inset, arrowhead), and the formation of F-ACTIN (arrowhead). Time-lapse video microscopy (middle, images selected from Vid. S1; intervals in h:min) and XY migration tracks (left, cell positions were recorded every 60 min for 7 h) showing that WT cells moved slowly at 2 ± 1 µm/h. (D) Inhibition of autophagosome formation by Atg5 shRNA dramatically increases localization of active RHOA at the plasma membrane of Tcirg1-null cells, which restored a normal small morphology and the formation of actin fibers (arrowhead).
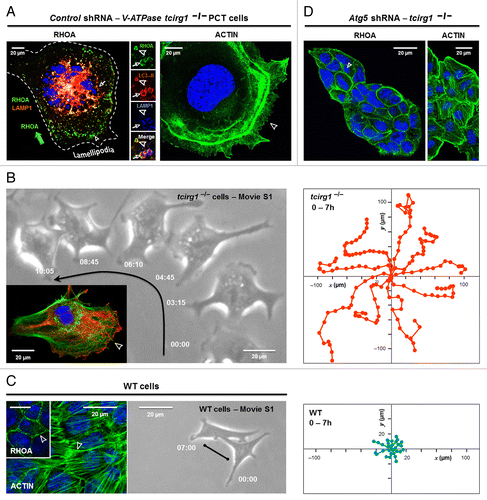
Figure 2. Autophagy is required for tumor A549 cell migration through the degradation of RHOA. (A–F) Consequences of autophagy defects on the RHOA pathway in tumor A549 cells. Depletion of ATG5 (B and E) and SQSTM1 (C and F) in A549 cells led to increased RHOA levels at the plasma membrane (A–C; left), a denser F-ACTIN reticulation (A–C; right) and a strong suppression of tumor cell migration (examined by time-lapse video microscopy [D–F] and wound closure assay [see also Fig. S1]). Shown are confocal images of A549 cells cotransfected with human shRNA and expressing proteins encoded by shRNA-resistant murine autophagy genes (red). (E and F) The migratory defects of ATG5-depleted A549 cells (middle) and SQSTM1-depleted A549 cells (right) were rescued upon RHO kinase inhibition with a low concentration of Y27632 (2.5 µM) that did not impede A549 tumor cell migration and only compromised rear retraction (D, lower panel). (See also Vids. S2–S6). (G) Proposed model by which autophagy is essential for the control of the RHOA pathway and the subsequent cell migration; once activated, the sequestration of the active RHOA within autophagic vesicles and the following degradation would limit the level of active RHOA present at the plasma membrane, thereby relieving inhibitory RHOA signaling. As a result, alterations in the autophagy pathway can have a profound impact upon cell motility: inhibition of autophagy degradation would promote rapid cell migration through the increased RHOA sequestration (TCIRG1 loss), whereas blocking autophagosome formation or sequestration would have the opposite effects (ATG5, ATG7, SQSTM1 shRNA). (H) Growing list of signaling proteins degraded by autophagy.
![Figure 2. Autophagy is required for tumor A549 cell migration through the degradation of RHOA. (A–F) Consequences of autophagy defects on the RHOA pathway in tumor A549 cells. Depletion of ATG5 (B and E) and SQSTM1 (C and F) in A549 cells led to increased RHOA levels at the plasma membrane (A–C; left), a denser F-ACTIN reticulation (A–C; right) and a strong suppression of tumor cell migration (examined by time-lapse video microscopy [D–F] and wound closure assay [see also Fig. S1]). Shown are confocal images of A549 cells cotransfected with human shRNA and expressing proteins encoded by shRNA-resistant murine autophagy genes (red). (E and F) The migratory defects of ATG5-depleted A549 cells (middle) and SQSTM1-depleted A549 cells (right) were rescued upon RHO kinase inhibition with a low concentration of Y27632 (2.5 µM) that did not impede A549 tumor cell migration and only compromised rear retraction (D, lower panel). (See also Vids. S2–S6). (G) Proposed model by which autophagy is essential for the control of the RHOA pathway and the subsequent cell migration; once activated, the sequestration of the active RHOA within autophagic vesicles and the following degradation would limit the level of active RHOA present at the plasma membrane, thereby relieving inhibitory RHOA signaling. As a result, alterations in the autophagy pathway can have a profound impact upon cell motility: inhibition of autophagy degradation would promote rapid cell migration through the increased RHOA sequestration (TCIRG1 loss), whereas blocking autophagosome formation or sequestration would have the opposite effects (ATG5, ATG7, SQSTM1 shRNA). (H) Growing list of signaling proteins degraded by autophagy.](/cms/asset/4cb5880a-6802-44e1-b836-616951a6cfcf/kaup_a_10927198_f0002.gif)
If our model is correct, one would expect that the inhibition of autophagy sequestration in autophagy-competent A549 tumor cells would be sufficient to impair cell motility (). We found that depletion of ATG5 or SQSTM1 (Fig. S1A) recapitulated the accumulation of RHOA at the cell surface (, left panels), higher levels of downstream phosphorylation of myosin regulatory light chain (P-MLC, Fig. S1B), a denser ACTIN network (; Fig. S1C) and consistently suppressed motility (; Fig. S1D; Vids. S2–S6), in comparison with control shRNA-transduced cells (). As expected, the inhibition of autophagosome formation by ATG7 depletion produced similar effects (Fig. S1). Reintroduction of ATG5 or SQSTM1 (rescue) back into these cells cancelled the stabilization of RHOA signaling as reflected by the loss of F-ACTIN, and the promotion of lamellipodia (). In keeping with this scenario, inhibiting RHO kinase by Y27632 treatment promoted cell spreading, formation of broad lamellipodia, and cell migration of ATG5-depleted cells and SQSTM1-depleted cells, showing that RHOA overactivation was directly responsible for the phenotype (; Vids. S3–S6). These data together provide support that SQSTM1-dependent autophagy is required for fine-tuning the RHOA pathway to ensure cell motility.
Recent compelling evidence suggests that autophagy negatively regulates several signaling pathways by degrading kinases,Citation20-Citation27 key downstream componentsCitation28,Citation29 and transcription factors ().Citation30-Citation35 However, this often occurs under stressful conditions such as nutrient starvation, extracellular matrix (ECM) detachment, HSP90 inhibition, proteasome inhibition, infections, and treatment with chemotherapy drugs.Citation20,Citation23-Citation27,Citation29,Citation31-Citation33,Citation35 Therefore, the ability of autophagy to degrade signaling proteins under basal growth conditions and thereby to ensure tumor-suppressive functions remains to be established.
Our data raise the importance of constitutive autophagy as a master regulator of RHOA-GTP. The regulation of RHOA is unique in that it involves GEFs and GTPase-activating proteins, along with the proteasome and autophagy (). Depending on its activation state, we provide the first lines of evidence that RHOA uses distinct routes for degradation: while the proteasome degrades the cytosolic and inactive forms, the autophagy pathway specifically degrades the membrane-associated and active pool of RHOA.Citation17 This is consistent with the recently reported degradation of two constitutively active RHO proteins, RHOH and RHOB, within lysosomesCitation36,Citation37 and the redistribution of the active RHOA to undefined intracellular puncta during cell migration,Citation19 or after activation with an autophagy inducer, lipopolysaccharide (LPS).Citation38 We therefore propose that basal autophagy acts as a tumor suppressor pathway that constitutively turns off RHOA activation (). In this model, the remarkable dynamics of autophagy together with its integration of extracellular cues might dictate the time and place where a RHO GTPase is active, and able to interact with its downstream substrates. Accordingly, we reported the targeting of autolysosomes at the leading edge of migrating cells, the same subcellular and temporal localizations where RHOA should be inactivated.
So far, the emerging mechanisms by which autophagy limits cell signaling involve both ubiquitination and SQSTM1 ().Citation27-Citation30,Citation32,Citation33,Citation35 Importantly, upon inhibition of autophagy by chloroquine treatment, we identified that RHOA is ubiquitinated and recognized by the autophagy receptor SQSTM1.Citation17 We therefore propose that RHOA, once activated, undergoes ubiquitination, recruiting the scaffold protein SQSTM1 that bridges RHOA to the autophagosome marker LC3 for selective autophagic targeting. In line with this concept, 2 recent connections suggest the existence of feedback loops between RHOA and the autophagic pathway in which LC3 dampens RHOA activation by inactivating the RHO-GEF activity of AKAP13/AKAP-Lbc,Citation39,Citation40 while active RHOA triggers the degradation of BECN1, a critical mediator of autophagy.Citation41 Together, this sophisticated crosstalk between autophagy and RHOA might commit cells to irreversible cell cycle exit and directed migration.
Future work is required to understand how, mechanistically, a plasma membrane protein is targeted to autophagy degradation. There are a number of ways, non-mutually exclusive, in which plasma membrane proteins can be targeted for degradation, the most common of which involves endocytosis followed by lysosomal degradation. Remarkably, autophagy was recently involved in the degradation of 2 internalized plasma membrane proteins, GABRA/GABAA receptors, and connexins.Citation42-Citation44 It is thus possible that active RHOA present at the plasma membrane are internalized into endosomes that fuse with autophagosomes. Another possibility is the recruitment of autophagy machinery (ATG5, LC3, and ATG7) to the RHOA-containing endosomes, leading to rapid trafficking and fusion with the lysosomes, as recently suggested.Citation45 At odds with endocytosis hypothesis, an intriguing finding of our study is the higher “retention” of RHOA at the plasma membrane of ATG5-depleted cells and SQSTM1-depleted cells (both in PCT cells and A549 cells), instead of an intracellular accumulation. In accordance, there is already evidence that the blockage of autophagy induces accumulation of GJA1 (connexin 43) at the plasma membrane by preventing its internalization (3-methyladenine, Atg7- and Atg5-null cells).Citation44 Similarly to RHOA, ubiquitination and SQSTM1 are also signals that target connexins for selective autophagy.Citation43,Citation44 Other lines of evidence are the observations that autophagy stimuli increase endocytosis, that plasma membrane participates in the formation of phagophores, and that some components of the autophagy machinery (at least ATG16L1) associate with the plasma membrane.Citation46 Altogether this might suggest that SQSTM1-dependent autophagy orchestrates endocytosis and degradation of signaling proteins. Understanding how autophagy and endocytosis are coordinated to allow timely degradation of signaling proteins will be our next challenge.
In conclusion, this unexpected link between RHOA and autophagy helps us to revisit under a new light the temporal and spatial control of RHOA,Citation18,Citation19,Citation47 the signaling and tumorigenic roles of SQSTM1,Citation48-Citation50 and the tumor-suppressive function of autophagy. Our findings also raise the possibility that inappropriate activation of autophagy in cancer cells can contribute to their rapid migration, by enhancing the degradation of RHOA. Of particular interest, when silencing autophagy, the A549 tumor cells failed to migrate, an observation that suggests important therapeutic implications. Metastasis is responsible for more than 90% of cancer deaths; interestingly, all cancers (melanoma, lung, pancreatic, and colorectal cancers) prone to metastasize are characterized by an “enhanced” autophagy, i.e. overexpression of SQSTM1 and punctate staining of LC3, in agreement with our study.Citation51-Citation57 Based on our result, we propose that inhibiting autophagy initiation might be useful for treatment of metastatic cancer. Given that inhibition of autolysosomal acidification/degradation might enhance cell migration (our study and refs. Citation58–Citation60), the therapeutic benefit of drugs such as chloroquine warrants careful investigation.
Materials and Methods
Cell culture and treatments
To inhibit the maturation of autophagosomes into degradative autolysosomes, renal cells derived from proximal convoluted tubules (PCT) of wild-type (WT, Tcirg1+/+) or tcirg1−/− (lysosomal v-ATPase a3-deficient, Jackson Laboratory) mice were isolated and immortalized with the pSV3 neo vector, as described previously.Citation17 PCT cells were chosen, as they express the highest level of v-ATPase,Citation61 the a3 subunit is localized in the lysosomal limiting membrane.Citation62 In agreement with the other reported defect of v-ATPase (mutations in PS1),Citation63 V-ATPase Tcirg1 depletion raises the lysosomal pH and thereby blocks autolysosomal degradation downstream of fusion, as reflected by the accumulation of the autolysosomes and/or autophagy substrates LC3-II, SQSTM1 and RHOA-GTP,Citation17 according to.Citation64 Likewise, we have checked that Tcirg1 depletion does not affect the activity of proteasomes, which degrade several Rho GTPases, their upstream regulators, and downstream targets.Citation17 Alternatively, we prevented autophagosome formation in the human lung cancer A549 cell line by ATG5 shRNA (Sigma, SHCLNV-NM_004849) or ATG7 shRNA (Sigma, SHCLNV-NM_NM_006395) transduction, and the recruitment of autophagy substrates within the autophagic vesicles by SQSTM1, through shRNA treatment (Sigma, SHCLNV-NM_NM_003900).Citation17 ShRNA-mediated protein downregulation and autophagy impairment were monitored by immunoblotting (ATG5, clone 7C6, Nanotools; ATG7, clone D12B11, Cell Signaling Technology Inc.; LC3-II, Nanotools clone 5F10 and SQSTM1, BD Transduction Laboratories™, 610833).
Analysis of the RHOA pathway
The activity of the RHOA pathway was monitored by i) the recruitment of RHOA to membranes; ii) the downstream Ser19-phosphorylation of myosin regulatory light chain (P-MLC, Cell Signaling Technology, western blotting; 3671); iii) the reticulation of ACTIN cytoskeleton; and iv) cell migration. Where indicated, a low dose of the selective RHO Kinase inhibitor Y27632 (2.5 μM; Calbiochem, 688002) was added.
Immunofluorescence staining
To determine whether RHOA is membrane-associated and thus activated, cells seeded on glass coverslips were fixed with ice-cold 10% TCA for 15 min, a method that preserves membrane-associated, but not cytoplasmic, RHOA.Citation65 The cells were then washed in PBS, and permeabilized in 0.3% Triton X-100 in PBS for 5 min. To detect the translocation of RHOA to autolysosomes (LC3+ and LAMP1+), cells were then incubated with anti-RHOA (clone 26C4; 1:100; Santa Cruz Biotechnology), anti-LC3 (1:500, clone 5F10, Nanotools) and anti-LAMP1 (1:500, BD PharMingen, 553792) antibody labeling. Alternatively, cells were fixed in PBS with 3.7% paraformaldehyde for 15 min at room temperature before staining of the ACTIN cytoskeleton with TRITC-phalloidin (1 h; room temperature; 1:1,000; Sigma, P1951). Slides were mounted with Pro Long Gold antifade reagent (Molecular Probes, P36931) with DAPI after 3 washes with PBS, and examined using a confocal microscope (Zeiss LSM5; Carl Zeiss).
Time-lapse video microscopy
For monitoring single-cell migration, exponentially growing cells cultured in complete growth medium were imaged on an inverted microscope (Axio Observer.Z1; Carl Zeiss) equipped with a CO2-equilibrated chamber (37 °C and 5% CO2) and a motorized X-Y stage. Three different fields per condition were photographed and a total of 18 fields were followed in 3 independent experiments. Images were captured every 5 min with a × 20 ph2 (Tcirg1-null) and × 32 ph1 (WT) phase-contrast objective during 18 h using MetaMorph 2.0 software (Molecular Devices). Videos were generated from the acquired images with ImageJ.
Scratch wound assay
Confluent cell monolayers were scratched with a pipet tip to create a uniform wound area. Cells were washed to remove debris, and fresh complete medium was then applied. Three random images of wounds were captured immediately after wounding and 48 h later, migration was quantified as the percentage of the wound area that was closed using Visilog 5.3 software®. The values are means of 3 scratches from 2 independent clones (n = 6) ± s.d.
| Abbreviations: | ||
| Atg | = | autophagy-related |
| GEF | = | guanine nucleotide exchange factor |
| LAMP | = | lysosomal-associated membrane protein |
| MAP1LC3 (LC3) | = | microtubule-associated protein 1 light chain 3 |
| shRNA | = | short hairpin RNA |
| SQSTM1/p62 | = | sequestosome 1 |
| TCIRG1/a3 | = | T-cell immune regulator 1, ATPase, H+ transporting, lysosomal V0 subunit A3 |
| v-ATPase | = | vacuolar-ATPase |
| WT | = | wild type |
Additional material
Download Zip (19.3 MB)Acknowledgments
We thank Nathalie Rochet, Nathalie Singer, Etienne Boulter, and Jean Albrengues for helpful discussions. Grant support: “Institut National de la Santé et de la Recherche Médicale,” “Agence de l'Environnement et de la Maîtrise de l'Energie” (A B and A C: convention ADEME n° 08 62 C 0044), Agence régionale santé Provence Alpes Côte d'Azur and Direction régionale de l’Environnement, de l’aménagement et du logement Provence Alpes Côte d’Azur (A B: plan régional santé environnement PRSE PACA n°6.3.3.3 and 6.3.3.4), “Association pour la Recherche contre le Cancer” (ARC Grants n° SL220110603478), French national research agency (“Investments for the Future” LABEX SIGNALIFE # ANR-11-LABX-0028-01, and “STEATOX” # ANR-13-CESA-0009-01) and NIH (DJK: GM053396).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961 - 7; http://dx.doi.org/10.1038/nrc2254; PMID: 17972889
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Lock R, Debnath J. Extracellular matrix regulation of autophagy. Curr Opin Cell Biol 2008; 20:583 - 8; http://dx.doi.org/10.1016/j.ceb.2008.05.002; PMID: 18573652
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19:797 - 806; http://dx.doi.org/10.1091/mbc.E07-10-1092; PMID: 18094039
- Livesey KM, Tang D, Zeh HJ, Lotze MT. Autophagy inhibition in combination cancer treatment. Curr Opin Investig Drugs 2009; 10:1269 - 79; PMID: 19943199
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809 - 20; PMID: 14638851
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003; 100:15077 - 82; http://dx.doi.org/10.1073/pnas.2436255100; PMID: 14657337
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21:1621 - 35; http://dx.doi.org/10.1101/gad.1565707; PMID: 17606641
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21:1367 - 81; http://dx.doi.org/10.1101/gad.1545107; PMID: 17510285
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005; 437:1043 - 7; http://dx.doi.org/10.1038/nature04217; PMID: 16222300
- Doye A, Mettouchi A, Bossis G, Clément R, Buisson-Touati C, Flatau G, Gagnoux L, Piechaczyk M, Boquet P, Lemichez E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002; 111:553 - 64; http://dx.doi.org/10.1016/S0092-8674(02)01132-7; PMID: 12437928
- Wang HR, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, Wrana JL. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 2003; 302:1775 - 9; http://dx.doi.org/10.1126/science.1090772; PMID: 14657501
- Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 2005; 307:1603 - 9; http://dx.doi.org/10.1126/science.1105718; PMID: 15761148
- Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol 2006; 8:485 - 91; http://dx.doi.org/10.1038/ncb1400; PMID: 16622418
- Chen Y, Yang Z, Meng M, Zhao Y, Dong N, Yan H, Liu L, Ding M, Peng HB, Shao F. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol Cell 2009; 35:841 - 55; http://dx.doi.org/10.1016/j.molcel.2009.09.004; PMID: 19782033
- Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, Burridge K. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol 2010; 12:477 - 83; http://dx.doi.org/10.1038/ncb2049; PMID: 20400958
- Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano S, Ilie M, Rubera I, Tauc M, Barale S, et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res 2013; 73:4311 - 22; http://dx.doi.org/10.1158/0008-5472.CAN-12-4142; PMID: 23704209
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature 2009; 461:99 - 103; http://dx.doi.org/10.1038/nature08242; PMID: 19693013
- Heasman SJ, Carlin LM, Cox S, Ng T, Ridley AJ. Coordinated RhoA signaling at the leading edge and uropod is required for T cell transendothelial migration. J Cell Biol 2010; 190:553 - 63; http://dx.doi.org/10.1083/jcb.201002067; PMID: 20733052
- Qing G, Yan P, Xiao G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK). Cell Res 2006; 16:895 - 901; http://dx.doi.org/10.1038/sj.cr.7310109; PMID: 17088896
- Niida M, Tanaka M, Kamitani T. Downregulation of active IKK beta by Ro52-mediated autophagy. Mol Immunol 2010; 47:2378 - 87; http://dx.doi.org/10.1016/j.molimm.2010.05.004; PMID: 20627395
- Kim JE, You DJ, Lee C, Ahn C, Seong JY, Hwang JI. Suppression of NF-kappaB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation. Cell Signal 2010; 22:1645 - 54; http://dx.doi.org/10.1016/j.cellsig.2010.06.004; PMID: 20600852
- Fliss PM, Jowers TP, Brinkmann MM, Holstermann B, Mack C, Dickinson P, Hohenberg H, Ghazal P, Brune W. Viral mediated redirection of NEMO/IKKγ to autophagosomes curtails the inflammatory cascade. PLoS Pathog 2012; 8:e1002517; http://dx.doi.org/10.1371/journal.ppat.1002517; PMID: 22319449
- Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K, Stevens C, Brunton VG, Langdon WY, Vidal M, et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol 2012; 14:51 - 60; http://dx.doi.org/10.1038/ncb2386; PMID: 22138575
- Sandilands E, Serrels B, Wilkinson S, Frame MC. Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells. EMBO Rep 2012; 13:733 - 40; http://dx.doi.org/10.1038/embor.2012.92; PMID: 22732841
- Hsueh YS, Yen CC, Shih NY, Chiang NJ, Li CF, Chen LT. Autophagy is involved in endogenous and NVP-AUY922-induced KIT degradation in gastrointestinal stromal tumors. Autophagy 2013; 9:220 - 33; http://dx.doi.org/10.4161/auto.22802; PMID: 23196876
- Goussetis DJ, Gounaris E, Wu EJ, Vakana E, Sharma B, Bogyo M, Altman JK, Platanias LC. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012; 120:3555 - 62; http://dx.doi.org/10.1182/blood-2012-01-402578; PMID: 22898604
- Paul S, Kashyap AK, Jia W, He YW, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-κB. Immunity 2012; 36:947 - 58; http://dx.doi.org/10.1016/j.immuni.2012.04.008; PMID: 22658522
- Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T, Fu W, Zhang J, Wu W, Zhang X, et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol 2010; 12:781 - 90; http://dx.doi.org/10.1038/ncb2082; PMID: 20639871
- Chang CP, Su YC, Hu CW, Lei HY. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ 2013; 20:515 - 23; http://dx.doi.org/10.1038/cdd.2012.146; PMID: 23175187
- Sukhdeo K, Mani M, Hideshima T, Takada K, Pena-Cruz V, Mendez G, Ito S, Anderson KC, Carrasco DR. β-catenin is dynamically stored and cleared in multiple myeloma by the proteasome-aggresome-autophagosome-lysosome pathway. Leukemia 2012; 26:1116 - 9; http://dx.doi.org/10.1038/leu.2011.303; PMID: 22051532
- Isakson P, Bjørås M, Bøe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010; 116:2324 - 31; http://dx.doi.org/10.1182/blood-2010-01-261040; PMID: 20574048
- Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARα oncoprotein. Autophagy 2011; 7:401 - 11; http://dx.doi.org/10.4161/auto.7.4.14397; PMID: 21187718
- Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H, Yan J, Lv X, Chen X, Hu ZW. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res 2012; 72:3238 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-11-3832; PMID: 22719072
- Jia L, Gopinathan G, Sukumar JT, Gribben JG. Blocking autophagy prevents bortezomib-induced NF-κB activation by reducing I-κBα degradation in lymphoma cells. PLoS One 2012; 7:e32584; http://dx.doi.org/10.1371/journal.pone.0032584; PMID: 22393418
- Pérez-Sala D, Boya P, Ramos I, Herrera M, Stamatakis K. The C-terminal sequence of RhoB directs protein degradation through an endo-lysosomal pathway. PLoS One 2009; 4:e8117; http://dx.doi.org/10.1371/journal.pone.0008117; PMID: 19956591
- Schmidt-Mende J, Geering B, Yousefi S, Simon HU. Lysosomal degradation of RhoH protein upon antigen receptor activation in T but not B cells. Eur J Immunol 2010; 40:525 - 9; http://dx.doi.org/10.1002/eji.200939556; PMID: 19950172
- Cetin S, Ford HR, Sysko LR, Agarwal C, Wang J, Neal MD, Baty C, Apodaca G, Hackam DJ. Endotoxin inhibits intestinal epithelial restitution through activation of Rho-GTPase and increased focal adhesions. J Biol Chem 2004; 279:24592 - 600; http://dx.doi.org/10.1074/jbc.M313620200; PMID: 15169791
- Baisamy L, Cavin S, Jurisch N, Diviani D. The ubiquitin-like protein LC3 regulates the Rho-GEF activity of AKAP-Lbc. J Biol Chem 2009; 284:28232 - 42; http://dx.doi.org/10.1074/jbc.M109.054668; PMID: 19696020
- Bauer PO, Wong HK, Oyama F, Goswami A, Okuno M, Kino Y, Miyazaki H, Nukina N. Inhibition of Rho kinases enhances the degradation of mutant huntingtin. J Biol Chem 2009; 284:13153 - 64; http://dx.doi.org/10.1074/jbc.M809229200; PMID: 19278999
- Yoo BH, Wu X, Li Y, Haniff M, Sasazuki T, Shirasawa S, Eskelinen EL, Rosen KV. Oncogenic ras-induced down-regulation of autophagy mediator Beclin-1 is required for malignant transformation of intestinal epithelial cells. J Biol Chem 2010; 285:5438 - 49; http://dx.doi.org/10.1074/jbc.M109.046789; PMID: 19778902
- Rowland AM, Richmond JE, Olsen JG, Hall DH, Bamber BA. Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J Neurosci 2006; 26:1711 - 20; http://dx.doi.org/10.1523/JNEUROSCI.2279-05.2006; PMID: 16467519
- Fong JT, Kells RM, Gumpert AM, Marzillier JY, Davidson MW, Falk MM. Internalized gap junctions are degraded by autophagy. Autophagy 2012; 8:794 - 811; http://dx.doi.org/10.4161/auto.19390; PMID: 22635056
- Bejarano E, Girao H, Yuste A, Patel B, Marques C, Spray DC, Pereira P, Cuervo AM. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol Biol Cell 2012; 23:2156 - 69; http://dx.doi.org/10.1091/mbc.E11-10-0844; PMID: 22496425
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450:1253 - 7; http://dx.doi.org/10.1038/nature06421; PMID: 18097414
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010; 12:747 - 57; http://dx.doi.org/10.1038/ncb2078; PMID: 20639872
- Miller AL, Bement WM. Regulation of cytokinesis by Rho GTPase flux. Nat Cell Biol 2009; 11:71 - 7; http://dx.doi.org/10.1038/ncb1814; PMID: 19060892
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008; 13:343 - 54; http://dx.doi.org/10.1016/j.ccr.2008.02.001; PMID: 18394557
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062 - 75; http://dx.doi.org/10.1016/j.cell.2009.03.048; PMID: 19524509
- Kadandale P, Stender JD, Glass CK, Kiger AA. Conserved role for autophagy in Rho1-mediated cortical remodeling and blood cell recruitment. Proc Natl Acad Sci U S A 2010; 107:10502 - 7; http://dx.doi.org/10.1073/pnas.0914168107; PMID: 20498061
- Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res 2012; 18:370 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-11-1282; PMID: 22080440
- Han C, Sun B, Wang W, Cai W, Lou D, Sun Y, Zhao X. Overexpression of microtubule-associated protein-1 light chain 3 is associated with melanoma metastasis and vasculogenic mimicry. Tohoku J Exp Med 2011; 223:243 - 51; http://dx.doi.org/10.1620/tjem.223.243; PMID: 21415575
- Sato K, Tsuchihara K, Fujii S, Sugiyama M, Goya T, Atomi Y, Ueno T, Ochiai A, Esumi H. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res 2007; 67:9677 - 84; http://dx.doi.org/10.1158/0008-5472.CAN-07-1462; PMID: 17942897
- Yoshioka A, Miyata H, Doki Y, Yamasaki M, Sohma I, Gotoh K, Takiguchi S, Fujiwara Y, Uchiyama Y, Monden M. LC3, an autophagosome marker, is highly expressed in gastrointestinal cancers. Int J Oncol 2008; 33:461 - 8; PMID: 18695874
- Fujii S, Mitsunaga S, Yamazaki M, Hasebe T, Ishii G, Kojima M, Kinoshita T, Ueno T, Esumi H, Ochiai A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci 2008; 99:1813 - 9; PMID: 18616529
- Rolland P, Madjd Z, Durrant L, Ellis IO, Layfield R, Spendlove I. The ubiquitin-binding protein p62 is expressed in breast cancers showing features of aggressive disease. Endocr Relat Cancer 2007; 14:73 - 80; http://dx.doi.org/10.1677/erc.1.01312; PMID: 17395976
- Jo YK, Kim SC, Park IJ, Park SJ, Jin DH, Hong SW, Cho DH, Kim JC. Increased expression of ATG10 in colorectal cancer is associated with lymphovascular invasion and lymph node metastasis. PLoS One 2012; 7:e52705; http://dx.doi.org/10.1371/journal.pone.0052705; PMID: 23285162
- Chi C, Zhu H, Han M, Zhuang Y, Wu X, Xu T. Disruption of lysosome function promotes tumor growth and metastasis in Drosophila. J Biol Chem 2010; 285:21817 - 23; http://dx.doi.org/10.1074/jbc.M110.131714; PMID: 20418542
- Tu C, Ortega-Cava CF, Chen G, Fernandes ND, Cavallo-Medved D, Sloane BF, Band V, Band H. Lysosomal cathepsin B participates in the podosome-mediated extracellular matrix degradation and invasion via secreted lysosomes in v-Src fibroblasts. Cancer Res 2008; 68:9147 - 56; http://dx.doi.org/10.1158/0008-5472.CAN-07-5127; PMID: 19010886
- Taddei TH, Kacena KA, Yang M, Yang R, Malhotra A, Boxer M, Aleck KA, Rennert G, Pastores GM, Mistry PK. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am J Hematol 2009; 84:208 - 14; http://dx.doi.org/10.1002/ajh.21362; PMID: 19260119
- Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada GH, Bourgoin S, Casanova J, Wildeman A, Bechoua S, Ausiello DA, et al. V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat Cell Biol 2006; 8:124 - 36; http://dx.doi.org/10.1038/ncb1348; PMID: 16415858
- Toyomura T, Murata Y, Yamamoto A, Oka T, Sun-Wada GH, Wada Y, Futai M. From lysosomes to the plasma membrane: localization of vacuolar-type H+ -ATPase with the a3 isoform during osteoclast differentiation. J Biol Chem 2003; 278:22023 - 30; http://dx.doi.org/10.1074/jbc.M302436200; PMID: 12672822
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010; 141:1146 - 58; http://dx.doi.org/10.1016/j.cell.2010.05.008; PMID: 20541250
- Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem 2006; 281:36303 - 16; http://dx.doi.org/10.1074/jbc.M607031200; PMID: 16963441
- Yüce O, Piekny A, Glotzer M. An ECT2-centralspindlin complex regulates the localization and function of RhoA. J Cell Biol 2005; 170:571 - 82; http://dx.doi.org/10.1083/jcb.200501097; PMID: 16103226